

Abstract
In addition to nitric oxide (NO) and prostacyclin (PGI2), the endothelium generates the endothelium-derived hyperpolarizing factor (EDHF). We set out to determine whether an EDHF-like response can be detected in wild-type (WT) and endothelial NO synthase knockout mice (eNOS −/−) mice. Vasodilator responses to endothelium-dependent agonists were determined in vivo and in vitro. In vivo, bradykinin induced a pronounced, dose-dependent decrease in mean arterial pressure (MAP) which did not differ between WT and eNOS −/− mice and was unaffected by treatment with Nω-nitro-l-arginine methyl ester and diclofenac. In the saline-perfused hindlimb of WT and eNOS −/− mice, marked Nω-nitro-l-arginine (l-NA, 300 μmol/liter)- and diclofenac-insensitive vasodilations in response to both bradykinin and acetylcholine (ACh) were observed, which were more pronounced than the agonist-induced vasodilation in the hindlimb of WT in the absence of l-NA. This endothelium-dependent, NO/PGI2-independent vasodilatation was sensitive to KCl (40 mM) and to the combination of apamin and charybdotoxin. Gap junction inhibitors (18α-glycyrrhetinic acid, octanol, heptanol) and CB-1 cannabinoid-receptor agonists (Δ9-tetrahydrocannabinol, HU210) impaired EDHF-mediated vasodilation, whereas inhibition of cytochrome P450 enzymes, soluble guanylyl cyclase, or adenosine receptors had no effect on EDHF-mediated responses. These results demonstrate that in murine resistance vessels the predominant agonist-induced endothelium-dependent vasodilation in vivo and in vitro is not mediated by NO, PGI2, or a cytochrome P450 metabolite, but by an EDHF-like principle that requires functional gap junctions.
The endothelium plays an important role in the control of vascular tone. At least three different vasodilating agents are synthesized by the endothelium upon exposure to mechanical forces, such as shear stress, or to receptor-dependent agonists, such as acetylcholine (ACh) and bradykinin. Whereas the properties of two of these vasodilators, nitric oxide (NO) and prostacyclin (PGI2), have been extensively investigated, the nature and mechanism of action of the third vasodilator, the endothelium-derived hyperpolarizing factor (EDHF), is still controversial (1–7).
The purpose of the present study was to characterize EDHF-mediated responses in vivo, in isolated arteries, and in the perfused hindlimb of the mouse. To address the hypothesis that NO might be EDHF, we also determined endothelium-dependent relaxation in endothelial NO synthase (eNOS) knockout (eNOS −/−) mice.
Materials and Methods
Mice.
Homozygous eNOS −/− mice were obtained from the colonies at Heinrich-Heine-Universität, Düsseldorf (genetic background C57BL/b6) and Beth Israel Deaconess Medical Center, Boston, via Iffa Credo, L'Arbresle, France (genetic background C57BL/b6/SV12) (8, 9). Male C57BL/b6 wild-type (WT) control mice (8–12 weeks old) were purchased from Charles River, Sulzfeld, Germany, and Iffa Credo, France. Mice were housed in conditions that conformed with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication no. 85-23). Lack of eNOS expression in eNOS −/− mice was confirmed by Western blot analysis of samples from aortic segments as described (10).
In Vivo Measurements.
Mice were anaesthetized with sodium pentobarbital (90 mg/kg, i.p., 200 μl). One carotid artery was catheterized to monitor blood pressure. Bradykinin was applied as a bolus (100 μl) through the jugular vein. Increasing doses were applied every 10 min as the transient hypotension produced by bradykinin was rapidly and fully reversible. Bradykinin rather than ACh was used, to prevent ACh-induced arrhythmias. Inhibitors of NO synthase (Nω-nitro-l-arginine methyl ester, l-NAME, 30 mg/kg) and of cyclooxygenase (diclofenac, 10 mg/kg) or saline solution (100 μl) were injected intraperitoneally at least 30 min before the application of bradykinin. Animals showing any sign of hemorrhage or animals with a systolic blood pressure lower than 60 mmHg were excluded.
Organ Bath Experiments.
Mice were killed, and arteries were dissected, cut into segments (2–3 mm), and mounted on wire triangles. Segments were placed in organ chambers, connected to force transducers, and gradually stretched to 1 g. Studies were performed in modified Krebs–Henseleit buffer (mmol/liter: NaCl 118.3; KCl 4.7; CaCl2 1.8, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, EDTA 0.026, glucose 11.1; pH 7.40 aerated with 95% O2/5% CO2). Diclofenac (10 μmol/liter) was present in all experiments. Rings were contracted with phenylephrine and relaxed with cumulative concentrations of ACh, bradykinin, or the NO donor sodium nitroprusside (SNP) in the presence or absence of the NOS inhibitor Nω-nitro-l-arginine (l-NA, 300 μmol/liter).
Perfused Hindlimb.
After sacrifice, the aorta at the thoraco-abdominal transition was prepared, a Teflon i.v. catheter (0.67 mm, 24 gauge, brand Durflo; Terumo, Leuven, Belgium) was introduced and advanced to one of the iliac arteries and tied with a 6/0 prolene stitch (Ethicon, Norderstedt, Germany). The inferior caval vein was slit open longitudinally to prevent venous congestion. A roller pump was used to constantly perfuse the hindlimb with filtered Krebs–Henseleit solution containing diclofenac (10 μmol/liter). A pressure transducer and a compliance chamber were connected to a side port of the perfusion system. Flow rate was gradually increased to achieve a perfusion pressure of approximately 120 mmHg. At this perfusion pressure a considerable amount of spontaneous myogenic tone is present, which made preconstriction unnecessary. When a substance produced a pronounced change in resistance, flow rate was adjusted to reduce perfusion pressure (e.g., K+) or phenylephrine was used to increase pressure (e.g., octanol, heptanol, 18α-glycyrrhetinic acid, HU210, Δ9-tetrahydrocannabinol). When a stable pressure plateau was reached, endothelium-dependent (ACh, bradykinin) or -independent vasodilators (SNP) were applied in increasing concentrations as a bolus (100 μl) in glucose solution (50 g/liter), in the presence or absence of inhibitors. For inhibitor studies, agonist-induced responses were recorded in the absence and then in the presence of the inhibitor to facilitate paired comparison of the results. To avoid artifacts attributable to the tachyphylaxis/desensitization of the endothelial B2 kinin receptor, these experiments were performed using ACh rather than bradykinin. Vasodilator responses were measured as changes in perfusion pressure. Agonist-induced vasodilations were always normalized to the effects elicited by SNP to ascertain the specificity of the effect observed.
The term “EDHF” used in this study refers to the KCl-sensitive, l-NA- and diclofenac-resistant component of endothelium-dependent vasodilation.
Statistics.
Values given are mean ± SEM. Relaxations/vasodilator responses were expressed as percent changes from the initial precontraction levels/perfusion pressure or the percent vasodilator responses as compared with the maximal vasodilator response induced by SNP. Statistical significance was tested by using two-way ANOVA for repeated measures followed by the Newman–Keuls test.
Results
Bradykinin-Induced Vasodilation in Vivo.
Mean arterial pressure (MAP) was significantly lower in WT than in eNOS −/− mice. Diclofenac and l-NAME induced a slight but significantly increase in MAP in WT and had no effect in eNOS −/− mice (Table 1).
Table 1.
Baseline characteristics of the anesthetized mice
| Mice | MAP, mmHg | Heart rate, BPM | n |
|---|---|---|---|
| WT | |||
| Control | 68.0 ± 2.7† | 428 ± 8† | 22 |
| l-NAME + diclofenac | 78.3 ± 3.8† | 377 ± 5*† | 28 |
| eNOS −/− | |||
| Control | 114.8 ± 13.8† | 489 ± 18† | 16 |
| l-NAME + diclofenac | 98.0 ± 10.1† | 472 ± 9† | 26 |
BPM, beats per minute.
, P < 0.05 vs. respective control;
, P < 0.05 WT vs. eNOS −/−.
Bolus applications of normal saline or bradykinin had a small negative chronotropic effect, which was never greater than 10% (data not shown). Bradykinin induced a dose-dependent transient decrease in MAP which was identical in anesthetized WT and eNOS −/− mice. Pretreatment with l-NAME and diclofenac had no effect on the depressor response to bradykinin in either strain (Fig. 1).
Figure 1.
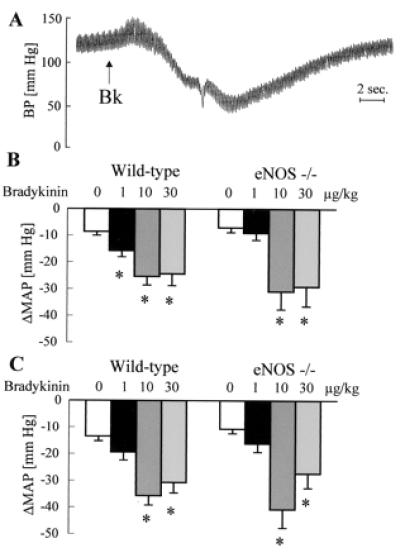
Effect of bradykinin on arterial blood pressure in anesthetized mice. (A) Original tracing showing the effect of a bolus application of bradykinin (Bk; 10 μg/kg, i.v.) on blood pressure (BP) in a diclofenac- (10 mg/kg, i.p.) and l-NAME- (30 mg/kg, i.p.) treated mouse. (B and C) Effect of bolus application of saline or bradykinin (doses indicated above the bars) on MAP in the absence (B) or in the presence (C) of diclofenac and l-NAME; n ≥ 5 each group; ∗, P < 0.05.
Endothelium-Dependent Relaxation in Conduit Arteries.
In the presence of diclofenac, but in the absence of l-NA, endothelium-dependent relaxation in response to ACh was observed in arterial ring preparations from WT mice (maximal relaxation to ACh: aorta 81% ± 2%, carotid 85% ± 6%, and femoral artery 85% ± 4%; n ≥ 5). l-NA completely inhibited these ACh-induced relaxations.
No relaxation in response to ACh was observed in rings from vessels derived from eNOS −/− mice (see Fig. 3B). Bradykinin also failed to induce any relaxation in conduit arteries from either WT or eNOS −/− mice, even in the absence of l-NA, suggesting that the bradykinin receptor is not functionally expressed in these vessels (data not shown).
Figure 3.
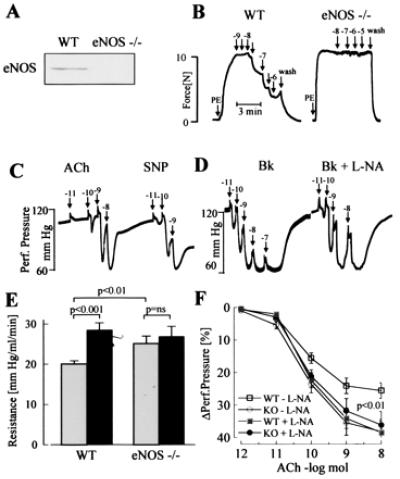
Effects of ACh, SNP, and l-NA on vasodilator responses of eNOS −/− mice. (A) Expression of eNOS in mouse aortic rings from WT and eNOS −/− mice as detected by Western blot analysis. (B) Original tracings showing relaxations of isolated aortic rings from WT and eNOS −/− mice. After constriction with phenylephrine (PE), cumulative dose–response curves to ACh were obtained. Numbers above the arrows indicate the final concentration of the agonist (log mol/liter). Tracings are representative of data obtained in six additional experiments. (C and D) Original tracings showing changes in hindlimb perfusion pressure in eNOS −/− mice upon bolus application of ACh or SNP (C) or bradykinin (Bk) (D) in the presence of diclofenac and presence or absence of l-NA. Numbers above the arrows indicate the dose of the agonist applied as a bolus (log mol). (E) Peripheral resistance of the perfused hindlimb of WT and eNOS −/− mice in the absence (shaded columns) and presence (filled columns) of l-NA. ns, Not significant. (F) Vasodilator responses to bolus applications (100 μl) of ACh in the perfused hindlimb of WT (□,■) and eNOS −/− mice (○,●) in the absence (open symbols) and presence (filled symbols) of l-NA (n ≥ 8).
Endothelium-Dependent Dilation in the Perfused Hindlimb.
In WT animals, in the presence of diclofenac but in the absence of l-NA, ACh (Fig. 2) as well as bradykinin (maximal relaxation: 81.6% ± 5%), induced a dose-dependent vasodilator response in the hindlimb. Agonist-induced vasodilator responses were preserved after perfusion with l-NA (applied for at least 30 min) (ACh: Fig. 2, bradykinin maximal relaxation 116% ± 6%). Responses to ACh as well as to SNP were increased by l-NA, as was perfusion pressure and peripheral resistance. A reduction in perfusion rate, to obtain identical levels of perfusion pressure, had no effect on peripheral resistance or on vasodilation elicited by ACh or SNP (Fig. 2). Treatment with Triton X-100 (three bolus injections 100 μl, 0.1%) to destroy the endothelium markedly attenuated vasodilation in response to ACh (remaining response: 21.0% ± 3.7%, n = 5), but had little effect on SNP-induced vasodilation (81.0% ± 13.6%, n = 5), indicating that the effect of ACh was endothelium dependent.
Figure 2.
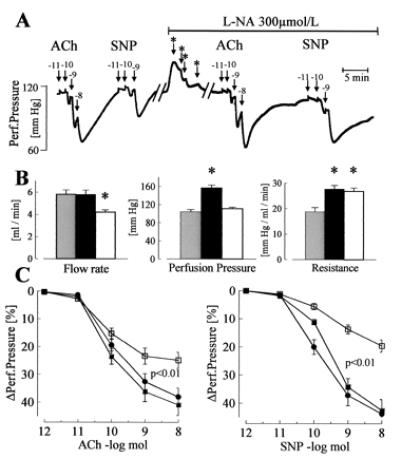
Dilator responses to endothelium-dependent and -independent agonists in the isolated perfused hindlimb of WT mice. (A) Original tracing showing the effect of bolus applications (100 μl) of ACh or SNP on perfusion pressure in the absence or presence of l-NA (300 μmol/liter). Numbers above the arrows indicate the dose of the agonist applied with the bolus (log mol); ∗ indicates a reduction in flow rate to maintain the perfusion pressure at a constant level. (B) Parameters of the perfused hindlimb in WT mice under basal conditions (shaded columns), during application of l-NA (filled columns), and after reduction of flow rate to adjust perfusion pressure during l-NA application (open columns). (C) Vasodilator responses to bolus applications (100 μl) of ACh and SNP in WT mice under control conditions (□), during the infusion of l-NA (■), and during infusion of l-NA and flow rate adjustment (●); n = 8 each group; ∗, P < 0.05. All experiments were performed in the presence of diclofenac (10 μmol/liter).
In the hindlimb of eNOS −/− mice, vasodilation in response to ACh and bradykinin was observed in the presence of diclofenac and was unaffected by l-NA. In contrast to WT mice, l-NA infusion did not increase vascular resistance in eNOS −/− mice (change in perfusion pressure by l-NA: WT mice, 43% ± 7%; eNOS −/− mice, 2.3% ± 1.7%; P < 0.001; n > 5). ACh-induced vasodilation and peripheral resistance were identical in eNOS −/− and WT mice treated with l-NA (Fig. 3).
Characterization of the l-NA-Resistant Vasodilation in the Perfused Hindlimb of WT Mice.
Increasing the K+ concentration in the perfusate to 40 mmol/liter completely prevented endothelium-dependent vasodilator responses. The combination of the K+ channel blockers charybdotoxin (100 nmol/liter) and apamin (100 nmol/liter) also inhibited ACh-induced relaxation, whereas charybdotoxin alone only slightly impaired it (Fig. 4A). Apamin by itself, the K+ channel blocker iberiotoxin (100 nmol/liter), or the combination of apamin, iberiotoxin, glibenclamide (1 μmol/liter), and 4-aminopyridine (5 mmol/liter) did not significantly inhibit the vasodilation elicited by ACh. When experiments were performed in the absence of l-NA, ACh-induced vasodilation was largely unaffected by K+ or inhibition of K+ channels with charybdotoxin. Only the combination of charybdotoxin and apamin had a small, but significant, inhibitory effect (data not shown). The ACh-induced l-NA- and diclofenac-resistant vasodilation in the hindlimb was not affected by the adenosine antagonist 8-cyclopentyl-1,3-dipropylxanthine (1 μmol/liter, data not shown) (11), the CYP inhibitors 17-octadecynoic acid (10 μmol/liter), miconazole (10 μmol/liter), and sulfaphenazole (10 μmol/liter), or a combination of phospholipase A2 and D inhibitors (Fig. 4B).
Figure 4.
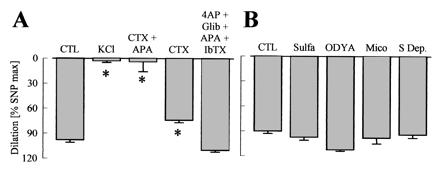
Effects of K+ channel blockade and cytochrome P450 (CYP) inhibition on ACh-induced vasodilator responses in the hindlimb of WT mice. Experiments were performed in presence of diclofenac (10 μmol/liter) and l-NA (300 μmol/liter). (A) CTL = control, potassium channel blockade with charybdotoxin (CTX, 100 nmol/liter), apamin (APA, 100 nmol/liter), 4-aminopyridine, (4AP, 5 mmol/liter), glibenclamide, (Glib, 1 μmol/liter), and iberiotoxin (IbTX, 100 nmol/liter). (B) Inhibition of CYP enzymes with 17-octadecynoic acid (ODYA, 10 μmol/liter), miconazole (Mico, 10 μmol/liter), and sulfaphenazole (Sulfa, 10 μmol/liter) or of arachidonic acid release (S Dep, substrate depletion) by using the combination of the phospholipase A2 inhibitors arachidonyl trifluoromethyl ketone (AACOCF3, 3 μmol/liter) and ONO-RS-082 (10 μmol/liter) with the diacylglycerol-lipase inhibitor THC-80267 (10 μmol/liter). ∗, P < 0.05; n ≥ 4.
In the presence of l-NA, inhibition of the soluble guanylyl cyclase with 1H-oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; 1 μmol/liter) (12) or mesoporphyrin IX (10 μmol/liter) (13) attenuated ACh-, cromakalim-, and nifedipine-induced vasodilation, all to the same extent. SNP-induced vasodilation was nearly abolished by the inhibitors. Catalase (400 units/ml), which inactivates the soluble guanylyl cyclase activator hydrogen peroxide (H2O2), and tin as well as zinc protoporphyrin IX (10 μmol/liter), which inactivate heme oxygenase (14), did not influence the responses to ACh (data not shown).
In the presence but not absence of l-NA, 18α-glycyrrhetinic acid (aGA, 30 μmol/liter), an inhibitor of gap junctional communication (15), inhibited vasodilation by more than 50%, whereas aGA had no effect in the absence of l-NA (Fig. 5). The unspecific gap junction inhibitors heptanol (1 mmol/liter) and octanol (1 mmol/liter) also markedly impaired vasodilation in the presence of l-NA (data not shown).
Figure 5.
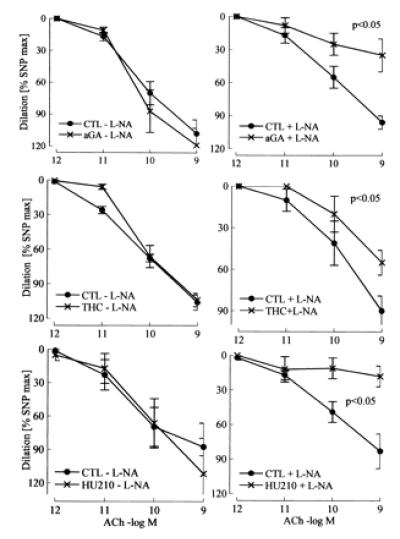
Effect of gap junction uncouplers and CB1 cannabinoid receptor agonists on ACh-induced vasodilator responses in the hindlimb of wild-type mice. Effects of the gap junction uncoupler 18α-glycyrrhetinic acid (aGA; 30 μmol/liter) and the CB1-receptor agonists Δ9-tetrahydrocannabinol (THC; 3 μmol/liter) and HU210 (10 μmol/liter) in the presence (+) or absence (−) of l-NA (300 μmol/liter). Diclofenac (10 μmol/liter) was continuously present during the experiment. CTL, control. n ≥ 5.
The nonselective CB receptor agonist Δ9-tetrahydrocannabinol (3 μmol/liter) and the selective CB1 receptor agonist HU210 (10 μmol/liter) both attenuated endothelium-mediated vasodilator response in the presence but not absence of l-NA (Fig. 5).
Identical results were obtained when eNOS −/− mice were used.
Discussion
In the present study we have demonstrated, both in vivo and in vitro, a pronounced agonist-induced l-NA- and diclofenac-resistant, endothelium-dependent vasodilation in resistance arteries of WT and eNOS −/− mice. This NO- and PGI2-independent relaxation was inhibited by depolarizing concentrations of K+ and was sensitive to K+ channel blockers, suggesting that these responses could be attributed to EDHF but not to endothelium-derived NO (4, 16).
Endothelium-dependent relaxations and vasodilations were observed in isolated conduit arteries and in the perfused hindlimb. However, whereas diclofenac- and l-NA-resistant dilator responses were detected in the perfused hindlimb, this was not the case in conduit arteries. The latter finding is consistent with a previous report that agonist-induced endothelium-dependent hyperpolarization is absent in murine conduit arteries (17). The most likely explanation for these observations is that EDHF is a more prominent vasodilator in smaller vessels (3). Indeed, EDHF-like dilations could be demonstrated only in perfused vascular preparations in the present study. This may simply reflect the fact that EDHF responses observed in different vascular beds from the same species or between species are highly variable (18). Alternatively, fluid shear stress may facilitate agonist-induced EDHF production in mice.
Endothelium-dependent relaxations resistant to l-NA have been linked with hyperpolarization on the basis of the observation that both phenomena are inhibited by K+ channel blockers and depolarizing concentrations of K+ (4, 16). Although hyperpolarization was not measured in the present study, our data strongly suggest that an EDHF mediates l-NA-resistant endothelium-dependent vasodilation in the mouse. Indeed, the sensitivity of the l-NA- and diclofenac-resistant vasodilation to the combination of charybdotoxin and apamin is a characteristic of EDHF-mediated responses (7, 19).
The major drawback of studies aiming to determine the physiological relevance of EDHF is that the contribution of this factor to endothelium-dependent vasodilation can be estimated only as the l-NA-and diclofenac-resistant portion of relaxation. This approach is problematic in vivo and in intact organs because of the pronounced vasopressor effect of NOS inhibitors (20, 21), and it relies on the complete suppression of NOS activity by the inhibitor used, which may not always be the case (22). In certain situations, high concentrations of NO can activate large conductance Ca2+-activated K+ channels either directly (23), or by a cGMP-dependent mechanism (24). In the present study we can completely exclude the participation of endothelium-derived NO in the EDHF-like dilation because identical results were obtained in WT and eNOS −/− mice. Furthermore, as the hypotension and hindlimb vasodilations were still observed in the presence of a nonspecific NOS inhibitor, a compensatory role of neuronal NO, as suggested in the cerebral circulation of eNOS −/− mice (25, 26) is unlikely to account for our observations. Finally, the hindlimb vasodilation was highly sensitive to the combination of charybdotoxin and apamin, but not to iberiotoxin, ruling out the involvement of large conductance K+ channels (27) and their potential activation by residual NO. The involvement of guanylyl cyclase and heme oxygenase in EDHF-mediated vasodilation has been reported in porcine pulmonary arteries (14). In the present study, inhibition of these enzymes had no effect on the non-NO/PGI2-mediated vasodilation.
It has been proposed that EDHF may serve as a backup vasodilator in situations associated with an altered bioavailability of endothelium-derived NO (28, 29). In the present study, l-NA induced a marked increase in perfusion pressure in the hindlimb of WT but not eNOS −/− mice, highlighting the important role of basal shear stress-induced NO production in the regulation of vascular tone and resistance. Indeed, the resistance of the l-NA-perfused hindlimb in eNOS −/− and WT mice was not different. Although a recent in vitro study suggested that PGI2 may compensate for the lack of basal NO release (30), our data indicate that such a mechanism is not sufficient to adjust resistance to a normal level in eNOS −/− mice in vivo. A role for PGI2 in hindlimb vasodilation having been excluded by the inclusion of diclofenac, the similarity in the resistance of eNOS −/− and WT mice treated with l-NA implies that, under the experimental conditions (nonpulsatile flow) used, EDHF is also not able to compensate for NO as a modulator of basal vascular tone. In addition, our observation that the combination of charybdotoxin and apamin induced only a small increase in resistance in WT mice argues against a significant basal release of EDHF in the present model.
On the other hand, EDHF appears to be at least as important as endothelium-derived NO in mediating agonist-induced vasodilation in the mouse, as indicated by the failure of either eNOS gene deletion or eNOS inhibition to attenuate agonist-induced vasodilator response in vivo or in vitro. In the perfused hindlimb, only the inhibition of both NO and EDHF impaired vasodilator responses to ACh, suggesting that in this vascular bed, EDHF and endothelium-derived NO can completely compensate for the lack of each other. Given such a situation, no difference in EDHF-mediated responses between acute inhibition of eNOS and eNOS knock-out would be expected, and indeed this was the case in the present study.
Both acute and chronic NO withdrawal enhanced, rather than impaired, ACh-induced hindlimb vasodilator response in the present study. When vasodilation was elicited by SNP, this difference was even more pronounced. Such an effect may reflect that, in the perfused hindlimb, NO continuously released from the endothelium induces a sustained reduction in vascular tone, thereby shifting the dose–response curve for the applied vasodilator to the right. Just such a phenomenon has previously been demonstrated in the rat aorta (31). Indeed, a constant infusion of high concentrations of SNP (100 nmol/liter) was required to antagonize the increase in perfusion pressure induced by l-NA in WT mice. This procedure also prevented the l-NA-induced increase in ACh-induced vasodilator response (R.P.B., unpublished observations).
We have previously reported that EDHF is a CYP-epoxygenase metabolite in porcine coronary arteries (32). In the present study, we found no evidence to suggest that a CYP metabolite of arachidonic acid is an EDHF in the mouse hindlimb. The only clear inhibition of EDHF-mediated vasodilator responses was observed with gap junctional inhibitors, which are reported to inhibit EDHF-mediated relaxation of the rabbit mesenteric artery (33, 34) It is interesting to note that gap junction uncouplers have almost no effect on EDHF-mediated relaxation in porcine coronary arteries (I.F., R.P.B., and R.B., unpublished observation). This observation implies that two distinct EDHFs account for the l-NA- and diclofenac-resistant endothelium-dependent vasodilation in mouse resistance vessels and porcine coronary arteries. Very recently, evidence for the presence of a CYP-derived EDHF has been presented in segments of the isolated mouse gracilis vessels in WT and eNOS −/− mice (35). It is currently unclear whether the specific genetic background of the animals studied, the lack of shear stress, the method of application of endothelial agonists (infusion vs. bolus), or the experimental environment used (isolated vessels vs. in situ preparation) underlies the somewhat contradictory results obtained. The fact that not only CYP inhibitors but also iberiotoxin completely inhibited the EDHF-type relaxation in the study by Huang et al. (35), however, indicates that two profoundly different EDHFs may be generated within the mouse vasculature.
In addition to the concept of a CYP-derived EDHF, it has been suggested that potassium might be an EDHF in some vessels (36). Because ouabain and barium, the inhibitors used in this study (36), exert severe side effects on myogenic tone (R.P.B., unpublished observation) we could not address this hypothesis in the perfused hindlimb.
Apart from gap junction uncouplers, the l-NA- and diclofenac-resistant portion of vasodilation in the present study was sensitive to CB1 receptor agonists. Such compounds impair EDHF-mediated relaxations in rabbit arteries without affecting Ca2+-signaling (37). Anandamide, another nonselective CB1 receptor agonist, which inhibits EDHF-mediated relaxation (37), also inhibits gap junctional communication and intercellular coupling in astrocytes (38). These findings raise the possibility that CB1 agonists interfere with the signal transduction cascade leading to the synthesis and release of EDHF and/or the propagation of hyperpolarization via gap junctions (37).
In summary, our study does not support a role for CYP enzymes, soluble guanylyl cyclase, or eNOS in the agonist-induced EDHF-mediated dilation in the mouse hindlimb, but it highlights a role for functional gap junctions and calcium-dependent K+ channels. Moreover, we have demonstrated that endothelial stimulation induces the release of an EDHF that is a potent endogenous vasodilator of murine resistance vessels, both in vivo and in vitro. This EDHF is able to completely compensate for the lack of NO in l-NA-treated WT or eNOS −/− mice.
Acknowledgments
The study was supported by a grant from the ADUMED-Stiftung and a young investigators grant from the Klinikum der Johann Wolfgang Goethe-Universität to R.P.B., as well as the Sonderforschungsbereich SFB 553, TP B1.
Abbreviations
- ACh
acetylcholine
- PGI2
prostacyclin
- EDHF
endothelium-derived hyperpolarizing factor
- eNOS
endothelial NO synthase
- WT
wild type
- l-NAME
Nω-nitro-l-arginine methyl ester
- SNP
sodium nitroprusside
- l-NA
Nω-nitro-l-arginine
- MAP
mean arterial pressure
- CYP
cytochrome P450
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Furchgott R F, Vanhoutte P M. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 2.Furchgott R F. Biosci Rep. 1999;19:235–251. doi: 10.1023/a:1020537506008. [DOI] [PubMed] [Google Scholar]
- 3.Quilley J, Fulton D, McGiff J C. Biochem Pharmacol. 1997;54:1059–1170. doi: 10.1016/s0006-2952(97)00039-7. [DOI] [PubMed] [Google Scholar]
- 4.Cohen R A, Vanhoutte P M. Circulation. 1995;92:3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- 5.Edwards G, Weston A H. Prog Drug Res. 1998;50:107–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- 6.Campbell W B, Harder D R. Circ Res. 1999;84:484–488. doi: 10.1161/01.res.84.4.484. [DOI] [PubMed] [Google Scholar]
- 7.Waldron G J, Cole W C. Clin Exp Pharmacol Physiol. 1999;26:180–184. doi: 10.1046/j.1440-1681.1999.03006.x. [DOI] [PubMed] [Google Scholar]
- 8.Gödecke A, Decking U K M, Ding Z, Hirchenhain J, Bidmon H, Gödecke S, Schrader J. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 9.Huang P L, Huang Z, Mashimo H, Block K D, Moskowitz M A, Becan J A, Fishman M C. Nature (London) 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 10.Bouloumie A, Bauersachs J, Linz W, Schölkens B A, Wiemer G, Fleming I, Busse R. Hypertension. 1997;30:934–941. doi: 10.1161/01.hyp.30.4.934. [DOI] [PubMed] [Google Scholar]
- 11.Bruns R F, Fergus J F, Badger E W, Bristol J A, Santay L A, Hartman J D, Hays S J, Huang C C. Naunyn-Schmiedebergs Arch Pharmacol. 1987;335:59–63. doi: 10.1007/BF00165037. [DOI] [PubMed] [Google Scholar]
- 12.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- 13.Ignarro L J, Ballot B, Wood K S. J Biol Chem. 1984;259:6201–6207. [PubMed] [Google Scholar]
- 14.Zakhary R, Gaine S P, Dinerman J L, Ruat M, Flavahan N A, Snyder S H. Proc Natl Acad Sci USA. 1996;93:795–798. doi: 10.1073/pnas.93.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davidson J S, Baumgarten I M, Harley E H. Biochem Biophys Res Commun. 1986;134:29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- 16.Félétou M, Vanhoutte P M. Drug News Perspect. 1999;12:217–222. [Google Scholar]
- 17.Chataigneau T, Félétou M, Huang P L, Fishman M C, Duhault J, Vanhoutte P M. Br J Pharmacol. 1999;126:219–226. doi: 10.1038/sj.bjp.0702300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Triggle C R, Dong H, Waldron G J, Cole W C. Clin Exp Pharmacol Physiol. 1999;26:176–179. doi: 10.1046/j.1440-1681.1999.03007.x. [DOI] [PubMed] [Google Scholar]
- 19.Corriu C, Félétou M, Canet E, Vanhoutte P M. Br J Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amezcua J L, Palmer R M J, Souza B M, Moncada S. Br J Pharmacol. 1989;97:1119–1124. doi: 10.1111/j.1476-5381.1989.tb12569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamontage D, Pohl U, Busse R. Pflügers Arch. 1991;418:255–270. doi: 10.1007/BF00370525. [DOI] [PubMed] [Google Scholar]
- 22.Cohen R A, Plane F, Najibi S, Huk I, Malinski T, Garland C J. Proc Natl Acad Sci USA. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolotina V M, Najibi S, Palacino J J, Pagano P J, Cohen R A. Nature (London) 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 24.Archer S L, Huang J M C, Hampl V, Nelson D P, Shultz P J, Weir E K. Proc Natl Acad Sci USA. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng W, Ma J, Ayata C, Hara H, Huang P L, Fishman M C, Moskowitz M A. Am J Physiol. 1996;271:H1145–H1150. doi: 10.1152/ajpheart.1996.271.3.H1145. [DOI] [PubMed] [Google Scholar]
- 26.Meng W, Ayata C, Waeber C, Huang P L, Moskowitz M A. Am J Physiol. 1998;274:H411–H415. doi: 10.1152/ajpheart.1998.274.2.H411. [DOI] [PubMed] [Google Scholar]
- 27.Chataigneau T, Félétou M, Duhault J, Vanhoutte P M. Br J Pharmacol. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- 29.McCulloch A I, Bottrill F E, Randall M D, Hiley C R. Br J Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun D, Huang A, Smith C J, Stackpole C J, Connetta J A, Shesely E G, Koller A, Kaley G. Circ Res. 1999;85:288–293. doi: 10.1161/01.res.85.3.288. [DOI] [PubMed] [Google Scholar]
- 31.Jackson W F, Busse R. Naunyn-Schmiedebergs Arch Pharmacol. 1991;344:345–350. doi: 10.1007/BF00183010. [DOI] [PubMed] [Google Scholar]
- 32.Fisslthaler B, Popp R, Kiss L, Potente M, Harder D R, Fleming I, Busse R. Nature (London) 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 33.Taylor H J, Chaytor A T, Evan W H, Griffith T M. Br J Pharmacol. 1998;125:1–3. doi: 10.1038/sj.bjp.0702078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaytor A T, Evans W H, Griffith T M. J Physiol (London) 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang A, Sun D, Smith C J, Connetta J A, Shesely E G, Koller A, Kaley G. Am J Physiol. 2000;278:H762–H768. doi: 10.1152/ajpheart.2000.278.3.H762. [DOI] [PubMed] [Google Scholar]
- 36.Edwards G, Dora K A, Gardener M J, Garland C J, Weston A H. Nature (London) 1998;396:269–216. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 37.Fleming I, Schermer B, Popp R, Busse R. Br J Pharmacol. 1999;126:949–960. doi: 10.1038/sj.bjp.0702381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venance L, Piomelli D, Glowinski J, Giaume C. Nature (London) 1995;376:590–594. doi: 10.1038/376590a0. [DOI] [PubMed] [Google Scholar]