

Abstract
Cyclooxygenase (COX)-derived prostaglandins (PGs) regulate numerous maternal–fetal interactions during pregnancy. PGs stimulate uterine contractions and prepare the cervix for parturition, whereas in the fetus, PGs maintain patency of the ductus arteriosus (DA), a vascular shunt that transmits oxygenated placental blood to the fetal systemic circulation. However, the origin and site of action of these PGs remain undefined. To address this, we analyzed mice lacking COX-1 (null mutation) or COX-2 (pharmacologic inhibition) or pups with a double null mutation. Our results show that COX-1 in the uterine epithelium is the major source of PGs during labor and that COX-1−/− females experience parturition failure that is reversible by exogenous PGs. Using embryo transfer experiments, we also show that successful delivery occurs in COX-1−/− recipient mothers carrying wild-type pups, establishing the sufficiency of fetal PGs for parturition. Although patency of the DA is PG dependent, neither COX-1 nor COX-2 expression was detected in the fetal or postnatal DA, and offspring with a double null mutation died shortly after birth with open DAs. These results suggest that DA patency depends on circulating PGs acting on specific PG receptors within the DA. Collectively, these findings demonstrate the coordinated regulation of fetal and maternal PGs at the time of birth but raise concern regarding the use of selective COX inhibitors for the management of preterm labor.
The timing of parturition is synchronized with fetal maturity to prevent premature birth (1). Prostaglandins (PGs) are associated with parturition, stimulating such diverse functions as uterine relaxation, contraction, and postpartum involution (2). Evidence that PGs participate in parturition stems from the observation of increased PG levels during labor, increased uterine contractility after PG administration, and delay in the onset of labor by inhibitors of PG synthesis (3–5).
PGs are lipid mediators derived from the hydrolysis of cellular phospholipids. PG G/H synthase or cyclooxygenase (COX) catalyzes the conversion of arachidonic acid to PGH2, the precursor to all prostanoids and thromboxanes. COX exists as two isoforms, COX-1 and COX-2, which share structural homology but arise from different loci (6). COX-1 is widely expressed and is considered a constitutive isoform. However, its expression in the mouse uterus is regulated by estrogen and progesterone (7). COX-1−/− mice are a valuable model for studying labor, because they have delayed parturition and decreased pup survival (8, 9). In contrast, COX-2 is rapidly induced in response to various cellular stimuli and is critical for ovulation, fertilization, and embryo implantation (10, 11). Studies on parturition in COX-2−/− mice are precluded by their infertility (10–12).
The delivery of oxygenated blood from the placenta to the fetus depends on patency of the ductus arteriosus (DA), a vascular shunt coupling the main pulmonary artery with the aorta. This shunt bypasses the uninflated fetal lung and must remain open during pregnancy and labor. After delivery, fetal adaptation requires the establishment of spontaneous breathing and increased oxygen tension, leading to DA closure. The mechanisms that regulate ductal patency and closure are unclear. PGs are implicated in this process, because infusion of PGs maintains DA relaxation, and treatment with nonsteroidal antiinflammatory drugs (NSAIDs) induces its closure (13, 14). PGE2 is essential for DA regulation in mice, because targeted deletion of the EP4 subtype of PGE2 receptor results in neonatal death with an open DA and pulmonary congestion (15, 16).
NSAIDs reduce uterine contractions and forestall preterm delivery but place the fetus at risk for in utero DA closure (17, 18). Therefore, isoform-selective COX inhibitors have been used in an attempt to prevent fetal complications (19–21). However, the specificity of COX expression during parturition is controversial, and the effects of selective COX inhibitors on the developing fetus are unknown. We used genetic and pharmacologic approaches to evaluate the contribution of COX-1- and COX-2-derived PGs to parturition and the relationship of maternal and fetal PGs during adaptation to extra-uterine life. Our results confirm that maternal COX-1 contributes significantly to parturition in the mouse, but that fetal PGs are sufficient to initiate labor. COX expression was absent in the DA, implying that PGs from maternal, placental, or fetal sources maintain DA patency via PG receptors within the ductus. Contrary to expectation, COX double null offspring have persistent patency of the DA rather than DA constriction, as occurs with NSAID treatment. Together, our findings suggest that selective COX inhibitors should be used with caution in the management of preterm labor until the regulation of DA patency is more fully understood.
Methods
Animal Models and Embryo Transfer.
All experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the Animal Care and Use Committee at the University of Kansas Medical Center. Wild-type and COX-1−/− mice were generated from heterozygous matings on a mixed 129/Bl6 genetic background (8). The COX-1−/− genotype was reestablished on a CD-1 genetic background (Charles River Breeding Laboratories). COX-1−/− and COX-2−/− mice were mated to generate COX-1−/−/COX-2−/+ breeding pairs. COX-1−/−/COX-2−/− double null offspring were generated by mating COX-1−/−/COX-2−/+ mice, with rescue of parturition failure by cesarean section or treatment with RU-486 (300 μg/mouse; Sigma). Some COX-1−/−/COX-2−/+ females were housed in cages with l liter per min 100% oxygen, providing 50–80% FiO2, from day 19 to postpartum day 1. The presence of a vaginal plug was considered day 1 of pregnancy. Adult and newborn mice were killed by cervical transection. Uterine and fetal tissues were collected and flash frozen at specified times for in situ hybridization. For determination of PG content, the amniotic sacs of day 19 uteri were opened, and pups were removed. Segments of uteri with attached placentae and fetal membranes were flash frozen and stored at −80°C. For embryo transfer experiments, wild-type and COX-1−/− recipient females were mated with vasectomized males to induce pseudopregnancy. On the morning of day 4, blastocysts from wild-type donor females were recovered in Whitten's media, washed, and transferred into uteri of wild-type or COX-1−/− recipients. Parturition events were continuously videorecorded under infrared filtered light in rooms with 0700–1900 day–night light cycles. The time of parturition was designated as the complete delivery of the first pup.
Determination of Prostaglandin Content.
Levels of PGE2, PGF2α, and 6-keto-PGF1α (the stable hydrolysis product of PGI2) were quantified by gas chromatographic/negative ion chemical ionization mass spectrometric assays. Briefly, a weighed amount of uterine tissue that had been stored at −80°C was pulverized in liquid nitrogen. The nitrogen was evaporated and the lipids extracted with 5 ml of ice-cold methanol containing indomethacin (10−6 M). To the methanol was added 4 ng of [2H4] PGE2 and 4 ng of [2H4] 6-keto-PGF1α (Cayman Chemicals, Ann Arbor, MI), and the prostanoids were purified, derivatized, and analyzed as described (22). The precision and accuracy of the assay were 6% and 95% for PGE2 and PGF2α and 4% and 96% for 6-keto-PGF1α. Interday variability for each assay is <10%.
Response to Drug Administration.
PGF2α, PGE2, and cPGI, a stable analog of PGI2 (Cayman Chemicals) were stored in 100% ethanol at −20°C. A range of doses was diluted in sesame oil and optimal dosage determined. Control animals received a similar volume of ethanol in oil. PGs were given at 1600 h on day 19, and the timing of parturition and pup vitality was observed by infrared videorecording over the next 24–96 h. Indomethacin (Sigma) or a selective COX-2 inhibitor (celecoxib; Searle) was reconstituted in 1% methylcellulose and gavage fed at 0800 and 1800 h on selected days. A range of celecoxib doses, 10–600 mg/kg/dose twice per day (b.i.d.), was given to one group of wild-type CD-1 females (Fig. 2B). In other experiments, wild-type CD-1 females received indomethacin 1.0 mg/kg/dose b.i.d. (three doses, begun on day 18) or 10 mg/kg/dose b.i.d. (three doses, begun on day 18, or five doses, begun on day 17), or celecoxib 600 mg/kg/dose b.i.d. (three doses, begun on day 18, or five doses, begun on day 17), such that the treatments of all of the animals were completed by the morning of day 19. An additional group of animals received celecoxib 600 mg/kg/dose b.i.d. until the morning of day 20 (three doses, begun on day 19, or seven doses, begun on day 17) to complete the dose-ranging studies. Parturition outcomes were videorecorded or animals were killed to determine the status of the fetal DA. Visual assessment of DA constriction was estimated in 25% increments by a single observer (J.R.).
Figure 2.

The contribution of COX-1, COX-2, and fetal PGs to parturition. (A) Response of COX-1−/− females to prostaglandin administration during delayed parturition. The length of gestation (bars) and pup survival (○) in wild-type and vehicle-treated COX-1−/− females are shown. *, P < 0.01, **, P < 0.001, compared with vehicle-treated COX-1−/−. Single s.c. injections were given on the evening of expected delivery and responses monitored by infrared videorecording. Alterations in gestational length are shown with corresponding survival rates. (B) Response of wild-type females to selective COX-2 inhibition on the morning of expected delivery. *, P < 0.05, compared with vehicle. Drug or vehicle was administered twice daily by gavage feeding and responses monitored by infrared videorecording. (C) Effect of fetal-derived PGs on parturition. The outcome of pregnancy after transfer of wild-type blastocysts to COX-1−/− and wild-type pseudopregnant females is shown. Parturition failure was reduced and pup survival was improved in COX-1−/− mothers with wild-type pups compared with COX-1−/− mothers with COX-1−/− or COX-1+/− litters. *, P < 0.05, compared with COX-1−/− mother with COX-1−/− pups. ET, embryo transfer.
In Situ Hybridization.
In situ hybridization was performed as described (11). Portions of day 19 uteri containing two to three pups were collected and flash frozen. For cardiac sections, the fetal heart was briefly fixed with 10% buffered formalin marked with blue dye, and portions of the right ventricle, pulmonary outflow tract, and DA were dissected free and flash frozen. Ten-micrometer sections of cardiac and uterine/fetal tissues were mounted onto the same polyl-lysine-coated slides, fixed in cold 4% paraformaldehyde, acetylated, and hybridized at 45°C for 4 h in formamide hybridization buffer containing 35S-labeled COX-1 or COX-2 cRNA probes (7, 23). RNase A-resistant hybrids were detected by autoradiography after 3- to 10-day exposure by using Kodak NTB-2 liquid emulsion. Parallel sections were hybridized with the corresponding sense probe.
Statistical Analysis.
PG content (mean ± SEM) and strain specificity results were compared by Student's t test. Third order orthogonal polynomial regression yielded best-fitting curves for strain-specific pup survival. The effects of gestational length and genetic background on pup survival were analyzed by multiple linear regression and univariate ANOVA. The difference in DA constriction between groups was tested by Mann–Whitney U rank comparisons.
Results
COX-1 and COX-2 Are Differentially Expressed During Parturition.
The distribution of COX-1 and COX-2 mRNAs during parturition was examined by in situ hybridization. On the morning of expected delivery (day 19), COX-1 mRNA accumulation was restricted to the uterine epithelium encircling the fetus [Fig. 1A and supplemental Figs. 4 and 5 (see www.pnas.org)]. Epithelial folding and redundancy were consistently observed at the site of placental attachment. The luminal epithelium in the interimplantation regions also showed high levels of COX-1 expression. The labyrinthine layer of the placenta showed comparatively lower levels of COX-1 expression, as well as the fetal epidermis, intestine, liver, and lung (supplemental Fig. 4). In contrast, COX-2 mRNA accumulation was primarily noted in the uterine decidual cells adjacent to the giant cell layer of the placenta (Fig. 1B and supplemental Fig. 5). Low levels of COX-2 mRNA were also detected in the labyrinthine layer of the placenta and the fetal liver (supplemental Fig. 4). COX-2 expression was undetectable in the uterine epithelium or myometrium but was consistently present in the yolk sac. The amniotic membrane was devoid of both COX-1 and COX-2 mRNA. In situ hybridization revealed no alteration in the pattern or intensity of COX-2 signals in COX-1−/− uteri (data not shown), suggesting there is no compensatory COX-2 up-regulation in COX-1−/− mice.
Figure 1.
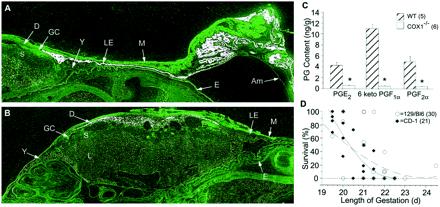
The etiology of prostaglandins in parturition. (A and B) In situ hybridization of 35S-labeled COX-1 (A) and COX-2 (B) in wild-type uteri on the morning of expected delivery (×10). Top includes implantation and interimplantation segments with the intact amniotic sac. Sections of the labyrinthine (L), spongiotrophoblast (S), and giant cell (GC), layers of the placenta and uterine luminal epithelium (LE) myometrium (M) and decidual (D) of the uterus are shown, along with fetal epidermis (E), amniotic membrane (Am), and yolk sac (Y). Sense probes were negative at sites of specific hybridization. (C) Prostaglandin concentrations of wild-type and COX-1−/− uteri on the morning of expected delivery, determined by GC mass spectrometry. *, P < 0.005. (D) Delayed parturition in 51 COX-1−/− litters on two genetic backgrounds. Mice with a 129/Bl6 background delivered later with less surviving pups, whereas CD-1 background mice had more severe lethality when parturition was delayed (129/Bl6 = dashed, R2 = 0.48; CD-1 = solid, R2 = 0.90). Overlapping points exist which represent the outcome of more than one litter.
To determine whether these expression patterns affected the spectrum of PGs produced during labor, we measured the PG content of wild-type and COX-1−/− uterine tissues after the removal of pups. Wild-type uteri with intact placentae and fetal membranes showed higher levels of prostacyclin than PGE2 or PGF2α (Fig. 1C). In contrast, uteri from COX-1−/− mice contained significantly reduced levels of these PGs. Collectively, these results show nonoverlapping COX-1 and COX-2 expression with the majority of PGs originating from COX-1, and that decidual COX-2 expression does not compensate for the lack of COX-1-derived PGs for parturition.
Absence of COX-1 or COX-2 Differentially Alters Parturition.
Parturition failure in COX-1−/− females (8, 9) and reduced uterine contractions in response to NSAIDs (18, 24–26) suggest that PGs are required for parturition. To define this mechanism further, we evaluated the response of COX-1−/− mice to treatment with specific PGs. The targeted COX-1 deletion was reestablished in an outbred strain of mice (CD-1) to improve breeding vigor and to examine strain-specific modifications of the phenotype. As compared with wild-type mice, the length of gestation in 129/Bl6 or CD-1 COX-1−/− mice was prolonged by 1–2 days [129/Bl6: 19.4 ± 0.7 (+/+) vs. 21.7 ± 1.2 (−/−) days; CD-1: 19.6 ± 0.5 (+/+) vs. 20.5 ± 0.9 (−/− days]. The pup survival rate was significantly reduced when gestation was prolonged beyond 21 days (Fig. 1D). By subgroup, COX-1−/− mice on a CD-1 background had more surviving pups than 129/Bl6 mice (6.2 ± 5.3 vs. 2.9 ± 3.6, P < 0.05) and delivered at an earlier gestational age (20.5 ± 0.9 vs. 21.7 ± 1.2 days, P < 0.01). Multivariate analysis showed that the increased number of surviving pups was the result of the shorter gestational period rather than the strain difference. Thus, CD-1 COX-1−/− females were as likely as 129/Bl6 COX-1−/− females to experience parturition failure, although the phenotype was more severe in the CD-1 background when delivery occurred beyond 21 days. There was no difference in the duration of parturition (time from first to last pup) between wild-type and COX-1−/− mice.
We examined the dose-dependent rescue of parturition failure in COX-1−/− mice by PGF2α, PGE2, and carbaprostacyclin (cPGI), a stable analogue of PGI2. As stated above, wild-type females delivered earlier and had higher pup survival (89.9% vs. 27.7%, P < 0.001) than COX-1−/− females (Fig. 2A). Doses of PGF2α (1.0 mg/kg), PGE2 (5.0 mg/kg), or cPGI (100 mg/kg) were chosen, which resulted in the shortest gestation and greatest pup survival (Fig. 2A). At higher doses, PGF2α was particularly effective in promoting the expulsion of pups, but survival rates were greatly diminished. In contrast, a wide range of cPGI doses (0.4–100 mg/kg) did not significantly affect gestational period or pup survival, corresponding to its role in smooth muscle relaxation. Thus, COX-1−/− mice provided a model for parturition in which the response to individual PGs was assessed.
The significance of COX-2 expression in the decidua was examined by using pharmacologic inhibition, because COX-2−/− females are infertile. In the mouse, selective inhibition of COX-2 has been demonstrated at a dose of 10 mg/kg of celecoxib (27). The gestational period (20.0 ± 0.5 days) of wild-type CD-1 mice at this dose was not significantly different from untreated mice (Fig. 2B). At a higher dose (600 mg/kg), celecoxib treatment resulted in delayed parturition (19.6 ± 0.5 days vs. 20.4 ± 0.1 days, P < 0.05) and decreased pup survival. Thus, celecoxib can prolong gestation and induce adverse effects on the fetus similar to nonselective NSAIDs (18, 24–26), but only at doses that may also inhibit COX-1 activity.
Fetal PGs Contribute to Parturition.
The failure of parturition in COX-1−/− mice allowed us to assess the contribution of fetal-derived PGs during labor. In contrast to an earlier report (8), we found no improvement in pup survival when COX-1−/− females were pregnant with heterozygous pups (by mating to COX-1+/+ males) (Fig. 2C). However, transfer of wild-type blastocysts to the uteri of pseudopregnant COX-1−/− females reduced the parturition delay and resulted in improved pup survival. Although the transfer procedure altered the timing of delivery, there was no difference between COX-1−/− and wild-type mothers who received wild-type blastocysts, suggesting that COX-1-derived fetal PGs are sufficient to induce parturition.
Regulation of the Fetal Ductus Arteriosus.
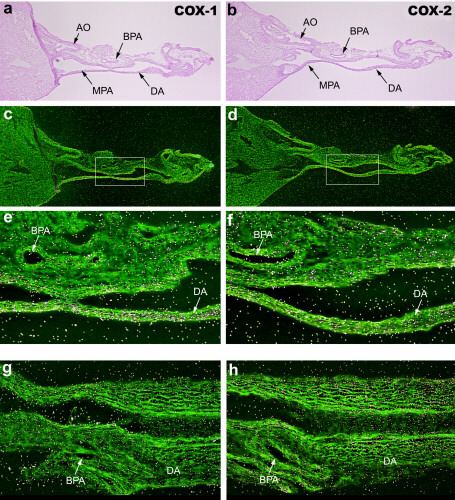
Fetal perfusion and oxygenation depend on patency of the DA, yet the source of PGs that regulate ductal tone is unknown. By in situ hybridization of DA and uterine sections on the same slide, we observed low to undetectable levels of COX-1 and COX-2 expression in the DA vessel wall (Fig. 3 G and H) compared with that of uterine and placental tissues (Fig. 1 A and B). COX-1 and COX-2 mRNAs were also absent in the newborn DA (1 h of age) at the time of its constriction (supplemental Fig. 6; see www.pnas.org). The absence of COX-1 and COX-2 expression in the fetal and neonatal DA was confirmed by reverse transcription–PCR (supplemental Fig. 7). Overall, there was little evidence to support significant COX expression in the pulmonary artery, DA, or aorta.
Figure 3.
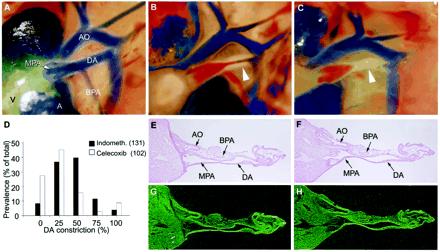
Prostaglandin effects on the fetal ductus arteriosus. (A–C) DA closure in untreated wild-type pups after delivery; immediate newborn period (A), 30 min–1 h (B), and 1–3 h of age (C) (×40). Arrowhead indicates closing DA. (D) Effect of COX inhibitors on in utero DA patency. Wild-type pups exposed to indomethacin or celecoxib before delivery were scored for the degree of DA constriction. (E–H) Brightfield (E and F) and darkfield (G and H) in situ hybridization images of 35S-labeled COX-1 (G) and COX-2 (H) mRNA in the fetal ductus arteriosus before delivery on the morning of day 19 (×40). Atria (A), ventricle (V), main pulmonary artery (MPA), branch pulmonary artery (BPA), ductus arteriosus (DA), and aorta (AO) are shown.
In all wild-type pups, complete DA closure was observed within 1–3 h of birth (Fig. 3 A–C). The DA of surviving COX-1−/− pups was closed by 3 h of life, suggesting that DA constriction occurs normally when COX-1-derived PGs are absent throughout development. To define further the role of COX-1 or COX-2 in DA patency, the effects of maternal treatment with indomethacin or a selective COX-2 inhibitor were evaluated, because maternal ingestion of NSAIDs is associated with fetal DA constriction (15, 17, 18). Wild-type pups whose mothers received indomethacin showed more constriction of the DA than pups exposed to high maternal doses (600 mg/kg) of celecoxib (P < 0.001) (Fig. 3D). Developmental maturity also affected in utero DA constriction, because pups from mothers with celecoxib-induced delayed parturition (day 20, n = 36) had more DA constriction than celexoxib-exposed pups examined on the day of expected delivery (day 19, n = 53) (P < 0.01). In contrast to an earlier report (15), complete in utero DA closure was observed only infrequently. Thus, PGs do not appear to be significantly produced within the DA, and the effects of NSAIDs or selective COX inhibitors on the fetal DA are likely via reduction in circulating PGs.
Interbreeding of COX-1−/−/COX-2−/+ mice produced double null offspring in an approximate Mendelian distribution [14 litters: 24% COX-1−/−/COX-2+/+, 49% COX-1−/−/COX-2−/+, 26% COX-1−/−/COX-2−/−]. These pups were similar in body weight (1.5 ± 0.2 g, COX-1−/−/COX-2−/− vs. 1.4 ± 0.3 g, COX-1−/−/COX-2+/+) and general appearance at birth, but COX-1−/−/COX-2−/− pups consistently died within 24 h of birth. Although the cause of death is unknown, all double null pups who survived for 8 h had widely patent DAs, whereas DAs of their littermates were tightly closed. COX-1−/−/COX-2−/− pups were able to suckle and were appropriately nested by the mother. The increased mortality of COX-1−/−/COX-2−/− pups was not improved by Caesarian section and placement with a foster mother (six litters) or by perinatal treatment with increased environmental oxygen (seven litters). COX-1−/−/COX-2−/+ and COX-1−/−/COX-2+/+ littermates routinely survived to adulthood and were fertile. Thus, the absence of both COX isoforms throughout development may be different from COX inhibition by NSAID treatment, because the former results in death with a patent DA, whereas the latter causes DA constriction.
Discussion
This study demonstrates that COX-1- and COX-2-derived PGs differentially regulate maternal–fetal interactions during late gestation and reveals the sufficiency of fetal PGs for parturition. These experiments comprehensively map the sites of PG synthesis in the whole embryo and uterus and demonstrate the impact of selective COX-1 or COX-2 loss on parturition. Although parturition in the mouse is primarily regulated by COX-1, blood flow within the fetus appears to be controlled by circulating PGs acting on specific PG receptors in the DA. Surprisingly, COX double null mice are viable throughout development but die as neonates because of dysregulation of the DA.
The importance of PGs for parturition is highlighted by the phenotype of COX-1−/− mice. Our observation of delayed parturition and reduced pup survival in COX-1−/− mice on two genetic backgrounds suggests that this phenotype is unaffected by strain-specific modification and confirms that COX-1 is the primary source of PGs for parturition. The etiology of pup mortality is not clear but is similar to human fetal demise during postterm delivery, where infant mortality is proportional to the length of gestation (28). Parturition failure is the suspected cause of pup mortality in homozygous females carrying COX-1−/− pups (8). However, there are conflicting reports on the outcome of COX-1−/− mothers mated with heterozygous or wild-type males (8, 9). Our results clearly show that pup survival is determined by the length of gestation, and that COX-1−/− females can deliver viable offspring if conditions permit delivery before day 21 of gestation. Further, the transfer of wild-type embryos to COX-1−/− females illustrates a significant role for fetal PGs in parturition. The opposite experiment was performed by Gross et al., who showed that wild-type females could deliver COX-1−/− offspring without difficulty (9). Thus, although the uterine epithelium appears to be the predominant source of PGs during labor, our results demonstrate that the fetus can provide the necessary PGs for timely parturition. This effect appears to be dose dependent, because heterozygous offspring cannot induce parturition in COX-1−/− females (Fig. 2C and ref. 9). The actions of fetal PGs are unknown but are presumed to induce ovarian luteolysis and possibly contribute to uterine contractility and cervical maturation.
In wild-type mice, distinct cell-specific expression patterns were noted for COX-1 and COX-2. COX-1 is expressed in the uterine luminal epithelium around the fetus, with extension throughout the interimplantation sites. Although COX-1 expression has been described in the mouse decidua (9), we and others (29) have shown that COX-1 is predominantly expressed in the uterine epithelium. Polarized cells secrete PGs apically or basolaterally (30). Thus, COX-1-derived PGs in the luminal epithelium may act in a paracrine manner to induce muscular contractions for delivery of pups. Accordingly, PG receptors in the myometrium are regulated to enhance the contractile response and relax the lower uterine segment during parturition (31–34). Alternatively, Tsuboi et al. suggest that COX-1-derived PGs are responsible for luteolysis, whereas COX-2-derived PGs are produced at a later stage of labor and are associated with the final steps of parturition (29). In our studies, COX-2 expression was highest in decidual cells. The decidual layer is of maternal origin and marks the region where placental separation occurs during parturition. Despite COX-2 expression in this critical region, selective inhibition of COX-2 by celecoxib did not inhibit parturition except at doses that may also inhibit COX-1. The differential expression of COX-1 and COX-2 suggests their distinct contribution to parturition. However, there are conflicting reports on the relative importance of COX-1 (9, 35, 36) and COX-2 (37–42) expression during labor. In humans, COX-2 up-regulation is more strongly associated with the onset of labor (2–5) and is found primarily in the placenta and fetal membranes (38–42). In addition, delay in the onset of human premature labor has been reported in response to nimesulide, an NSAID that preferentially inhibits COX-2 (20). We speculate that the uterine epithelium may assume the role of the human amniotic epithelium as the primary source of PGs for parturition in the mouse.
COX-1−/− uteri show compensatory up-regulation of COX-2 before implantation (23). During parturition, the low level of PGs produced in COX-1−/− uteri is not compensated by decidual COX-2, suggesting that the compensatory mechanism is ineffective during labor. Prostacylin is the major PG in mouse and human labor, yet its specific functions have not been identified (43, 44). Mice harboring a mutation of the prostacyclin receptor (IP) have normal fertility and parturition (45). However, there is recent evidence that prostacyclin's effects in the mouse uterus are mediated by the nuclear receptor PPARδ rather than the cell surface receptor IP (12). Carbaprostacyclin was unable to rescue the parturition failure of COX-1−/− mice (Fig. 2A). Although prostacyclin is typically associated with smooth muscle relaxation, stimulation of the uterus has been observed at higher doses (44). Its preference for cell-surface or nuclear receptors during parturition is unknown. In contrast, single doses of PGE2 or PGF2α effectively rescued the phenotype of COX-1−/− mice. Studies in FP−/− mice show that PGF2α acts on ovarian FP receptors with induction of luteolysis and a corresponding fall in P4 levels (29). However, PGE2 is considered to be luteotrophic and is unlikely to act via this pathway. Thus, the response of COX-1−/− mice to PGE2 may be related to direct activation of myometrial or cervical PGE receptors or to an unanticipated role as a luteolysin. The differential response of COX-1−/− mice to individual PGs and other uterotonic agents is currently under study.
Premature birth accounts for the majority of human neonatal deaths and long-term disability, and its incidence continues to rise even though preterm labor is recognized more frequently (46). Although premature labor can be inhibited with NSAIDs, their use is associated with DA closure in utero (17, 18). Initial enthusiasm for the use of selective COX-2 inhibitors in human premature labor (20) has been dampened by reports of their adverse effects on the fetus (47). The use of selective COX inhibitors to treat premature labor might offer additional benefit if the origin and specificity of peripartum PGs were known. Despite evidence for PG production by the DA in vitro (48), we observed minimal COX-1 or COX-2 expression in the mouse DA during labor or with postnatal closure. Selective inhibition of COX-2 had less effect on in utero DA constriction than nonselective NSAIDs. These results, together with our uterine studies and the presence of PG receptors in the DA (15, 16, 48) collectively suggest that COX-1-derived PGs from fetal, placental, or maternal sources act on the DA in an endocrine, rather than paracrine or autocrine manner. The timely closure of the DA in COX-1−/− mice further suggests that the absence of COX-1-derived PGs during development may be compensated by other mechanisms, whereas acute inhibition of PG synthesis affects both parturition and DA patency. In this regard, both COX-1−/− and COX-2−/− pups survive without a DA phenotype (8, 10), whereas we observed that COX-1−/−/COX-2−/− double null pups die on the first day of life with an open DA. These findings are similar to those in mice lacking the PGE2 receptor subtype EP4 (15, 16). If PGs are required to maintain DA patency, then these results suggest that an acute withdrawal of PGs acting on PG receptors in the DA is distinct from the absence of PG signaling throughout development. Furthermore, oxygen administration to double null pups did not improve their survival, suggesting that dysregulation of PG ligand–receptor interactions in double null mice cannot be overcome by the multiple factors that mediate oxygen-induced DA constriction (13, 48). Until the mechanisms for maintaining DA patency are better understood, the use of selective COX inhibitors for the treatment of premature labor will involve some risk to fetal survival. The development of agonists or antagonists to specific PG receptors may lead to more effective means of preserving DA patency during the treatment of preterm labor once the sequence of molecular events in parturition and fetal adaptation is known.
Supplementary Material
Acknowledgments
We are grateful to Robert Langenbach (NIEHS, Research Triangle Park, NC) and James Trzaskos (DuPont Merck, Wilmington, DE) for providing the original COX-1 and COX-2 null mice and to John Belmont for his statistical support. This work was supported by National Institutes of Health grants RR11825, HD37677 (J.R.), DK48831, GM15431 (J.D.M.), and HD12304, HD29968 (S.K.D.), and by National Institute of Child Health and Human Development center grants in Reproductive Biology (HD3394) and Mental Retardation and Developmental Disabilities (HD02528). J.D.M. is the recipient of a Burroughs Wellcome Clinical Scientist Award in Translational Research.
Abbreviations
- PG
prostaglandin
- COX
cyclooxygenase
- DA
ductus arteriosus
- NSAIDs
nonsteroidal antiinflammatory drugs
- cPGI
carbaprostacyclin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Smith R. Sci Am. 1999;280:68–75. doi: 10.1038/scientificamerican0399-68. [DOI] [PubMed] [Google Scholar]
- 2.Challis J R G. In: Maternal-Fetal Medicine. Creasy R K, Resnik R, editors. Philadelphia: Saunders; 1999. pp. 484–497. [Google Scholar]
- 3.O'Brien W F. Clin Perinatol. 1995;22:973–984. [PubMed] [Google Scholar]
- 4.Olson D M, Mijovic J E, Sadowsky D W. Semin Perinatol. 1995;19:52–63. doi: 10.1016/s0146-0005(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 5.Gibb W. Ann Med. 1998;30:235–241. doi: 10.3109/07853899809005850. [DOI] [PubMed] [Google Scholar]
- 6.Smith W L, DeWitt D L. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty I, Das S K, Wang J, Dey S K. J Mol Endocrinol. 1996;16:107–122. doi: 10.1677/jme.0.0160107. [DOI] [PubMed] [Google Scholar]
- 8.Langenbach R, Morham S G, Tiano H F, Loftin C D, Ghanayem B I, Chulada P C, Mahler J F, Lee C A, Goulding E H, Kluckman K D, et al. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 9.Gross G A, Imamura T, Luedke C, Vogt S K, Olson L M, Nelson D M, Sadovsky Y, Muglia L J. Proc Natl Acad Sci USA. 1998;95:11875–11879. doi: 10.1073/pnas.95.20.11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dinchuk J E, Car B D, Focht R J, Johnston J J, Jaffee B D, Covington M B, Contel N R, Eng V M, Collins R J, Czerniak P M, et al. Nature (London) 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 11.Lim H, Paria B C, Das S K, Dinchuk J E, Langenbach R, Trzaskos J M, Dey S K. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- 12.Lim H, Gupta R A, Ma W G, Paria B C, Moller D E, Morrow J D, DuBois R N, Trzaskos J M, Dey S K. Genes Dev. 1999;13:1561–1574. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clyman R I. Semin Perinatol. 1987;11:64–71. [PubMed] [Google Scholar]
- 14.Hammerman C. Clin Perinatol. 1995;22:457–479. [PubMed] [Google Scholar]
- 15.Nguyen M, Camenisch T, Snouwaert J N, Hicks E, Coffman T M, Anderson P A W, Malouf N N, Koller B H. Nature (London) 1997;390:78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- 16.Segi E, Sugimoto Y, Yamasaki A, Aze Y, Oida H, Nishimura T, Murata T, Matsuoka T, Ushikubi F, Hirose M, et al. Biochem Biophys Res Comm. 1998;246:7–12. doi: 10.1006/bbrc.1998.8461. [DOI] [PubMed] [Google Scholar]
- 17.Sharpe G L, Thalme B, Larsson K S. Prostaglandins. 1974;8:363–368. doi: 10.1016/0090-6980(74)90110-5. [DOI] [PubMed] [Google Scholar]
- 18.Moise K J. Am J Obstet Gynecol. 1993;168:1350–1353. doi: 10.1016/s0002-9378(11)90763-7. [DOI] [PubMed] [Google Scholar]
- 19.Bennett P R, Slater D. In: Improved Non-Steroid Anti-Inflammatory Drugs: COX-2 Enzyme Inhibitors. Vane J, Botting J, Botting R, editors. London: Kluwer; 1996. pp. 167–188. [Google Scholar]
- 20.Sawdy R, Slater D, Fisk N, Edmonds D K, Bennett P. Lancet. 1997;350:265–266. doi: 10.1016/S0140-6736(05)62229-5. [DOI] [PubMed] [Google Scholar]
- 21.Yousif M, Thulesius O. J Pharm Pharmacol. 1998;50:618–685. doi: 10.1111/j.2042-7158.1998.tb06905.x. [DOI] [PubMed] [Google Scholar]
- 22.DuBois R N, Awad J, Morrow J, Roberts L J, Bishop P R. J Clin Invest. 1994;93:493–498. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reese J, Brown N, Paria B C, Morrow J, Dey S K. Mol Cell Endocrinol. 1999;150:23–31. doi: 10.1016/s0303-7207(99)00033-7. [DOI] [PubMed] [Google Scholar]
- 24.Aiken J W. Nature (London) 1972;240:21–25. doi: 10.1038/240021a0. [DOI] [PubMed] [Google Scholar]
- 25.Lewis R B, Schulman J D. Lancet. 1973. 1159–1161. [DOI] [PubMed] [Google Scholar]
- 26.Novy M J, Cook M J, Manaugh L. Am J Obstet Gynecol. 1974;118:412–416. doi: 10.1016/s0002-9378(16)33801-7. [DOI] [PubMed] [Google Scholar]
- 27.Smith C J, Zhang Y, Koboldt C M, Muhammad J, Zweifel B S, Shaffer A, Talley J J, Masferrer J L, Seibert K, Isakson P C. Proc Natl Acad Sci USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Resnik R, Calder A. In: Maternal-Fetal Medicine. Creasy R K, Resnik R, editors. Philadelphia: Saunders; 1999. pp. 532–539. [Google Scholar]
- 29.Tsuboi K, Sugimoto Y, Iwane A, Yamamoto K, Yamamoto S, Ichikawa A. Endocrinology. 2000;141:315–324. doi: 10.1210/endo.141.1.7236. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs A L, Decker G L, Glasser S R, Julian J, Carson D D. Endocrinology. 1990;126:2125–2136. doi: 10.1210/endo-126-4-2125. [DOI] [PubMed] [Google Scholar]
- 31.Smith G C S, Baguma-Nibasheka M, Wu W X, Nathanielsz P W. Am J Obstet Gynecol. 1998;179:1545–1552. doi: 10.1016/s0002-9378(98)70022-5. [DOI] [PubMed] [Google Scholar]
- 32.Brodt-Eppley J, Myatt L. Biol Reprod. 1998;59:878–883. doi: 10.1095/biolreprod59.4.878. [DOI] [PubMed] [Google Scholar]
- 33.Brodt-Eppley J, Myatt L. Obstet Gynocol. 1999;93:89–93. doi: 10.1016/s0029-7844(98)00378-0. [DOI] [PubMed] [Google Scholar]
- 34.Senior J, Marshall K, Sangha R, Clayton J K. J Pharmacol. 1993;108:501–506. doi: 10.1111/j.1476-5381.1993.tb12832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bennett P R, Henderson D J, Moore G E. Am J Obstet Gynecol. 1992;167:212–216. doi: 10.1016/s0002-9378(11)91660-3. [DOI] [PubMed] [Google Scholar]
- 36.Myatt L, Langdon G, Brockman D E. Prostaglandins. 1994;48:285–296. doi: 10.1016/0090-6980(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 37.Zuo J, Lei Z M, Rao C H V, Pietrantoni M, Cook V D. J Clin Endocrinol Metab. 1994;79:894–899. doi: 10.1210/jcem.79.3.8077379. [DOI] [PubMed] [Google Scholar]
- 38.Macchia L, Di Paola R, Guerrese M C, Chiechi L M, Tursi A, Caiaffa M F, Haeggstrom J Z. Biochem Biophys Res Comm. 1997;233:496–501. doi: 10.1006/bbrc.1997.6492. [DOI] [PubMed] [Google Scholar]
- 39.Slater D, Berger L, Newton R, Moore G, Bennett P. Biochem Biophys Res Commun. 1994;198:304–308. doi: 10.1006/bbrc.1994.1043. [DOI] [PubMed] [Google Scholar]
- 40.Hirst J J, Teixeria F J, Zakar T, Olson D M. J Clin Endocrinol Metab. 1995;80:517–523. doi: 10.1210/jcem.80.2.7852513. [DOI] [PubMed] [Google Scholar]
- 41.Slater D M, Berger L C, Newton R, Moore G E, Bennett P R. Am J Obstet Gynecol. 1995;172:77–82. doi: 10.1016/0002-9378(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 42.Mijovic J E, Zakar T, Nairn T K, Olson D M. J Clin Endocrinol Metab. 1997;83:1358–1367. doi: 10.1210/jcem.83.4.4692. [DOI] [PubMed] [Google Scholar]
- 43.Moonen P, Klok G, Kierse M J N C. Prostaglandins. 1984;28:309–321. doi: 10.1016/0090-6980(84)90019-4. [DOI] [PubMed] [Google Scholar]
- 44.Novy M J, Liggins G C. Semin Perinatol. 1980;4:45–66. [PubMed] [Google Scholar]
- 45.Murata T, Uskikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y, Ichikaw A, Aze Y, Tanaka T, Yosida N, et al. Nature (London) 1997;388:678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 46.Goldenberg R L, Rouse D J. N Engl J Med. 1998;339:313–320. doi: 10.1056/NEJM199807303390506. [DOI] [PubMed] [Google Scholar]
- 47.Peruzzi L, Gianoglio B, Porcellini M G, Coppo R. Lancet. 1999;354:1615. doi: 10.1016/S0140-6736(99)03105-0. [DOI] [PubMed] [Google Scholar]
- 48.Smith G C. Pharmacol Rev. 1998;50:35–58. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}