

Abstract
Genomewide association studies depend on the extent of linkage disequilibrium (LD), the number and distribution of markers, and the underlying structure in populations under study. Outbreeding species generally exhibit limited LD, and consequently, a very large number of markers are required for effective whole-genome association genetic scans. In contrast, several of the world's major food crops are self-fertilizing inbreeding species with narrow genetic bases and theoretically extensive LD. Together these are predicted to result in a combination of low resolution and a high frequency of spurious associations in LD-based studies. However, inbred elite plant varieties represent a unique human-induced pseudooutbreeding population that has been subjected to strong selection for advantageous alleles. By assaying 1,524 genomewide SNPs we demonstrate that, after accounting for population substructure, the level of LD exhibited in elite northwest European barley, a typical inbred cereal crop, can be effectively exploited to map traits by using whole-genome association scans with several hundred to thousands of biallelic SNPs.
Keywords: barley, linkage disequilibrium, oligo pool assay
Linkage disequilibrium (LD), the nonrandom association of alleles at distinct loci in a sample population, is now routinely exploited to map disease genes in humans (1–4). In crop plants, the potential of exploiting LD in population-based association mapping, with the objective of estimating the position of a gene conferring a specific trait or phenotype by using LD between alleles of genetically mapped markers, has become a focus of considerable interest. A major attraction of association mapping is the potential to locate genes responsible for a wide range of traits in sample populations by using preexisting phenotypic data collected during crop improvement and cultivar registration programs. In addition, as association mapping exploits all historical recombination events that have occurred during establishment of the sample population (2, 5, 6) mapping resolution may greatly exceed that possible in small biparental experimental crosses.
As spurious associations between phenotypes and marker loci can be caused by population structure, the extent and structure of LD within a sample population must be known before selecting an appropriate association mapping strategy (1). This is particularly true when looking at an inbreeding crop species such as barley where such complicating factors are expected to be more prevalent. In maize, a naturally outcrossing species, LD decays rapidly over 1–2 kb (7) and in diverse populations of Arabidopsis, an inbreeding species, LD extends to ≈1 cM (250 kb), although isolated populations exhibit LD >50 cM (8). Barley in addition to being an inbreeder has, as well as other crop plants, undergone a severe population bottleneck during domestication (9). In addition, the progenitors of contemporary European spring and winter barley varieties were selected from a relatively small number of successful European landraces during the initiation of modern breeding practice >100 years ago (10, 11). These bottlenecks have impacted on the patterns of variation within cultivated European barleys, and complete LD has been observed across contiguous genomic sequences up to 212 kb in length (refs. 12 and 13 and unpublished results). In landrace accessions, LD decayed over ≈90 kb and, in wild barley, Hordeum vulgare ssp. spontaneum, LD did not extend beyond the genic regions (12). In a genomewide association study using 123 mapped amplified fragment length polymorphism markers in modern two-row spring barley varieties LD was observed between markers as far apart as 10 cM (14). Barley cultivars thus potentially provide attractive, extant genetic resources that may make it possible to observe and quantify the consequences of human intervention on their gene pool during both domestication and more contemporary breeding activities.
Whole-genome association studies in crop plants, with the exception of rice, are currently limited by the number of markers available, their format, and cost. To progress toward applying practical whole-genome association studies in barley, we established that the elite northwest European barley gene pool exhibits an average frequency of 2.6 haplotypes per locus by resequencing a collection of PCR-amplified alleles from a representative collection of elite barley genotypes. Based on this information we then developed a pilot oligonucleotide pool assay containing SNPs in 1,524 barley unigenes for use with Illumina GoldenGate BeadArray technology (15, 16). We used this platform to study genomewide molecular diversity and the extent of LD in a collection of elite cultivars. As the barley genome is not sequenced, using SNPs derived from unigenes provides a mechanism to link markers via sequence homology directly to the rice genome sequence, providing access to putative regional gene content and considerable added value. Our results provide experimental support for the feasibility of low- to medium-resolution genomewide association mapping studies in inbreeding plant species using a readily obtainable number of biallelic SNP markers.
Results and Discussion
Haplotype Structure Within the Elite Barley Gene Pool.
To estimate the number and frequency of SNPs that we would require to estimate patterns of diversity across the barley genome and to guide development of a SNP panel suited for whole-genome association genetic scans, we resequenced amplicons from a random set of 88 genes located throughout the barley genome from 24 barley lines that included 19 elite European spring and winter barley varieties. In total we obtained >35 kb of high-quality aligned sequence. The full data set (24 genotypes) contained 367 polymorphic sites (Pi = 0.0021), whereas the European subset (19 genotypes) contained only 193 polymorphic sites (Pi = 0.00167). On average, 3.4 haplotypes were observed per locus in the full data set; however, the European elite germplasm exhibited only 2.6 haplotypes per locus (range 2–6) (see Supporting Text, which is provided as supporting information on the PNAS web site). Based on this finding and previous observations relating to the extent of LD and haplotype diversity in cultivated barley (12, 13, 17, 18) we concluded that assaying a single SNP per gene (locus), at approximately one gene per cM across the 1,100-cM barley genome would provide suitable coverage for LD mapping, with the caveat that SNP haplotype information for certain genome regions rather than a single SNP may subsequently have to be factored into our analysis. We therefore chose to develop an Illumina Golden Gate bead array containing 1,524 SNPs from 1,524 barley unigenes by using putative SNPs identified in EST assemblies and validated SNPs from resequenced allelic amplicons (P.R.B., L.R., N.R., D.F.M., M.K.R., Steve Wanamaker, N.S., J.T.S., Raymond D. Fenton, Jayati Mandal, Pascal Condamine, Kavitha Madishetty, R.K.V., A.G., R.W., and T.G.C., unpublished work) as a platform to explore genome diversity and assess the feasibility of performing informative whole-genome scans.
Genomewide SNP Diversity.
We selected 102 of the most successful recent North European barley cultivars, key progenitors that are prominent in European barley pedigrees and a small number of more “exotic” lines (Table 2, which is published as supporting information on the PNAS web site) for genotypic analysis. Most European barley cultivars include in their pedigrees landrace varieties from four regions: England (variety Spratt Archer), Sweden (Gull), Moravia (Binder), and Bavaria (Isaria), which we included along with successful varieties from throughout the 20th century (10, 11). The majority of the barley grown in the Europe is two-row spring-sown, which provides better quality for the production of malt for beer and whisky manufacture. Winter barley was not widely grown in Europe until middle of the 20th century and, as a result, had a much narrower germplasm base (10, 11). However, it is higher yielding and, to introduce premium malting quality traits, breeders have frequently used spring barley varieties in their winter barley breeding programs.
Of the 1,524 barley SNPs present on our pilot oligonucleotide pool assay, 1,391 assays (91%) were successful in the 102 barley lines and 1,029 of these loci were of known position on the barley genetic linkage map. Less than 0.3% heterozygous genotypes were observed, which is consistent with the highly inbred nature of cultivated barley. Heterozygous loci were generally clustered in the genome (data not shown), suggesting that they arose from occasional outbreeding events followed by loss of heterozygosity. An examination of allele frequency distributions at all 1,391 loci (Fig. 4, which is published as supporting information on the PNAS web site) showed that the full data set contained a large number of SNPs with a minor allele frequency (MAF) of <0.1, reflecting the broad diversity present in the small number of non-European compared with European accessions. SNPs with a MAF of <0.1 were excluded from our further analyses to avoid problems of spurious LD (1). Of the remaining 656 informative SNPs we observed an average nucleotide diversity of 0.257 in the 102 cultivars (Table 1).
Table 1.
Characteristics of the 1,029 mapped SNP loci in barley germplasm subsets
| Germplasm set | No. of accessions | Nucleotide diversity, Pi | Sites with MAF > 0.1 for LD analysis | Pairwise LD tests (P < 0.001) |
|---|---|---|---|---|
| Full | 102 | 0.25702 | 658 | N/D |
| European | 89 (91)* | 0.22620 | 612 | 29,139 |
| Winter | 38 | 0.23719 | 572 | 3,523 |
| Spring two-row | 51 (53** | 0.15337 | 449 | 2,122 |
N/D, not determined.
*Two North American lines, Harrington and Betzes, clustered with European two-row spring varieties and were subsequently analyzed as part of European varieties.
Population Substructure.
Principal coordinate (PCO) analysis of the complete genotype dataset (Fig. 1) identified three major subgroups: (i) a compact group containing mostly European two-row spring barley; (ii) a group containing mostly European two- and six-row winter barleys; and (iii) a diverse set of accessions from North America and Japan. Two North American spring two-row barley cultivars, Harrington and Betzes, clustered with European germplasm, probably because of the presence of European varieties in their pedigrees (Supporting Text), and were therefore included in further analysis as part of the European two-row spring germplasm. Analysis of population structure using a linkage model and allowing for admixture in the program Structure (19) supported partitioning of the genotype data into spring and winter barley populations (data not shown). A maximum parsimony dendrogram further confirmed the partitioning of the germplasm and resolved the major groups according to their pedigrees, country of origin, and breeder (Fig. 5, which is published as supporting information on the PNAS web site).
Fig. 1.

PCOs of the SNP genotype data of 102 barley varieties. The full genotype data set is partitioned into foreign material (triangles), European two-row spring (squares), and winter (diamonds) varieties. The complete list of varieties is available in Table 1.
Comparison of European spring and winter barley populations showed that the majority of sites were polymorphic in both spring and winter barley groups (Fig. 6, which is published as supporting information on the PNAS web site). Average nucleotide diversity was less in the germplasm subgroups compared with the whole set with the winter barley group showing a higher number of unique SNPs and higher nucleotide diversity than the spring group (Table 1). A single fixed difference between European spring and winter barley subpopulations was identified.
To investigate whether we could identify potential signatures of selection within and between the germplasm groups we examined the polymorphism information content (PIC) of the 1,029 genetically mapped loci along each chromosome. We observed that the PIC values were extremely variable among individual chromosomes and among different germplasm groups (Fig. 2a). At a whole chromosome level 6H was the most diverse. Overall there was reduced diversity in the European two-row spring barley accessions, which was most apparent for chromosome 1H where the winter genotypes revealed significantly greater diversity all along its length. However, in specific chromosomal regions different germplasm subgroups exhibited contrasting levels of diversity. For example, reduced diversity in winter barley in the central part of chromosome 5H (90–120 cM) could be attributed to the small number of founding genotypes that contributed the winter seasonal growth habit locus Vrn-H1 (10, 11, 20). Similarly, the abrupt decrease of diversity on the short arm of chromosome 3H (40–60 cM) observed in all groups, coincides with the locus for nonshattering of ears after ripening. Seed shattering is important for seed dispersal in the wild relatives of other cultivated plants (21), but was lost in barley because of mutations in two closely linked loci Btr1 and Btr2 (22). Hence, the observed loss of diversity at this locus in all germplasm groups could potentially be explained by a domestication bottleneck that predated population stratification by more contemporary breeding practices. In other regions, highly significant reductions in diversity were observed, but obvious candidate traits could not be identified. For illustration, the area flanking the centromeric portion of chromosome 7H (40–100 cM) exhibits a profound reduction in diversity in the spring barley group, which correlates with the presence of a large single haplotype block across the entire spring germplasm subset that is suggestive of strong selection. However, there is no obvious reason from breeding history or trait mapping studies why this region should have progressed so far toward fixation. Given the small effective population size further investigation will be required to resolve such observations and confirm that the observed patterns are not simply explained by drift and chance effects.
Fig. 2.

Diversity and LD in barley genome. (a) Distribution of PIC along the linkage map. PIC was averaged across a window of 25 adjacent loci with a step of one and plotted against the linkage map. Data points are colored by group of germplasm. (b) Decay of LD (r2) as a function of genetic distance (cM) between pairs of loci on individual chromosomes in European two-row spring barley. Loci with MAF < 0.1 were excluded from analysis. Only LD values with P < 0.001 are shown.
Extent of LD in Barley Genome.
One of the primary objectives of our study was to determine whether whole-genome association genetic scans would be possible in barley by using a number of genetic markers that is feasible to generate in a modestly funded crop plant research program. We therefore derived measures of LD (r2) in the European germplasm set (n = 91) by using the 612 markers with a MAF > 0.1. As expected, the extent of LD we observed was strongly affected by population structure. Highly significant intrachromosomal LD (P > 0.001; r2 > 0.5) extended over >60 cM (mean 3.9 cM; median 1.16 cM) in the combined set of European spring and winter barley with 20.4% of all significant (P > 0.001) associations (r2 > 0.5) being interchromosomal (Fig. 7a, which is published as supporting information on the PNAS web site). In the spring two-row subset (n = 53), LD extended only up to 15 cM (mean 1.53 cM; median 0.8 cM), and the proportion of interchromosomal associations was reduced to 2% (Figs. 2b and 7b). The extent of LD varied across the chromosomes with a clear relationship between genetic distance and LD. In contrast, there was no obvious relationship with physical distance as the strong LD observed across the centromeric regions, which exhibit significantly reduced recombination, extend over distances of hundreds of megabases (23, 24) (Fig. 3).
Fig. 3.

LD matrix of European two-row spring barley. Data points are colored by magnitude of LD (r2). LD is plotted along the cumulative linkage map of the barley genome with chromosome starting at multiples of 200 cM. (Inset) Variation in LD along the barley chromosome 3H, which correlates with the recombination (genetic distance) and physical map of centromeric region of 3H (23). BIN 6 of the linkage map encompasses ≈600 Mb of chromosome 3H including the centromere (CEN) (23, 24), and r2 exhibits strong LD across the whole region.
Genomewide Association Mapping in Barley.
To investigate whether genomewide association mapping was possible by using a collection of markers in the range used here, we examined whether we could correctly position any of the unmapped genes in our data set via an LD-mapping approach in the set of 53 European two-row spring barley varieties. From the original 1,391 polymorphic SNPs in our genotyping data set, 362 remained unmapped as they did not segregate in any of the three experimental mapping populations used to establish marker order. However, 85 of these showed MAF > 0.1 in the 53 European two-row spring barley varieties. We therefore considered these to be the equivalent of 85 simple Mendelian traits that segregated in our population. Given the small population sample size and nonrandom distribution of marker loci, we established a quantitative threshold for LD mapping by attempting to remap 140 loci with known linkage map locations using LD at different r2 value cut-offs (Table 3, which is published as supporting information on the PNAS web site). Comparison of the locations of these loci predicted by LD mapping with their known map locations indicated that an r2 > 0.5 was a reasonable threshold providing >50% accuracy with ≈5% false-positive calls.
We calculated pairwise LD (r2) for each of the 85 unmapped and 449 mapped loci (MAF > 0.1) and assumed that strong LD indicated linkage (Fig. 8, which is published as supporting information on the PNAS web site). When this threshold was applied, 43 of the 85 unmapped loci could be assigned a putative map location (Table 4, which is published as supporting information on the PNAS web site). When a locus with an unknown map location and the mapped loci with which it was in LD were compared with TIGR rice pseudomolecules version 4 using a BLASTx homology search (www.tigr.org/tdb/e2k1/osa1/pseudomolecules/info.shtml), 80.1% (34 of 42, because one mapped locus was not homologous to rice) colocated to the same region of the rice genome, supporting the results of LD mapping. We consider these results encouraging because although conservation of gene order between barley and rice is often very good, it is also known to have exceptions (25, 26).
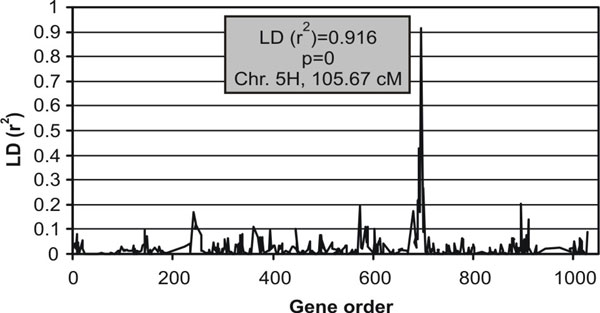
We then attempted to identify regions of the barley genome conferring the phenotype “seasonal growth habit” by LD mapping. We used the mixed linear model implemented in TASSEL to correlate the known seasonal growth habits of the full European barley set with the genotypes of 612 mapped SNPs. Population structure data were supplied as the proportion of winter alleles in the genomes of analyzed accessions based on the program Structure at K=2 (19). A single, highly significant (P = 0) association with LD (r2) of 0.16 was detected on chromosome 5H coinciding with the locus ABC14350, which showed fixed polymorphism between spring and winter populations (Fig. 9, which is published as supporting information on the PNAS web site). Winter growth habit is frequently ascribed simply as the requirement for vernalization to promote flowering. In barley, the vernalization requirement is explained by two loci, Vrn-H1 and Vrn-H2, with winter types encoded only by Vrn-H2,vrn-H1 genotypes (20, 27). HvBM5A, a homologue of the wheat vernalization gene Vrn1 and a candidate gene for Vrn-H1 also maps to the long arm of 5H (20, 28). To check whether vernalization requirement could explain the association of locus ABC14350 with seasonal growth habit, we mapped HvBM5A on one of the populations used in our study (Morex × Barke) by using intronic SNPs that differ between some spring alleles (data not shown). The results showed that ABC14350 was some 27 cM proximal of HvBM5a and was, thus, not tightly linked to the vernalization locus. However, the position of the ABC14350 did coincide with the location of a major quantitative trait locus determining winter hardiness that contains a cluster of Arabidopsis CBF gene homologs (C-Repeat/DRE-binding factor) that are key regulators of the Arabidopsis cold acclimation signaling pathway (29–31). Although members of this CBF gene cluster have not been mapped in the populations used in this study, the region flanking ABC14350 is syntenic to a region on the distal end of rice chromosome 9 that contains the rice orthologues of the barley 5H CBF cluster. It is therefore likely that the complete association found between ABC14350 and seasonal growth habit reflects this region's involvement in the control of winter hardiness. Failure to find significant associations with Vrn-H1 and Vrn-H2 probably relates to the epistatic control of this trait, where some spring varieties have the “wrong” allele at one of the Vrn loci. For example, Armelle (spring) has a winter allele at Vrn-H1 and Emir (spring) a winter allele at Vrn-H2 on chromosome 4H. Seasonal growth habit is further complicated by a poorly defined “facultative” class that generally contains winter sown genotypes that do not require vernalization to promote flowering, e.g., Panda (winter) carries a spring allele at Vrn-H2 (20).
The experimental results of our study allow us to draw a number of conclusions about the potential for whole-genome association scans in this type of genetic material. First, because LD mapping depends on correlations between allele frequencies of loci with unknown locations and those with known map location, a denser linkage map will improve the success of LD mapping, whereas a larger population size will improve the significance of associations. To address these issues we have designed two additional pilot oligonucleotide pool assays that can be used to genotype the same set of germplasm and a large set of European and North American barley accessions (n > 4,000) Second, although we used only pairwise LD in our analyses, more sophisticated analytical approaches assembling biallelic markers into haplotypes may increase the number and significance of the associations for a given trait. Third, population structure in elite germplasm is clearly of importance and may significantly reduce accuracy of LD mapping, if not taken into account. However, even though significant population structure corresponding to division based on seasonal growth habit was found, there was little evidence for strong structure within these groups. This finding may reflect the free access to elite varieties within the framework of Plant Variety Rights in the United Kingdom and Europe that different breeding companies can use in their own improvement programs. The history of repeated germplasm exchange between competing programs apparently has resulted in a reduction of structure in the population of elite germplasm.
Association genetics is becoming a routine procedure for identifying genomic regions, genes, and even SNPs responsible for certain traits (32–34). However, genomewide scans have been widely considered prohibitively expensive because of the large number of SNPs that need to be genotyped, often relegating association genetics to a secondary validation step in map-based cloning procedures after standard mapping in biparental populations. The presented results suggest that a relatively small number of SNPs, in the range of many hundreds to a few thousand, evenly spaced along the genetic linkage map, could be sufficient for an initial whole-genome association scan in inbreeding crop plants, such as barley, in an appropriate set of germplasm. Whether such numbers will be sufficient for detecting marker associations in the case of complex traits remains to be experimentally validated. Although association scans on such scale would generally not result in the identification of a small set of candidate genes for a given trait, the observed extent of LD would ensure resolution relevant for existing breeding programs and provide a rational basis for marker-assisted selection.
Methods
SNP Selection.
SNPs were selected from a set of experimentally validated markers (17) and from publicly available EST sequences derived from nine different barley genotypes.
Germplasm Selection.
The germplasm was chosen to represent a range of material of predominately northwestern European origin including a number of recent varieties on the United Kingdom's recommended list (Table 1 and Supporting Text). The material was composed of both spring- and winter-sown varieties including several that featured in the pedigree of the influential United Kingdom winter variety Maris Otter and some of its direct descendants (11, 35). The choice of material ensured a good representation of likely allelic variants found in the current elite United Kingdom gene pool and gave limited pedigree structure based around a winter by spring cross. In addition, a number of lines representing a wider range of germplasm were assayed through the inclusion of parents of mapping populations, Steptoe × Morex (36), Morex × Barke (N.S. and A.G., unpublished work), and Oregon Wolfe Barley Dominant × Recessive (37), as well as some donors of disease resistance.
Linkage Map.
A consensus linkage map of SNP loci was generated (P.R.B., L.R., N.R., D.F.M., M.K.R., Steve Wanamaker, N.S., J.T.S., Raymond D. Fenton, Jayati Mandal, Pascal Condamine, Kavitha Madishetty, R.K.V., A.G., R.W., and T.G.C.) and consisted of >1,100 SNP loci covering all seven barley chromosomes with only two gaps >10 cM. Integration with Steptoe × Morex restriction fragment length polymorphism map (36) and sequence comparison with rice indicated excellent coverage of the telomeric regions of barley chromosomes (Fig. 10, which is published as supporting information on the PNAS web site).
Genotyping and Curation of Genotypic Data.
DNA from a single 2-week-old plant was prepared by using a DNeasy Plant DNA miniprep kit (Qiagen, Hilden, Germany).
Genotyping was done by using Illumina GoldenGate BeadArrays (15, 16). Genotype data were manually supervised to correct for excessive emphasis on heterozygotes by GenCall software (Illumina, San Diego, CA), and only the most reliable calls were retained. Putative null alleles (high GenTrain score SNPs with low GenCall scores in some varieties) were identified as having GenCall scores <10% of the average GenCall score for each locus and were manually inspected. Only genotype calls that clearly had low intensity and were very different from the majority of calls were marked as null alleles.
Data Analysis.
PCO analysis of the full genotype data set was carried out in GenStat (VSN International, Herts, U.K.) using a simple matching similarity matrix. PIC for each locus was determined according to Botstein et al. (38) using a published formula (39). Genotype calls were converted to the actual SNP sequence, and DNASP 4.0 software (40) was used to study nucleotide diversity and divergence. SEQBOOT, DNAPARS, and CONSENSE programs from the PHYLIP package (41) were used to calculate the extended majority rule consensus tree. Population structure was studied by using STRUCTURE 2.1 (19, 42). Tassel 1.94 (www.maizegenetics.net/index.php?page=bioinformatics/tassel/index.html) was used to calculate LD (r2) and P values (two-tailed Fisher's exact test) in different germplasm subsets after removing minor alleles (<0.1). LD plots were generated in Microsoft (Redmond, WA) Excel. Association mapping of the seasonal growth habit was performed by using the mixed linear model implemented in Tassel 1.94 and the population structure estimates from Structure at K=2.
Supplementary Material
Acknowledgments
SNP genotyping data were collected by J. DeYoung and staff at the Southern California Genotypic Consortium Genotyping Laboratory at the University of California, Los Angeles, following the technical assistance of J. Mandal, R. D. Fenton, and P. Condamine, University of California, Riverside, in the final preparation of DNA samples. This study was funded by the Biotechnology and Biological Sciences Research Council and Scottish Executive Environment and Rural Affairs Department (R.W.), National Science Foundation Plant Genome Research Program DBI-0321756 (T.J.C.), and core funding from the Institute of Plant Genetics and Crop Plant Research (A.G.).
Abbreviations
- LD
linkage disequilibrium
- MAF
minor allele frequency
- PCO
principal coordinate
- PIC
polymorphism information content.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
See Commentary on page 18385.
References
- 1.Lander ES, Schork NJ. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 2.Ardlie KG, Kruglyak L, Seielstad M. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- 3.Hirschhorn JN, Daly MJ. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 4.Tishkoff SA, Verrelli BC. Annu Rev Genomics Hum Genet. 2003;4:293–340. doi: 10.1146/annurev.genom.4.070802.110226. [DOI] [PubMed] [Google Scholar]
- 5.Nordborg M, Tavare S. Trends Genet. 2002;18:83–90. doi: 10.1016/s0168-9525(02)02557-x. [DOI] [PubMed] [Google Scholar]
- 6.Flint-Garcia SA, Thornsberry JM, Buckler ES. Annu Rev Plant Biol. 2003;54:357–374. doi: 10.1146/annurev.arplant.54.031902.134907. [DOI] [PubMed] [Google Scholar]
- 7.Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES., IV Proc Natl Acad Sci USA. 2001;98:11479–11484. doi: 10.1073/pnas.201394398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, et al. Nat Genet. 2002;30:190–193. doi: 10.1038/ng813. [DOI] [PubMed] [Google Scholar]
- 9.Tanksley SD, McCouch SR. Science. 1997;277:1063–1066. doi: 10.1126/science.277.5329.1063. [DOI] [PubMed] [Google Scholar]
- 10.Fischbeck G. In: Diversity in Barley. von Bothmer R, van Hintum T, Knuepffer H, Sato K, editors. Amsterdam: Elsevier; 2003. pp. 29–52. [Google Scholar]
- 11.Fischbeck G. In: Barley Genetics VI: Proceedings of the 6th International Barley Genetics Symposium. Munck L, editor. Sweden: Munksgaard International, Helsinborg; 1992. pp. 885–901. [Google Scholar]
- 12.Caldwell KS, Russell JR, Langridge P, Powell W. Genetics. 2006;172:557–567. doi: 10.1534/genetics.104.038489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piffanelli P, Ramsay L, Waugh R, Benabdelmouna A, D'Hont A, Hinze K, Schulze-Lefert P, Panstruga R. Nature. 2004;430:887–891. doi: 10.1038/nature02781. [DOI] [PubMed] [Google Scholar]
- 14.Kraakman AT, Niks RE, Van den Berg PM, Stam P, Van Eeuwijk FA. Genetics. 2004;168:435–446. doi: 10.1534/genetics.104.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, et al. Cold Spring Harb Symp Quant Biol. 2003;68:69–78. doi: 10.1101/sqb.2003.68.69. [DOI] [PubMed] [Google Scholar]
- 16.Oliphant A, Barker DL, Stuelpnagel JR, Chee MS. BioTechniques. 2002;32(Suppl):356–361. [PubMed] [Google Scholar]
- 17.Rostoks N, Mudie S, Cardle L, Russell J, Ramsay L, Booth A, Svensson JT, Wanamaker SI, Walia H, Rodriguez EM, et al. Mol Genet Genomics. 2005;274:515–527. doi: 10.1007/s00438-005-0046-z. [DOI] [PubMed] [Google Scholar]
- 18.Russell J, Booth A, Fuller J, Harrower B, Hedley P, Machray G, Powell W. Genome. 2004;47:389–398. doi: 10.1139/g03-125. [DOI] [PubMed] [Google Scholar]
- 19.Falush D, Stephens M, Pritchard JK. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Zitzewitz J, Szucs P, Dubcovsky J, Yan L, Francia E, Pecchioni N, Casas A, Chen TH, Hayes PM, Skinner JS. Plant Mol Biol. 2005;59:449–467. doi: 10.1007/s11103-005-0351-2. [DOI] [PubMed] [Google Scholar]
- 21.Harlan JR. The Living Fields: Our Agricultural Heritage. Cambridge, UK: Cambridge Univ Press; 1998. [Google Scholar]
- 22.Takahashi R. In: Advances in Genetics. Demerec M, editor. New York: Academic; 1955. pp. 227–266. [Google Scholar]
- 23.Kuenzel G, Waugh R. Theor Appl Genet. 2002;105:660–665. doi: 10.1007/s00122-002-0913-5. [DOI] [PubMed] [Google Scholar]
- 24.Kuenzel G, Korzun L, Meister A. Genetics. 2000;154:397–412. doi: 10.1093/genetics/154.1.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caldwell KS, Langridge P, Powell W. Plant Physiol. 2004;136:3177–3190. doi: 10.1104/pp.104.044081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han F, Kilian A, Chen JP, Kudrna D, Steffenson B, Yamamoto K, Matsumoto T, Sasaki T, Kleinhofs AA. Genome. 1999;42:1071–1076. doi: 10.1139/g99-060. [DOI] [PubMed] [Google Scholar]
- 27.von Bothmer R, Sato K, Komatsuda T, Yasuda S, Fischbeck G. In: Diversity in Barley. von Bothmer R, van Hintum T, Knuepffer H, Sato K, editors. Amsterdam: Elsevier; 2003. pp. 9–27. [Google Scholar]
- 28.Szucs P, Karsai I, von Zitzewitz J, Meszaros K, Cooper LLD, Gu YQ, Chen THH, Hayes PM, Skinner JS. Theor Appl Genet. 2006;112:1277–1285. doi: 10.1007/s00122-006-0229-y. [DOI] [PubMed] [Google Scholar]
- 29.Francia E, Rizza F, Cattivelli L, Stanca AM, Galiba G, Toth B, Hayes PM, Skinner JS, Pecchioni N. Theor Appl Genet. 2004;108:670–680. doi: 10.1007/s00122-003-1468-9. [DOI] [PubMed] [Google Scholar]
- 30.Skinner JS, von Zitzewitz J, Szucs P, Marquez-Cedillo L, Filichkin T, Amundsen K, Stockinger EJ, Thomashow MF, Chen THH, Hayes PM. Plant Mol Biol. 2005;59:533–551. doi: 10.1007/s11103-005-2498-2. [DOI] [PubMed] [Google Scholar]
- 31.Skinner J, Szucs P, von Zitzewitz J, Marquez-Cedillo L, Filichkin T, Stockinger EJ, Thomashow MF, Chen THH, Hayes PM. Theor Appl Genet. 2006;112:832–842. doi: 10.1007/s00122-005-0185-y. [DOI] [PubMed] [Google Scholar]
- 32.Whitt SR, Wilson LM, Tenaillon MI, Gaut BS, Buckler ES., IV Proc Natl Acad Sci USA. 2002;99:12959–12962. doi: 10.1073/pnas.202476999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES. Nat Genet. 2001;28:286–289. doi: 10.1038/90135. [DOI] [PubMed] [Google Scholar]
- 34.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 35.Macaulay M, Ramsay L, Russell J, Marshall D, Waugh R, Thomas W. In: Advances in Applied Biology: Providing New Opportunities for Consumers and Producers in the 21st Century. Andrews M, Bancroft RD, Boatman ND, Bush MN, Dale MFB, Foulkes J, Glass CR, Jacobson R, Kelly C, Knight JD, et al., editors. Warwick, UK: Association of Applied Biologists; 2004. pp. 139–146. [Google Scholar]
- 36.Kleinhofs A, Kilian A, Saghai-Maroof M, Biyashev R, Hayes P, Chen F, Lapitan N, Fenwick A, Blake T, Kanazin V, et al. Theor Appl Genet. 1993;86:705–712. doi: 10.1007/BF00222660. [DOI] [PubMed] [Google Scholar]
- 37.Costa JM, Corey A, Hayes PM, Jobet C, Kleinhofs A, Kopisch Obusch A, Kramer SF, Kudrna D, Li M, Riera Lizarazu O, et al. Theor Appl Genet. 2001;103:415–424. [Google Scholar]
- 38.Botstein D, White RL, Skolnick M, Davis RW. Am J Hum Genet. 1980;32:314–331. [PMC free article] [PubMed] [Google Scholar]
- 39.Davila JA, Loarce Y, Ramsay L, Waugh R, Ferrer E. Hereditas. 1999;131:5–13. doi: 10.1111/j.1601-5223.1999.00005.x. [DOI] [PubMed] [Google Scholar]
- 40.Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 41.Felsenstein J. Cladistics. 1989;5:164–166. [Google Scholar]
- 42.Pritchard JK, Stephens M, Donnelly P. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}