

Abstract
A major feature of septic shock is the development of a vascular crisis characterized by nonresponsiveness to sympathetic vasoconstrictor agents and the subsequent irreversible fall in blood pressure. In addition, sepsis, like other inflammatory conditions, results in a large increase in the production of free radicals, including superoxide anions (O2⨪) within the body. Here we show that O2⨪ reacts with catecholamines deactivating them in vitro. Moreover, this deactivation would appear to account for the hyporeactivity to exogenous catecholamines observed in sepsis, because administration of a superoxide dismutase (SOD) mimetic to a rat model of septic shock to remove excess O2⨪ restored the vasopressor responses to norepinephrine. This treatment with the SOD mimetic also reversed the hypotension in these animals; suggesting that deactivation of endogenous norepinephrine by O2⨪ contributes significantly to this aspect of the vascular crisis. Indeed, the plasma concentrations of both norepinephrine and epinephrine in septic rats treated with the SOD mimetic were significantly higher than in untreated rats. Interestingly, the plasma concentrations for norepinephrine and epinephrine were inversely related to the plasma concentrations of adrenochromes, the product of the autoxidation of catecholamines initiated by O2⨪. We propose, therefore, that the use of a SOD mimetic represents a new paradigm for the treatment of septic shock. By removing O2⨪, exogenous and endogenous catecholamines are protected from autoxidation. As a result, both hyporeactivity and hypotension are reversed, generation of potentially toxic adrenochromes is reduced, and survival rate is improved.
Despite significant advances in the treatment of shock, the mortality rate is still very high. Persistent systemic vasodilation resulting in multiple organ failure is a frequent cause of death among patients who succumb to endotoxic shock (1–4). Diverse molecular mechanisms of inflammation and cellular damage have been implicated in the pathogenesis of septic shock and multiple organ failure (5–7), including those related to overt generation of cytokines, eicosanoids, and reactive oxygen species (ROS), such as nitric oxide (NO), superoxide anions (O2⨪) and peroxynitrite (ONOO−, the product formed from the interaction between NO and O2⨪). To date, inhibitors of the synthesis/release or actions of cytokines, eicosanoids, and NO have not been shown to have beneficial effects on shock or mortality in randomized controlled clinical trials (8). In retrospect, these results are not surprising in light of the fact that these mediators possess both proinflammatory and anti-inflammatory properties. Thus, although individual mediators of each of these structural classes have been causally implicated in septic shock, each plays important homeostatic functions in the complex host response to the septic challenge. Accordingly, their utility as appropriate therapeutic targets for immunopharmacologic suppression has been questioned (8). Based on our own data as well as results reported in the literature, we believe that O2⨪ plays a key role in the pathogenesis of hemodynamic instability and organ dysfunction during septic shock (9–14). O2⨪ is primarily produced by activated neutrophils and macrophages and has been associated with the inflammatory response that accompanies tissue damage in septic shock (15–17). Under normal conditions, the formation of O2⨪ is kept under tight control by endogenous superoxide dismutase (SOD) enzymes. These enzymes include the manganese enzyme in mitochondria, SOD2, and copper/zinc enzymes present in the cytosol, SOD1, or extracellular surfaces, SOD3. The importance of SOD2 is highlighted by the findings that, in contrast to SOD1 (18) and SOD3 (19), SOD2 knockout is lethal to mice (20–22). In disease states, the production of O2⨪ is increased at a rate that overwhelms the capacity of the endogenous SOD defense system, resulting in O2⨪-mediated damage. The proinflammatory properties of O2⨪ include endothelial cell damage and increased microvascular permeability (23–25), formation of chemotactic factors such as leukotriene B4 (26, 27), recruitment of neutrophils at sites of inflammation (28, 29), lipid peroxidation and oxidation, DNA single strand damage (30), release of cytokines such as TNF-α and IL-1β (31, 32), and formation of ONOO−, a potent cytotoxic and proinflammatory molecule (33–37). One of the most important features of shock that ultimately determines survival is the reversibility of inadequate organ perfusion secondary to loss of vasomotor tone, which in turn leads to reduced venous return, reduced cardiac output, and severe arterial hypotension (5). To overcome such hemodynamic derangements, standard treatment consists of prompt initiation of antibiotics, whereas hemodynamic abnormalities are addressed by i.v. fluid resuscitation and exogenously administered catecholamines, such as dopamine (DA) and especially norepinephrine (NE) to preserve or augment blood flow to vital organs (e.g., brain, heart, liver, and kidney). Despite such aggressive therapy, successful outcomes are limited because of the development of vascular hyporeactivity (i.e., the loss of normal vasoconstrictor responses) to exogenously administered DA and NE. This hyporeactivity hampers the ability of the clinician to sustain blood pressure, as exhibited in non-survivors of septic shock, in which blood pressure continues to drop despite administration of progressively larger doses of DA and NE. This inability to restore and maintain an appropriate blood pressure leads to severe hypoperfusion of critical organs and eventually death. Therefore, if development of sepsis-related vascular hyporeactivity to NE could be overcome, then the therapeutic administration of NE would effectively maintain blood pressure, which, in turn, would attenuate the severity of hypotension.
It is well established that O2⨪ autoxidizes catecholamines, including DA, NE, and epinephrine (Epi), to their respective dopachrome and adrenochrome products (38–42). This reactivity has been used in a well-documented assay to assess superoxide activity in which the linear production of adrenochrome of a catecholamine, such as epinephrine, is inhibited by SOD (43–46). Our hypothesis is that this autoxidation of catecholamines by O2⨪ deactivates them and this deactivation accounts for the loss of vasoconstrictor tone to NE in septic shock as well as the associated hypotensive response. This hypothesis was tested in the current study, by using Escherichia coli lipopolysaccharide (LPS)-induced shock in rats. To manipulate the levels of O2⨪ we used M40403, a recently described selective SOD mimetic (32).
M40403 is a stable low molecular weight, manganese-containing, nonpeptidic molecule with a catalytic rate constant of 2 × 107 M−1⋅s−1 at pH 7.0. It has the advantage over native manganese containing SOD of being a much smaller molecule (MW 483 vs. MW 30,000 for the mimetic and native enzyme, respectively; ref. 32). M40403 is cytoprotective and anti-inflammatory (32). In contrast to the native enzymes, these SOD mimetics are not deactivated by hydrogen peroxide or ONOO− (47). Furthermore, an important and unique property of M40403 and other structurally related mimetics is that they selectively remove O2⨪ at a high rate without interacting with other reactive oxygen species including NO, hydrogen peroxide, or O2⨪ (32, 47). In animal models of septic shock, the development of hyporeactivity is not unique for catecholamines but is seen in response to numerous vasoconstrictors, including endothelin, angiotensin II, and thromboxane A2 (48). However, these vasoconstrictors are not used clinically in an attempt to prevent hypotension.
The key findings of our work are that O2⨪ interacts with catecholamines to destroy their biological activity, resulting in loss of clinical vasoconstrictor responses (refractory hypotension) and hypotension. We propose, therefore, that the therapeutic use of NE in septic shock is severely limited in that, although being one of the most commonly used vasopressors, its vasoconstrictor activity is broken down and deactivated by O2⨪ after infusion. This deactivation of catecholamines by O2⨪ may also have broader implications in other disease states in which catecholamines and free radicals play a role.
Materials and Methods
Materials.
The composition of the Krebs' buffer was as follows: 119 mM NaCl/2.5 mM KCl/1.3 mM MgSO4/2.5 mM CaCl2/1.0 mM NaH2PO4/26.2 mM NaHCO3/10 mM Hepes (pH 7.4). DA, NE, Epi, hypoxanthine (HX), xanthine oxidase (XO), and LPS were all obtained from Sigma. The SOD mimetic, M40403, was synthesized as reported previously (32).
Catecholamine Studies.
The catecholamines NE, Epi, and DA were incubated in test tubes with Krebs' buffer containing HX (2 mM)/XO (1 unit/ml) for the generation of O2⨪. This system results in the generation of O2⨪ in the ratio of 2 molecules of O2⨪ to every 1 molecule of HX used (44). At the conclusion of the experiment the incubates were assayed for catecholamine content by HPLC with electrochemical detection using Varian star workstation software as described in detail below.
Anesthetized Rat Model: Surgical Procedure.
Male Sprague–Dawley rats (250–300 g) were anesthetized with inactin (100 mg/kg i.p.). The trachea was cannulated to facilitate respiration, and body temperature was maintained at 37°C by means of a heating pad. The left femoral vein was cannulated for administration of drugs. The left femoral artery was cannulated and connected to a pressure transducer to allow for the monitoring of blood pressure. LPS from E. coli (4 mg/kg, serotype 0111:B4) was administered as a bolus i.v. injection at a volume of 0.3 ml. Control animals received saline at the same volume and by the same route. In experiments involving blood samples, these were withdrawn from the arterial cannula.
Drug Administration.
Drugs were dissolved in isotonic saline. In all experiments LPS was given as a 0.3-ml bolus i.v. injection at T0. M40403 was given at T1 or T5 as an infusion i.v. at the rate of 1 ml/h. Control animals received the respective vehicle at the same volume and by the same route (i.v.). Where used, NE was given as a 0.3-ml bolus i.v. injection. Surviving animals were killed at 9 h after the administration of LPS as dictated by the animal experimental protocol.
Catecholamine Measurements.
Catecholamines in test tube samples or plasma samples were identified and quantified by HPLC with electrochemical detection (HPLC-EC) as previously published (49). The system consists of a Varian model 2510 solvent delivery system and a model 9090 autosampler (Varian) coupled to a C18 column and an ESA (Bedford, MA) Coulochem II detector. Separations were performed isocratically by using a filtered and degassed mobile phase consisting of 10% methanol/0.1 M sodium phosphate/0.2 mM sodium octyl sulfate/0.1 mM EDTA, adjusted to pH 2.8 with phosphoric acid. The HPLC system is coupled to a Pentium computer with which chromatograms were recorded and analyzed with Varian star workstation software.
Adrenochrome Measurements.
The detection and quantification of the sum of the noradrenochrome and adrenochrome was carried out by using an HPLC method using a Vydac (Hesperia, CA) C18 pharmaceutical 4.6 × 250 mm column and with a 5% acetonitrile + 95% SDS (10 mM) mobile phase (5 min elution), then 40% acetonitrile with 60% SDS plus 0.1% trifluoroacetic acid (TFA) (5 min elution) mobile phase, all eluted at 1 ml/min. Detection of the adrenochromes utilizes the visible fluorescence of their adrenolutin product formed via treatment with NaOH (1 M, 1 ml/min) as post column derivatization (50). The resultant adrenolutins are detected via the emission at 518 nm after excitation at 406 nm with linear detection response to parts per billion levels. Because the adrenochromes are unstable in plasma at 37°C (reacting in a 1st-order fashion with a t1/2 of 21 min with the nucleophilic components of the plasma proteins), it is important to slow this process by cooling the blood samples to 2–4°C and maintain that low temperature for all subsequent handling. The blood samples are processed in the following manner: 100 μl of plasma (obtained via centrifugation of the blood at 4°C to separate the cells) is added to 300 μl acetonitrile and centrifuged at 4°C to precipitate proteins. The supernatant (100 μl) is then injected directly into the HPLC system.
Statistical Analysis.
Results are expressed as mean ± SEM for n rats or incubations in the case of the test tube experiments. Statistical differences between treatments were determined by one-way ANOVA, followed by Student–Newman–Keuls test. Statistical differences were accepted when P < 0.05.
Results
O2⨪ Chemically Interacts with Catecholamines.
We initially conducted in vitro experiments to assess the interaction between O2⨪ and catecholamines. HX (2 mM)/XO (1 unit/ml) results in the generation of O2⨪ in the ratio of 2 molecules of O2⨪ to every 1 molecule of HX used (44). Exposing catecholamines (NE, Epi, or DA) to this O2⨪-generating system resulted in a significant decrease in the chemical detection of the catecholamines by HPLC (Table 1; n = 6). This decrease was prevented in a dose-dependent manner in the presence of the SOD mimetic, M40403 (10−7−10−6 M; Table 1; n = 6).
Table 1.
M40403 prevents the decomposition of catecholamines by superoxide in a dose-dependent manner
| NE, ng/ml | Epi, ng/ml | DA, ng/ml | |
|---|---|---|---|
| Control | 44 ± 1.2 | 40 ± 1.1 | 42 ± 2 |
| +HX/XO | 14.3 ± 1.5* | 18.3 ± 1.2* | 22 ± 1.1* |
| +M40403 (10−7M) | 27.1 ± 1.5† | 28.3 ± 1.1† | 29.5 ± 1.5† |
| +M40403 (5 × 10−7M) | 34.7 ± 1.2† | 32.7 ± 1.8† | 31.3 ± 2.4† |
| +M40403 (10−6M) | 39.3 ± 1.3† | 37.7 ± 2.4† | 38.8 ± 2.2† |
The chemical detection, as measured by HPLC, of NE, Epi, or DA is reduced after incubation with HX/XO. This reduction is prevented in a dose-dependent manner in the presence of the SOD mimetic M40403 (10−7-10−6 M; n = 6; ∗, P < 0.05 vs. control;
, P < 0.05 vs. HX/XO).
To assess whether this reaction with O2⨪ renders the catecholamines biologically inactive, we evaluated the ability of NE, before and after incubation with HX/XO, to raise mean arterial pressure (MAP) in anesthetized rats. NE (0–1 μg/kg), given as i.v bolus injections, raised the MAP of the rats in a dose-dependent manner (Fig. 1; n = 6). After incubation with HX/XO, the ability of NE to increase MAP was significantly attenuated (Fig. 1; n = 6). When M40403 (10−6 M) was included with HX/XO in the incubation mixture, the vasopressor actions of NE were significantly protected (Fig. 1; n = 6). HX/XO alone caused a slight but nonsignificant increase in MAP of 4 ± 3 mmHg (1 mmHg = 133 Pa). M40403 caused a slight but nonsignificant decrease in MAP of 3 ± 5 mmHg. These data clearly demonstrate that O2⨪ can deactivate NE in vitro and, as a consequence, abolish its biological activity, as evidenced by the loss of its vasopressor effects in vivo.
Figure 1.
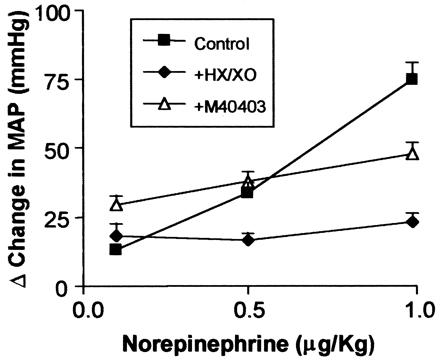
The ability of NE (0.1–1.0 μg/kg), given as bolus i.v injections to an anesthetized rat, to increase MAP (■) is prevented after incubation with HX/XO (⧫). This ability is protected by the inclusion of M40403 in the incubate (▵; n = 6). Incubation period was 5 min for each experiment.
The Role of the Deactivation of Catecholamines in the LPS-Induced Model of Endotoxic Shock.
Because sepsis is a disease in which large amounts of O2⨪ are produced (15–17, 51), optimum conditions exist for catecholamines to be deactivated in vivo in a similar manner to that which occurs in vitro. This deactivation may account for the hyporeactivity to exogenous NE that develops in this disease. If this hypothesis is correct, then the removal of O2⨪ generated in vivo should restore the reactivity to exogenous NE. Administration of E. coli LPS to rats leads to the development of hyporeactivity to exogenously administered NE, a phenomenon that typically occurs within the first 60 min. This model was therefore used to test our hypothesis. Bolus i.v injections of NE (0.1–1 μg/kg) raised the MAP of anesthetized rats in a dose-dependent manner (Fig. 2; n = 6). Two hours after the injection of LPS (4 mg/kg), the pressor responses to NE (0.1–1 μg/kg) were greatly reduced, indicative of the development of hyporeactivity (Fig. 2; n = 6). These responses to NE were restored by M40403 (0.25 mg/kg, given as a 15-min i.v infusion 1 h after LPS; Fig. 2). Pressor responses to NE in rats not treated with LPS were unaffected by the SOD mimetic (Fig. 2; n = 6). These data strongly support our hypothesis that the hyporeactivity that develops in sepsis to exogenously administered NE is caused by the deactivation of this catecholamine by O2⨪ produced in vivo.
Figure 2.
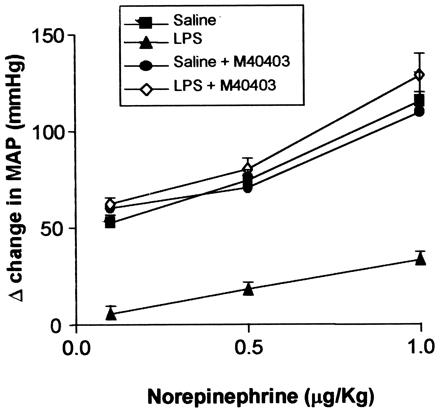
The MAP of the anesthetized rat is increased by the administration of NE (0.1–1 μg/kg; ■). LPS (4 mg/kg) administered to the anesthetized rat results in the development of hyporeactivity to NE (0.1–1 μg/kg) within 1 h (▴). This hyporeactivity is reversed by administration of M40403 (1 mg/kg) to the LPS-treated rat (◊). The reactivity to NE in saline-treated rats is not affected by M40403 (●; n = 6 for all).
Reversal of LPS-Induced Hypotension by M40403.
As a result of these findings, the following experiments were carried out to examine the possibility that the severe and irreversible hypotension that develops in sepsis is related to the deactivation of endogenous vasoconstrictor catecholamines by O2⨪. Intravenous administration of LPS (4 mg/kg) in rats led to a profound fall in blood pressure associated with a high mortality rate (90% mortality at 9 h, n = 10; Fig. 3). When the SOD mimetic, M40403 (0.25 mg/kg/h) was administered as an i.v infusion 1h post LPS for the duration of the experimental protocol, the development of hypotension was prevented and mortality rate greatly reduced (90% survival by 9 h, n = 10; Fig. 3). In addition, when the administration of M40403 was postponed until 5 h post LPS in this model, the severe hypotensive phase of this condition was reversed (Fig. 3; n = 10). The rate of survival in this case was 70% by 9 h.
Figure 3.
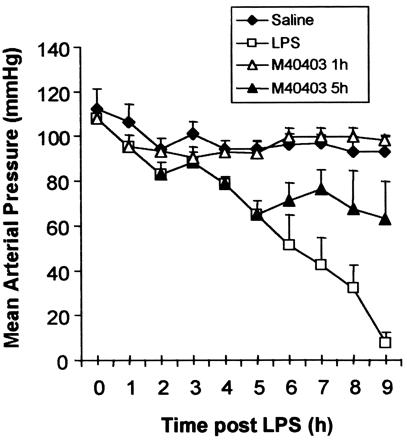
Administration of LPS (4 mg/kg i.v) results in the development of irreversible hypotension in the anesthetized rat (□; n = 10). Treatment with M40403 (0.25 mg/kg/h) at 1 h post LPS prevents this fall in MAP (▵; n = 10). M40403 (0.25 mg/kg/h) administered at 5 h post LPS reverses the fall in MAP (▴; n = 10). Control animals are represented by (⧫).
Measurement of Plasma Catecholamine and Adrenochrome Levels in the Presence or Absence of M40403.
As stated earlier, it has long been known that O2⨪ react with NE and Epi to form the corresponding adrenochrome products (38–46). Therefore, we analyzed plasma samples from rats treated with LPS for the presence of adrenochromes, as well as for catecholamines. Fig. 4 a–c shows that plasma levels of NE and Epi, as well as adrenochromes, increased after LPS treatment (n = 10). Levels of the catecholamines and of the adrenochromes could not be evaluated at the 9-h timepoint in these rats because the survival rate at this point was only 10% (n = 10). Inhibition of hypotension by M40403 correlated with increased plasma levels of catecholamines (Fig. 4 a and b) and a concomitant decrease (Fig. 4c; n = 10) in plasma levels of the adrenochromes.
Figure 4.
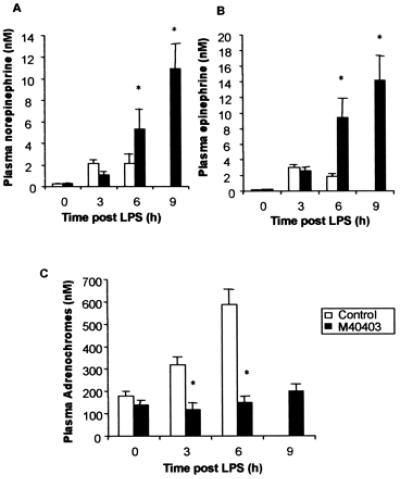
(A and B) Plasma concentrations of Epi and NE increase over time after administration of LPS (4 mg/kg, i.v; open bars). In rats treated with LPS and M40403 (0.25 mg/kg/h given at 1 h post LPS; filled bars), the plasma concentrations of the catecholamines are significantly higher (n = 10 for all; *, P < 0.05). (C) Plasma adrenochrome concentrations increase over time after administration of LPS (4 mg/kg, i.v; open bars). In rats treated with LPS and M40403 (0.25 mg/kg/h given at 1 h post LPS; filled bars), the plasma concentrations of adrenochromes are significantly lower. In these experiments, there were no surviving control rats left alive for a 9-h measurement (n = 10 for all; *, P < 0.05).
Discussion
The findings obtained in this study provide strong evidence for a pivotal role for the deactivation of catecholamines by O2⨪ in the pathogenesis of septic shock. They suggest that the observed hyporeactivity to exogenous NE in the clinic may be explained by the fact that patients basically receive a vasoconstrictor that is easily deactivated through in vivo generation of O2⨪. Thus, we have found that exposure (in vitro) of NE to HX/XO (an O2⨪-generating system) destroyed its biological activity, as evidenced by loss of NE-mediated vasoconstriction when given to rats. Furthermore, our results suggest that in vivo the deactivation of vasoconstrictor catecholamines may account at least in part for the hypotension that is seen after LPS administration. Indeed, M40403, significantly prevented the LPS-induced fall in blood pressure, and this prevention was associated with (i) increased levels of NE and Epi in plasma and (ii) decreased levels in the plasma of their corresponding adrenochromes (products of the interaction of O2⨪ and NE/Epi). Although it remains to be tested, it is possible that formation of adrenochromes accounts for the organ dysfunction associated with septic shock. Some evidence already exists to support a role for adrenochromes as specific mediators of cytotoxicity and cell damage, although their mechanism(s) of actions are not known at this stage (38, 40, 52, 53). More pertinent to the cardiovascular abnormalities of septic shock is the fact that adrenochromes have been shown to be cardiotoxic and cause myocardial necrosis (52–54). If true, such adrenochrome-mediated cardiotoxicity would have adverse consequences for subjects with preexisting compromise of ventricular function and systemic O2 delivery after coronary artery disease, hypertension, and other conditions. Moreover, the possibility exists that adrenochromes may have similarly toxic effects on other organ systems, which may well contribute to the morbidity and mortality of septic shock.
Another reactive oxygen species that has been extensively studied in septic shock is NO. The belief that the overproduction of NO is the major cause of circulatory failure in septic shock has caused much of the research in this field to be aimed at developing a selective inhibitor of inducible nitric oxide synthase (iNOS). This strategy, however, has been fraught with difficulties and has not yielded the benefits expected. One reason for this lack of progress may lie in the fact that, by the time iNOS is up-regulated in endotoxic shock, there is a corresponding down-regulation in the endothelial constitutive (ec) NOS (55, 56), which would mean that NO produced from iNOS is required for vital organ perfusion in this disease. Our results reported here, as well as those by other investigators, would support the idea that NO in itself is not the culprit of hemodynamic dysfunction in septic shock, but perhaps it is the interactions that NO has with other reactive species that results in the problems that arise. This new insight allows for the development of new therapeutic therapies aimed at preventing these interactions. Furthermore, if increased formation of NO from iNOS is responsible for the hypotension/hyporeactivity seen post LPS, then one might have expected an exacerbation of both phenomena with the SOD mimetic (e.g., the SOD mimetic increases the biological properties of NO by preventing its interaction with superoxide; ref. 57). This exacerbation was not the case. An added benefit of therapy with SOD mimetics in general would be the protection against organ injury caused directly by O2⨪ and ONOO− in this disease. Interestingly, NO also has the ability to chemically react with and deactivate catecholamines in vitro (58); however, it is less effective than either O2⨪ (this study) or ONOO−§ in this regard. This reaction has been reported to result in the nitration of catecholamines, resulting in the formation of 6-nitro-catecholamines (59). The results we report here also suggest that deactivation of catecholamines by NO in vivo is not as potent as that occurring with O2⨪ or ONOO−. Treatment with a SOD mimetic should, by removing O2⨪, ultimately afford protection to NO in these animals, and yet we still observed a significant increase in catecholamines in the plasma samples in the presence of M40403.
ONOO− (a highly reactive oxidant produced by the combination of O2⨪ and NO at rates approaching the diffusion limit; ref. 33) can also react with and deactivate catecholamines in vitro to form adrenochromes§ (60). However, the mechanisms by which O2⨪ and ONOO− interact with and deactivate catecholamines to adrenochromes differ. Thus, the reaction between O2⨪ and catecholamines is autocatalytic in nature and so self-generates more O2⨪ to continue the reaction (38–46). Conversely, the reaction between ONOO− and catecholamines is stoichometric in nature and so requires the generation of a new molecule of ONOO− for every reaction that takes place (60). At present it is not possible to assess how much ONOO− contributes in vivo to the deactivation of the catecholamines. What we do know, however, is that M40403, by selectively removing O2⨪, will attenuate the formation of ONOO− and hence ONOO−-mediated catecholamine deactivation and damage. Furthermore, ONOO− possesses a number of independent proinflammatory/cytotoxic mechanisms including (i) the initiation of lipid peroxidation, (ii) the inactivation of a variety of enzymes (most notably, mitochondrial respiratory enzymes and membrane pumps), and (iii) depletion of glutathione. Moreover, ONOO− can also cause DNA damage, resulting in the activation of the nuclear enzyme poly(ADP-ribose) synthetase (PARS), depletion of nicotinamide adenine dinucleotide (NAD), and adenosine triphosphate (ATP), which lead to irreversible cellular damage as evidenced in septic shock and other forms of circulatory shock (61). ONOO− also deactivates the native SOD, leading to a build up of O2⨪ (62, 63).
Our results indicate that deactivation of catecholamines may play a central role in the development of septic shock. If confirmed in more relevant animal models of shock, these results suggest that selective removal of O2⨪ by low molecular weight synthetic enzymes such as M40403 could be an attractive therapeutic approach to attenuate hyporeactivity and hypotension. In conclusion, our findings concerning the deactivation of catecholamines by O2⨪, resulting not only in the loss of their biological activity but also in the generation of cytotoxic products, have widespread implications, particularly for understanding the mechanisms of diseases in which both sympathetic mediators and free radicals play a role. For instance, adrenochromes have been implicated in the degeneration of dopaminergic neurons in Parkinson's disease (38, 64). It is important to note that vasoconstriction is but one of the many fundamentally important effects of the catecholamines. For instance, NE and Epi inhibit the release of proinflammatory cytokines including TNF-α and IL-1β whereas they potentiate the release of the anti-inflammatory cytokine IL-10 (65–67). Based on our findings, it is very likely that, in the presence of free radicals generated in inflammatory states, the biological and therapeutic efficacy of catecholamines will be reduced.
Acknowledgments
We thank Dr. D. Vinjamoori and Mr. J. Long, Analytical Sciences Center, Monsanto Co., St Louis, MO for their help with the adrenochrome assays. This study was supported in part by National Heart, Lung, and Blood Institute Grant 60260.
Abbreviations
- SOD
superoxide dismutase
- O2⨪
superoxide anions
- ONOO−
peroxynitrite
- NE
norepinephrine
- Epi
epinephrine
- LPS
Escherichia coli lipopolysaccharide
- DA
dopamine
- HX
hypoxanthine
- XO
xanthine-oxidase
- NO
nitric oxide
- iNOS
inducible NO synthase
- MAP
mean arterial pressure
Footnotes
Macarthur, H., Salvemini, D. & Westfall, T. C. (1999) FASEB J. 13, A757 (abstr.).
References
- 1.Siegel J H, Greenspan M, Del Guercio L R M. Ann Surg. 1967;165:504–551. doi: 10.1097/00000658-196704000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parker M M, Shelhamer J H, Bacharach S L. Ann Intern Med. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- 3.Groeneveld A B J, Bronsveld W, Thijs L G. Surgery. 1986;99:140–152. [PubMed] [Google Scholar]
- 4.Dellinger R P. Infect Dis Clin North Am. 1999;13:495–509. doi: 10.1016/s0891-5520(05)70088-x. [DOI] [PubMed] [Google Scholar]
- 5.Matuschak G M. In: Principles of Critical Care. 2nd Ed. Hall J B, Schmidt G A, Wood L D H, editors. New York: McGraw–Hill; 1998. pp. 221–248. [Google Scholar]
- 6.American College of Chest Physicians. Crit Care Med. 1992;20:864–875. [PubMed] [Google Scholar]
- 7.de Warra I, Jaccard C, Corradin S B, Chiolero R, Yersin B, Gallati H, Assicot M, Bohoun C, Baumgartner J D, Glauser M P, Heumann D. Crit Care Med. 1997;25:607–613. doi: 10.1097/00003246-199704000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Zeni F, Freeman B, Natanson C. Crit Care Med. 1997;25:1095–1100. doi: 10.1097/00003246-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Haglund U, Gerdin B. Circ Shock. 1991;34:405–411. [PubMed] [Google Scholar]
- 10.Moslen M T. Adv Exp Med Biol. 1994;366:17–27. doi: 10.1007/978-1-4615-1833-4_2. [DOI] [PubMed] [Google Scholar]
- 11.Horowitz S, Mantell L. In: Sepsis and Multiple Organ Failure. Fein A M, Abraham E M, Balk R A, editors. Baltimore: Williams & Wilkins; 1997. pp. 139–151. [Google Scholar]
- 12.Fukuyama N, Takebayashi Y, Hida M, Ishida H, Ichimori K, Nakazawa H. Free Radical Biol Med. 1997;22:771–774. doi: 10.1016/s0891-5849(96)00401-7. [DOI] [PubMed] [Google Scholar]
- 13.de Garavilla L, Chermak T, Valentine H L, Hanson R C. Circ Shock. 1992;25:139–148. [Google Scholar]
- 14.Omar B A, Flores S C, McCord J M. Adv Pharmacol. 1992;23:109–161. doi: 10.1016/s1054-3589(08)60964-3. [DOI] [PubMed] [Google Scholar]
- 15.Oldner A, Goiny M, Rudehill A, Ungerstedt U, Sollevi A. Crit Care Med. 1999;27:790–797. doi: 10.1097/00003246-199904000-00037. [DOI] [PubMed] [Google Scholar]
- 16.Cheng X S, Shimokawa H, Momii H, Oyama J, Fukuyama N, Egashira K, Nakazawa H, Takeshita A. Cardiovasc Res. 1999;42:651–659. doi: 10.1016/s0008-6363(98)00317-4. [DOI] [PubMed] [Google Scholar]
- 17.Lamarque D, Whittle B J R. Eur J Pharmacol. 1995;277:187–194. doi: 10.1016/0014-2999(95)00075-v. [DOI] [PubMed] [Google Scholar]
- 18.Reaume A G, Elliott J L, Hoffman E K, Kowall N W, Ferrante R J, Siwek D F, Wilcox H M, Flood D G, Beal M F, Brown R H, Jr, et al. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 19.Carlsson L M, Jonsson J, Edlund T, Marklund S L. Proc Natl Acad Sci USA. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Huang T T, Carlson E J, Melov S, Ursell P C, Olson J L, Noble L J, Yoshimura M P, Berger C, Chan P H, et al. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 21.Lebovitz R M, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk M M. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun A S, Zastawny T H, Dizdaroglu M, Goodman S I, Huang T T, Miziorko H, Epstein C J, Wallace D C. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Droy-LeFaix M T, Drouet Y, Geraud G, Hosfod D, Braquet P. Free Rad Res Commun. 1991;12–13:725–735. doi: 10.3109/10715769109145852. [DOI] [PubMed] [Google Scholar]
- 24.Esplugues J V, Whittle B J. Br J Pharmacol. 1989;97:1085–1092. doi: 10.1111/j.1476-5381.1989.tb12565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Z F, Hollyoak M, Barrow R E, He F, Muller M J, Herndon D N. J Burn Care Rehabil. 1995;16:111–117. doi: 10.1097/00004630-199503000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Fantone J C, Ward P A. Am J Pathol. 1982;107:395–418. [PMC free article] [PubMed] [Google Scholar]
- 27.Deitch E A, Bridges W, Berg R, Specian R D, Granger N. J. Trauma. 1990. 942–951. [DOI] [PubMed] [Google Scholar]
- 28.Boughton-Smith N K, Deakin A M, Follenfant R L, Whittle B J R, Garland L G. Br J Pharmacol. 1993;110:896–902. doi: 10.1111/j.1476-5381.1993.tb13897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvemini D, Wang Z Q, Wyatt P S, Bourdon D M, Marino M H, Manning P T, Currie M G. Br J Pharmacol. 1996;188:829–838. doi: 10.1111/j.1476-5381.1996.tb15475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dix T A, Hess K M, Medina M A, Sullivan R W, Tilly S L, Webb T L L. Biochemistry. 1996;35:4578–4583. doi: 10.1021/bi952010w. [DOI] [PubMed] [Google Scholar]
- 31.Volk T, Gerst J, Faust-Belbe G, Stroehmann A, Kox W J. Inflamm Res. 1999;48:544–549. doi: 10.1007/s000110050501. [DOI] [PubMed] [Google Scholar]
- 32.Salvemini D, Wang Z-Q, Zweier J L, Samouilov A, Macarthur H, Misko T P, Currie M G, Cuzzocrea S, Sikorski J A, Riley D P. Science. 1999;286:304–306. doi: 10.1126/science.286.5438.304. [DOI] [PubMed] [Google Scholar]
- 33.Beckman J S, Beckman T W, Chen J, Marshall P A, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ischiropoulos H, Zhu L, Beckman I S. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 35.Beckman J S, Crow L P. Biochem Soc Trans. 1993;21:330–334. doi: 10.1042/bst0210330. [DOI] [PubMed] [Google Scholar]
- 36.Salvemini D, Wang Z-Q, Stern M K, Currie M G, Misko T P. Proc Natl Acad Sci USA. 1998;95:2659–2663. doi: 10.1073/pnas.95.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misko T P, Highkin M K, Veenhuizen A W, Manning P T, Stern M K, Currie M G, Salvemini D. J Biol Chem. 1998;273:15646–15653. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- 38.Graham D G. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- 39.Bindoli A, Deeble D J, Rigobello M P, Galzigna L. Biochim Biophys Acta. 1990;1016:349–356. doi: 10.1016/0005-2728(90)90168-4. [DOI] [PubMed] [Google Scholar]
- 40.Bindolini A, Rigobello M P, Delbli D J. Free Radical Biol Med. 1992;13:391–395. doi: 10.1016/0891-5849(92)90182-g. [DOI] [PubMed] [Google Scholar]
- 41.Fridovich I. Annu Rev Biochem. 1975;44:147–159. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 42.Heikkila R E. In: Handbook of Methods for Oxygen Radical Research. Greenwald R A, editor. Boca Raton, FL: CRC; 1985. pp. 233–235. [Google Scholar]
- 43.Misra H P. In: Handbook of Methods for Oxygen Radical Research. Greenwald R A, editor. Boca Raton, FL: CRC; 1985. pp. 237–241. [Google Scholar]
- 44.Misra H P, Fridovich I. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 45.Heikkila R E, Cabbat F S. Anal Biochem. 1976;75:356–362. doi: 10.1016/0003-2697(76)90089-0. [DOI] [PubMed] [Google Scholar]
- 46.Sun M, Zigman S. Anal Biochem. 1978;90:81–89. doi: 10.1016/0003-2697(78)90010-6. [DOI] [PubMed] [Google Scholar]
- 47.Riley D P, Henke S L, Lennon P J, Weiss R H, Neumann W L, Rivers W J, Aston K W, Sample K R, Rahman H, Ling C-S, Shieh J-J, Busch D H, Szulbinski W. Inorg Chem. 1996;35:5213–5231. [Google Scholar]
- 48.Thiemermann C. Adv Pharmacol. 1994;28:45–79. doi: 10.1016/s1054-3589(08)60493-7. [DOI] [PubMed] [Google Scholar]
- 49.Chen X L, Westfall T C. Am J Physiol. 1994;266:C784–C793. doi: 10.1152/ajpcell.1994.266.3.C784. [DOI] [PubMed] [Google Scholar]
- 50.Dhalla, K. S., Ganguly, P. K., Rupp, H., Beamish, R. E. & Dhalla, N. S. Mol. Cell. Biochem.,87, 85–92. [DOI] [PubMed]
- 51.Taylor D E, Ghio A J, Piantadosi C A. Arch Biochem Biophys. 1995;316:70–76. doi: 10.1006/abbi.1995.1011. [DOI] [PubMed] [Google Scholar]
- 52.Yates J C, Beamish R E, Dhalla N S. Am Heart J. 1981;102:210–221. doi: 10.1016/s0002-8703(81)80012-9. [DOI] [PubMed] [Google Scholar]
- 53.Singal P K, Dhillon R E, Beamish R E, Kapur N, Dhalla N S. J Exp Path. 1982;63:167–175. [PMC free article] [PubMed] [Google Scholar]
- 54.Matthews S B, Hallett M B, Henderson A H, Campbell A K. Adv Myocardiol. 1985;6:367–381. [PubMed] [Google Scholar]
- 55.Yoshizumi M, Perrella M A, Burnett J C, Lee M E. Circ Res. 1993;73:205–209. doi: 10.1161/01.res.73.1.205. [DOI] [PubMed] [Google Scholar]
- 56.Liu S, Adcock I M, Old R W, Barnes P, Evans T W. Crit Care Med. 1996;24:1219–1225. doi: 10.1097/00003246-199607000-00026. [DOI] [PubMed] [Google Scholar]
- 57.Gryglewski R J, Palmer R M J, Moncada S. Nature (London) 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 58.Macarthur H, Mattammal M B, Westfall T C. Biochem Biophys Res Commun. 1995;216:686–692. doi: 10.1006/bbrc.1995.2676. [DOI] [PubMed] [Google Scholar]
- 59.Shintani F, Kinoshita T, Kanba S, Ishikawa T, Suzuki E, Sasakawa N, Kato R, Asai M, Nakaki T. J Biol Chem. 1996;271:13561–13565. doi: 10.1074/jbc.271.23.13561. [DOI] [PubMed] [Google Scholar]
- 60.Daveu C, Servy C, Dendane M, Marin P, Ducrocq C. Nitric Oxide. 1997;1:234–243. doi: 10.1006/niox.1997.0123. [DOI] [PubMed] [Google Scholar]
- 61.Szabo C. In: Pathophysiology and Clinical Application Of Nitric Oxide. Rubanyi G M, editor. New York: Harwood; 1999. pp. 69–98. [Google Scholar]
- 62.Yamakura F, Taka H, Fujimura T, Murayama K. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- 63.Macmillan-Crow L A, Thompson J A. Arch Biochem Biophys. 1999;66:82–88. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- 64.Baez S, Seguraaguilar J, Widersten M, Johansson A S, Mannervik B. Biochem J. 1997;324:25–28. doi: 10.1042/bj3240025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Poll T, Lowry S F. Am J Physiol. 1997;273:R1885–R1890. doi: 10.1152/ajpregu.1997.273.6.R1885. [DOI] [PubMed] [Google Scholar]
- 66.van der Poll T, Coyle S M, Barbosa K, Braxton C, Lowry S F. J Clin Invest. 1996;97:713–719. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van der Poll T, Jansen J, Endert E, Sauerwein H P, van Deventer S J H. Infect Immun. 1994;62:2046–2050. doi: 10.1128/iai.62.5.2046-2050.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]