

Abstract
Since our discovery of the catalytic reduction of dinitrogen to ammonia at a single molybdenum center, we have embarked on a variety of studies designed to further understand this complex reaction cycle. These include studies of both individual reaction steps and of ligand variations. An important step in the reaction sequence is exchange of ammonia for dinitrogen in neutral molybdenum(III) compounds. We have found that this exchange reaction is first order in dinitrogen and relatively fast (complete in <1 h) at 1 atm of dinitrogen. Variations of the terphenyl substituents in the triamidoamine ligand demonstrate that the original ligand is not unique in its ability to yield successful catalysts. However, complexes that contain sterically less demanding ligands fail to catalyze formation of ammonia from dinitrogen; it is proposed as a consequence of a base-catalyzed decomposition of a diazenido (Mo N
N NH) intermediate.
NH) intermediate.
Keywords: catalysis, fixation, nitrogenase
One of the most fascinating catalytic reactions in biology is the reduction of dinitrogen to ammonia by various nitrogenases, the first and most studied being one whose core contains seven irons and one molybdenum (1–4). “Alternative” nitrogenases are now known, one that contains vanadium instead of molybdenum (which functions when Mo is absent and V is available) and another that contains only iron (which functions when both Mo and V are absent) (5–7). The FeMo nitrogenase appears to be the most efficient (≈75% in electrons), yielding approximately only one dihydrogen per dinitrogen reduced. It also has been purified and studied for decades. Its structure has been determined in x-ray studies, and that structure has elicited a great deal of discussion concerning precisely where dinitrogen is reduced (8–11). Although a huge effort to understand how dinitrogen is reduced by various nitrogenases has been made over a period of >40 years, definitive conclusions concerning the site and mechanism of dinitrogen reduction in nitrogenase(s) remain elusive (12).
Two reports of the catalytic reduction of dinitrogen to ammonia with protons and electrons at room temperature and pressure have been published. The first is one in which dinitrogen is reduced to a mixture of hydrazine and ammonia (≈10:1) (13). Molybdenum is required, and dinitrogen reduction is catalytic with respect to molybdenum. The reaction is run in methanol in the presence of magnesium hydroxide and a strong reducing agent such as sodium amalgam. Few details concerning the mechanism of this reaction have been established. The second was reported by our group in 2003 (14). The catalysts are Mo complexes that contain the [(HIPTNCH2CH2)3N]3− ([HIPTN3N]3−) ligand, where HIPT is 3,5-(2,4,6-i-Pr3C6H2)2C6H3, or hexaisopropylterphenyl; an example, [HIPTN3N]MoN2, is shown in Fig. 1(15–17). The [HIPTN3N]3− ligand was designed to prevent any bimetallic reactions (aside from electron transfer), maximize steric protection of a metal coordination site in a monometallic species, and provide increased solubility of intermediates in nonpolar solvents. Eight of the proposed intermediates in a hypothetical “Chatt-like” reduction of dinitrogen (Fig. 2) have been isolated and characterized (Mo is [HIPTN3N]Mo). These intermediates include paramagnetic MoN2 (1; Fig. 1), diamagnetic [MoN2]− (2), diamagnetic MoNNH (3), diamagnetic {MoNNH2}BAr′4 (4; Ar′3,5-(CF3)2C6H3), diamagnetic Mo N (7), diamagnetic {MoNH}BAr′4 (8), paramagnetic {Mo(NH3)}BAr′4 (12), and paramagnetic Mo(NH3) (13). All are extremely sensitive to oxygen with the exception of 7.
N (7), diamagnetic {MoNH}BAr′4 (8), paramagnetic {Mo(NH3)}BAr′4 (12), and paramagnetic Mo(NH3) (13). All are extremely sensitive to oxygen with the exception of 7.
Fig. 1.
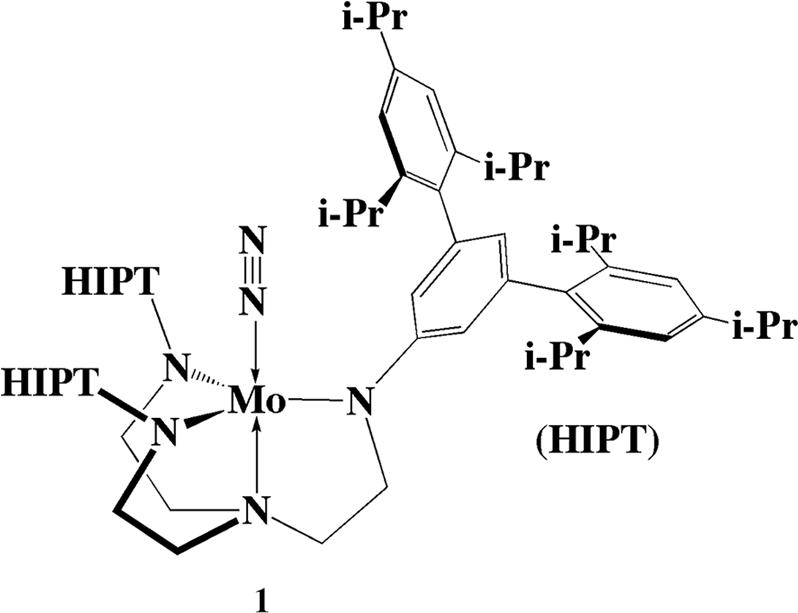
Drawing of [HIPTN3N]MoN2MoN2.
Fig. 2.

Proposed intermediates in the reduction of dinitrogen at a [HIPTN3N]Mo (Mo) center through the stepwise addition of protons and electrons.
Dinitrogen is reduced catalytically in heptane with [2,6-lutidinium]BAr′4 as the proton source and decamethylchromocene as the electron source in the presence of one of the complexes that is believed to be part of the catalytic cycle (14). Catalytic runs by several researchers with several different Mo derivatives (usually 1, 3, 7, or 12) reveal that a total of 7–8 equivalents (equiv) of ammonia are formed out of ≈12 possible (the number possible depending on the Mo derivative used), which suggests an efficiency of ≈65% based on the reducing equiv available. The efficiency of formation of ammonia from the gaseous dinitrogen that is present is 55–60%.
Ongoing studies of the catalytic reduction have begun to reveal why it succeeds and when, and increasingly why, it fails. In this work, we report some of the latest results concerning the parent catalytic system and contrast those with investigations that involve a “hybrid” system in which one of the three HIPT groups in the ligand has been replaced with a 3,5-(CF3)2C6H3 group.
Results and Discussion
What We Know About the Parent Dinitrogen Complex, [HIPTN3N]MoN2.
The synthesis of MoN2 begins with reduction of MoCl with magnesium to yield {MoN2}−. Several salts of this anion have been prepared, including a [n-Bu4N]+ salt. In salts that contain an alkali metal or magnesium, the metal to varying degrees is bound to the β nitrogen and to the central aryl ring in the HIPT group, depending on the metal cation and its degree of solvation by, e.g., THF. It is believed that {MoCl}− is formed upon reduction of MoCl and that dinitrogen then attacks this species to give chloride ion and MoN2. [Loss of chloride first to yield Mo, the “naked” species (14 in Fig. 2), is an alternative that now appears to be less likely, as we will see later.] One electron oxidation of {MoN2}− derivatives with zinc chloride produces MoN2 in good yield. It is proposed that when MoN2 is formed upon reduction of MoCl, it is readily reduced to {MoN2}− under the reaction conditions. MgBr(THF)3{MoN2} and {Mg(dme)3}{MoN2}2 have been characterized structurally (16). The dinitrogen complex is reduced (in PhF/0.1 M [Bu4N]BAr′4) to {MoN2}− reversibly at −2.11 V and is oxidized reversibly at −0.66 V. The MoN2+/0 potential in PhF (−0.66 V) is nearly 1 V higher than the Mo(NH3)+/0 redox couple (−1.63 V) in fluorobenzene. Several relevant potentials are collected in Table 1.
Table 1.
Electrochemical data obtained for various compounds referenced to Cp2Fe+/0
| Redox couple | E1/2 in THF† | E1/2 in PhF‡ |
|---|---|---|
| [(HIPTN3N)MoN2]0/− | −1.81 | −2.01 |
| [(HIPTN3N)Mo(NH3)]+/0 | −1.51 | −1.63 |
| [(HIPTN3N)MoN2]+/0 | −0.42 (Ipa) | −0.66 |
| Cp2Co+/0 | −1.33 | −1.33 |
| Cp2*Cr+/0 | −1.47 | −1.63 |
| Cp2*Co+/0 | −1.84 | −2.01 |
Data were taken in part from ref. 20. CV at 1.6-mm Pt working electrode or 3.0-mm glassy carbon working electrode at 22°C. CV scan rates were 10–200 mV/s.
†0.4 M [Bu4N]PF6 in THF.
‡0.1 M [Bu4N]BAr′4 in PhF.
A sample of Mo15N2 in C6D6 (the ratio of 15N2/14N2 was 92:8) was oxidized by FeCp2BAr′4 to {Mo15N2}+ under 14N2. After 15 min the cation was then reduced with CoCp2* to yield a mixture of Mo15N2 and Mo14N2 in a ratio of 18:82, according to IR spectra. Therefore, 15N2 must be replaced by 14N2 in {Mo15N2}+ to a significant degree in a matter of minutes. The dinitrogen stretching mode in {MoN2}+ in the IR spectrum in solution was found at 2,255 cm−1 in heptane. This result should be compared with a νNN = 1,990 cm−1 for MoN2 and 1,855 cm−1 for {MoN2}− as the tetrabutylammonium salt. We believe that 2,255 cm−1 is the highest known value for dinitrogen bound end on to a transition metal (18).‡ It should be compared with νNN for free dinitrogen in the Raman spectrum at 2,331 cm−1. The reduction of νNN by only 76 cm−1 in {MoN2}+ from the value for free dinitrogen suggests that N2 is very weakly bound to the metal. Note the 400-cm−1 difference between νNN in {MoN2}− and {MoN2}+. We could find no other example in the literature of νNN values in three compounds (anionic, neutral, and cationic) that are prepared by removing one electron sequentially. We can view the electrons as coming from the metal, i.e., Mo(II) in {MoN2}− is oxidized to Mo(III) in MoN2 and then to Mo(IV) in {MoN2}+. However, the oxidation state of Mo in {MoN2}− also could be viewed as Mo(IV), i.e., {MoN2}− is a deprotonated Mo(IV) diazenido (MoNNH) complex.
Oxidation of MoN2 with AgBPh4 in THF resulted in the formation of an orange compound, which was identified as cationic, paramagnetic {[HIPTN3N]Mo(THF)}BPh4 through a single-crystal x-ray diffraction study. The structure shown in Fig. 3 reveals that the THF is bound to the metal along the z axis with Mo–O(1T) = 2.1811(18) Å and N(4)–Mo–O(1T) = 173.97(7)°. The Mo–O distance is approximately the same as the Mo–N(4) distance [2.141(2) Å]. It should be noted that in {Mo(2,6-Lut)}BPh4 (17) (where 2,6-Lut is 2,6-lutidine), the 2,6-lutidine is bound “off-axis” to a significant degree (Namine − Mo − Nlut = 157°) in a “slot” created by two of the HIPT groups. The THF ligand is considerably less sterically demanding and therefore is bound strictly trans to the amine nitrogen donor. All structural information can be found in the supporting information, which is published on the PNAS web site.
Fig. 3.

Thermal ellipsoid drawing of the structure of {Mo(THF)}BPh4. (Hydrogen atoms, isopropyl groups, solvents of crystallization, and tetraphenylborate ion are not shown.)
Reduction of {Mo(2,6-Lut)}BPh4 with CrCp2* under dinitrogen in C6D6 has been observed to yield MoN2 and lutidine upon mixing (17). Examination of the cyclic voltammogram (CV) of {Mo(THF)}BPh4 (Fig. 4) reveals that reduction of {Mo(THF)}+ to Mo(THF) yields MoN2 in a few seconds since the complete MoN2 to {MoN2}− couple is observed; reoxidation of Mo(THF) to {Mo(THF)}+ is not observed on the reverse sweep, only the wave for oxidation of MoN2 to {MoN2}+. Therefore, it appears that dinitrogen readily replaces THF in Mo(THF), and apparently also in Mo(2,6-Lut), on the basis of the qualitative observations noted above. The orders in dinitrogen in these reactions have not been determined, but on the basis of similar reactions that involve interconversion of MoN2 and Mo(NH3) and reactions involving conversion of Mo15N2 into Mo14N2 under 14N2 (see below for both), it seems likely that replacement of L (a σ donor) in Mo(L) by dinitrogen is first order in dinitrogen, i.e., the intermediate is not the naked species, Mo, but a six-coordinate species, Mo(N2)(L). Six-coordinate distorted octahedral species are known in TMS- or C6F5-substituted triamidoamine Mo or W complexes when strongly binding ligands are present (CO or isonitriles) (19). A crystallographically characterized example is {[(C6F5NCH2CH2)3N]W(CO)2}Na(THF)3 (19). However, stable six-coordinate species have not yet been observed in [HIPTN3N]Mo systems. If we assume that the conversion of Mo(THF) into MoN2 is 95% complete in 3 s, that the reaction is in fact a displacement of THF by dinitrogen (see below), and finally that the concentration of dinitrogen in solution is 0.04 M at 1 atm (see supporting information), then the bimolecular rate constant for conversion of Mo(THF) into MoN2 is 25 M−1·s−1 at 22°C.
Fig. 4.

CV of {Mo(THF)}BPh4 in 0.1 M [n-Bu4N]BAr′4/PhF at a scan rate of 100 mV·s−1 at a glassy carbon electrode.
Replacement of 15N2 in Mo15N2 by 14N2 is not likely to be a reaction that is important to catalytic reduction of dinitrogen, but its mechanism turns out to be noteworthy. The conversion of Mo15N2 to Mo14N2 under 14N2 is readily followed by IR, either by following the decrease of the ν15N15N absorption in Mo15N2 or the increase of the ν14N14N absorption in Mo14N2 (Scheme 1). In C6D6 the reaction was reported to be first order in Mo and slow, with t1/2 ≈35 h at 1 atm and 22°C (16). The exchange reaction also was followed in heptane at (total) pressures of 30 psi (15 psi overpressure) where t1/2 = 32 h, and 55 psi (40 psi overpressure) where t1/2 = 30 h. These results suggest that the exchange of 15N2 for 14N2 is independent of dinitrogen pressure up to 55 psi, with kobs = 6 × 10−6 s−1. Therefore, dinitrogen exchange (Scheme 1) apparently is dissociative with the naked species (14; Fig. 2), or possibly some weakly “solvated” variation thereof, the likely intermediate. A bisdinitrogen intermediate, Mo(N2)2, must be a relatively high-energy species, in contrast to the Mo(N2)(L) intermediates noted above. The reason why replacement of 15N2 with 14N2 cannot be bimolecular in the simplest terms is that only three frontier orbitals are readily available in the coordination pocket, and an intermediate or transition state that contains two dinitrogens would require that at least four orbitals be used (2σ and 2π). On the other hand, replacement of dinitrogen by a simple σ bonding ligand, or vice versa, requires only three orbitals.
Scheme 1.

It is interesting to note that exchange of 15N2 in [HTBTN3N]Mo15N2 (where HTBT is hexa-t-butylterphenyl) under 14N2 is exceedingly slow in C6D6 (weeks) (20). The consumption of [HTBTN3N]Mo15N2 was followed by IR and shown to be transformed into [HTBTN3N]Mo14N2 in a reaction that is first order in [HTBTN3N]Mo15N2 with k = 2.6 × 10−7 s−1 at 22°C (t1/2 ∼ 750 h). At 5 atm overpressure of 14N2 the result was essentially the same (k = 3.1 × 10−7 s−1). Because νNN = 1,990 cm−1 in [HTBTN3N]Mo14N2, the same as in Mo14N2, the Mo–N2 bond strength in [HTBTN3N]Mo14N2 must be similar to that in Mo14N2. The only explanation for a rate of unimolecular exchange that is ≈20 times slower than in Mo15N2 appears to be that 15N2 cannot escape the binding cavity in [HTBTN3N]Mo15N2 as readily as it does in Mo15N2, and free 14N2 and free 15N2 cannot be within the cavity above the metal simultaneously. Other evidence suggests that the degree of steric crowding in [HTBTN3N]Mo compounds is so severe as to limit reactions required for reduction of dinitrogen (see below).
The reaction between MoN2 and ammonia (under dinitrogen) leads within 1–2 h to an equilibrium mixture of MoN2 and Mo(NH3) (Scheme 2). For example ammonia (0.28 atm, ≈21 equiv vs. Mo) was added to MoN2 in C6D6 under dinitrogen. The solubility of 15N2 at 22°C in C6D6 was measured vs. an internal 15N standard (CH315NO2), whereas the solubility of ammonia was measured by proton NMR (as described in the supporting information).
Scheme 2.

In C6D6 the solubility of ammonia was found to be 0.1 M atm−1, and the solubility of 15N2 was found to be 0.04 M atm−1. If we assume that there is no significant difference between the solubility of 15N2 and 14N2, then Keq ([MoN2][NH3]/[Mo(NH3)][N2]) can be determined readily. In a series of experiments of this type, Keq was established to be ≈0.1 (see supporting information for details). The fact that equilibrium is reached in 1–2 h rules out any rate-limiting unimolecular loss of dinitrogen from MoN2; dinitrogen most likely is replaced by ammonia in a bimolecular reaction. Because the reaction between Mo(NH3) and dinitrogen must pass through the same intermediate or transition state as the reaction between MoN2 and ammonia, the reaction between Mo(NH3) and dinitrogen must depend on dinitrogen pressure (this hypothesis is the case; see below). Second, the reaction between Mo(NH3) and dinitrogen is related to that between other Mo(L) species and dinitrogen noted above (where L is THF or 2,6-Lut), which are complete within seconds at 1 atm of dinitrogen. We assume that reactions that involve these other Mo(L) species are also first order in dinitrogen, but confirmation has been difficult for experimental reasons; the exchange is simply too fast using current techniques. Clearly, ammonia is much more strongly bound than THF or 2,6-Lut to Mo, as one might expect on the basis of their relative Brønsted basicities (NH3 > 2,6-Lut > THF).
Reduction of {Mo(NH3)}+ by CrCp2* in heptane or CoCp2* in THF is complete within seconds. Therefore, Mo(NH3) can be prepared and studied in situ. We are particularly interested in the rate of the forward reaction in Scheme 2. When we follow the conversion by taking aliquots from a vial for IR spectra and plot ln(1 − A/A∞) vs. time (where A is the absorbance for the MoN2 that is formed), we obtain a nearly straight line through two half-lives from which an observed first-order rate constant of ≈1 × 10−4 s−1 can be obtained. This experiment has been repeated several times (Table 2). The reaction also was followed by differential pulse voltammetry in THF (Table 2) in an open vial in the drybox; the rate of disappearance of the wave ascribed to the {Mo(NH3)}+/0 couple at −1.63 V vs. an internal ferrocene/ferrocenium standard was found to be first order in Mo, and the observed rate constant was found to be 1.0 × 10−4 s−1. When this exchange was followed by IR at an overpressure of 15 psi, kobs was found to be 2.4 × 10−4 s−1. The exchange clearly depends upon dinitrogen pressure, i.e., dinitrogen concentration in solution.
Table 2.
Data for conversion of Mo(III)(NH3) to Mo(III)(N2) complexes (by IR unless otherwise noted)
| Ligand system† | Reductant | Solvent | Overpressure, psi | Other | 104kobs, s−1 |
|---|---|---|---|---|---|
| [HIPTN3N]3−‡ | CrCp2* | Heptane | 1.1 | ||
| [HIPTN3N]3−§ | CoCp2* | THF | 1.0 | ||
| [HIPTN3N]3− | CrCp2* | THF | 30 | 2.4 | |
| [HIPTN3N]3− | CrCp2* | Heptane | 4 BPh3 | 3.3 | |
| [HIPTN3N]3− | CrCp2* | Heptane | 10 BPh3 | 9.0 | |
| [HTBTN3N]3− | CrCp2* | Heptane | 0.06 | ||
| [pBrHIPTN3N]3− | CrCp2* | Heptane | 1.0 | ||
| [CF3Hybrid]3− | CrCp2* | Heptane | 0.67 | ||
| [MeOHybrid]3− | CrCp2* | Heptane | 2.5 |
†BAr′4 salt unless otherwise noted.
‡BPh4 salt.
§By differential pulse voltammetry with 0.4 M [n-Bu4N]PF6.
The conversion of Mo(NH3) to MoN2 actually is not a simple reaction. A typical starting concentration of Mo(NH3) might be 0.02 M, so that at t1/2 the ammonia concentration (0.01 M if none leaves the solution) is much greater than what it would be at equilibrium. Therefore, ammonia back-reacts with MoN2 to yield Mo(NH3) many times before it diffuses out of benzene on a time scale of 1–2 h. (We know that equilibrium is not established for 1–2 h from the equilibrium studies discussed above.) Therefore, the apparent rate constant found in the bulk experiments is much less than what it would be if the ammonia were somehow completely removed as it formed. We also can calculate that if conversion of Mo(NH3) to MoN2 is observed in a closed vial or other small vessel, the equilibrium amount of Mo(NH3) remaining is not negligible, with the exact amount of course depending on the headspace in the closed vessel. How much ammonia is lost entirely will then depend on how often the vessel is opened and for how long, etc. In most runs in fact the plot or ln(1 − A/A∞) vs. time is curved toward the “end” of the run, consistent with an approach to an equilibrium and/or an incorrect value for A∞. We also find that results vary with conditions, e.g., solvent volume, as one might expect.
Two pieces of evidence suggest that kobs for conversion of Mo(NH3) to MoN2 is actually ≈10 times what it appears to be in “bulk” conversions. First, in a CV in which {Mo(NH3)}+ is first reduced to Mo(NH3), the reversible reduction of MoN2 to {MoN2}− can be observed at slow (10 mV·s−1) scan rates (17), consistent with replacement of a significant amount of ammonia in Mo(NH3) (say, ≈5%) with dinitrogen shortly after Mo(NH3) is formed (30 s to 1 min). These estimated values suggest that kobs for conversion of Mo(NH3) to MoN2 is in fact ≈10−3 s−1, with the half-life between 10 and 15 min. One could argue that in this CV experiment the tiny amount of ammonia in the diffusion layer near the electrode migrates into the bulk solution in which there is no ammonia rapidly enough so that a significant amount of MoN2 in fact can be observed at the electrode surface during the CV experiment.
The second piece of evidence is that conversion of Mo(NH3) to MoN2 is accelerated dramatically in the presence of BPh3. These reactions all show strictly linear plots of ln(1 − A/A∞) vs. time and reach a maximum kobs in the presence of ≈15 equiv of BPh3 that is again ≈10−3 s−1 (see Table 2 and supporting information). The ammonia adduct of BPh3 was shown not to react with MoN2, and BPh3 was shown not to react with Mo(NH3) in the absence of dinitrogen at room temperature over a period of hours. Therefore, we conclude that conversion of Mo(NH3) into MoN2 is accelerated because free ammonia is scavenged by BPh3. The “saturation” value for kobs for this reaction (10−3 s−1) is consistent with the rough estimate obtained in the CV experiment described above. If we assume that the true kobs is 10−3 s−1 and the concentration of dinitrogen in solution is 0.04 M, then the true bimolecular rate constant (k in Scheme 2) becomes 2.5 × 10−2 M−1·s−1, and k′ becomes 0.25 M−1·s−1. Therefore, although conversion of Mo(NH3) into MoN2 should be complete in <1 h, the back-reaction between ammonia and MoN2 to reform Mo(NH3) slows the rate of conversion of Mo(NH3) to MoN2 by ≈1 order of magnitude. It is interesting to note that the estimated rate constant for displacement of THF from Mo(THF) by dinitrogen to give MoN2 (25 M−1·s−1; see above) is 103 times larger than that for conversion of Mo(NH3) into MoN2 (0.025 M−1·s−1), largely because ammonia is simply a much better s base than THF.
What We Know About the Catalytic Conversion of Dinitrogen to Ammonia.
Dinitrogen is reduced catalytically in heptane with [2,6-lutidinium]BAr′4 [where Ar′ is 3,5-(CF3)2C6H3] as the proton source and decamethylchromocene as the electron source in the presence of several of the isolated complexes shown in Fig. 2 under the conditions described in 2003 (14). A total of 7–8 equiv of ammonia are formed out of ≈12 possible (depending on the Mo derivative used), which suggests an efficiency of ≈65% based on the reducing equiv available, with the efficiency of formation of ammonia from gaseous dinitrogen being 55–60%. A run employing Mo–15N15NH under 15N2 yielded entirely 15N-labeled ammonia. CoCp2, which is a weaker reducing agent than CrCp2* by 140 mV in THF and 300 mV in PhF (Table 1), also can be used as the reducing agent for catalytic dinitrogen reduction, although it is approximately half as efficient as CrCp2* (17).
An important question is the following: What product or products are formed besides ammonia? One possibility is hydrazine. We analyzed one run under standard conditions for hydrazine, as described in the supporting information. The amount of hydrazine formed was <0.01 equiv vs. the catalyst. This result contrasts with the only other known catalytic reduction of dinitrogen by Mo, where the major product is hydrazine (10 parts for each ammonia) (13).
A second likely possibility is that dihydrogen is formed. We find that dihydrogen is in fact formed, and the amount is that predicted if all remaining reducing equiv go into forming dihydrogen. A typical result is a 64% yield of ammonia (on the basis of electrons available) and a 33% yield of dihydrogen. Therefore, we believe that only ammonia and dihydrogen are formed under standard conditions (see supporting information).
If the Mo catalyst is left out of a standard run, we can examine the production of hydrogen in a “background” reaction. Several runs in heptane and one in THF all show that the yield of dihydrogen produced upon adding CrCp2* to [2,6-lutidinium]BAr′4 is only ≈60% of that expected in a period of 6 h. In contrast, if [2,4,6-trimethylpyridinium]BAr′4 (collidinium) is used as the acid source, then >95% of the expected hydrogen is obtained. Although we have not yet identified the initial reduction product, preliminary NMR evidence suggests that after workup of the 2,6-lutidinium reduction in air, the product is the bipyridine formed through coupling in the para position of 2,6-Lut. This bipyridine can be prepared through reduction of 2,6-Lut with sodium followed by treatment of the reaction with SO2 (21). However, the electron balance for production of dihydrogen and ammonia in catalytic runs suggest that this bipyridine is not formed to any significant extent under catalytic conditions. Relatively slow coupling of 2,4,6-collidinium can account for the near quantitative yield of hydrogen noted above. A similar 4,4′ coupling upon reduction of N-alkylated 2,6-lutidinium salts has been known for some time (22–24).
The yield of ammonia depends dramatically on the nature of the acid used, as shown in Table 3. Use of [2,4,6-collidinium]BAr′4 yields results analogous to those obtained in runs that employ [2,6-lutidinium]BAr′4. However, other pyridinium salts yield less to no ammonia. [2,6-diphenylpyridinium]BAr′4 and [3,5-dimethylpyridinium]BAr′4 in fact produce no ammonia from dinitrogen. In Table 3, we also show that addition of a large amount of 2,6-Lut or THF to a standard reaction both significantly reduce the yield of ammonia. We believe that the pyridinium salt and pyridine that build up as acid is consumed play several complex roles that cannot be deconvoluted at this stage. Finally, we have shown that [Et3NH]BAr′4 is not a successful acid; no ammonia is produced from dinitrogen in a standard catalytic reaction.
Table 3.
Variation of the ammonia yield with the nature of the acid (BAr′4 salt)
| Other | BH+ | Total NH3, equiv | % vs. theory | % from N2 |
|---|---|---|---|---|
| 2,4,6-Me3C6H2NH+ | 7.2/12 | 60 | 52 | |
| 2,4-Me2C6H3NH+ | 5.1/12 | 42 | 34 | |
| 2,6-Et2C6H3NH+ | 3.7/12 | 31 | 23 | |
| 2,6-Ph2C6H3NH+ | 0.4/6 | 7 | 0 | |
| 3,5-Me2C6H3NH+ | 1.0/12 | 9 | 0 | |
| +145 2,6-Lut | 2,6-Me2C6H3NH+ | 1.5/6 | 25 | 8 |
| +150 THF | 2,6-Me2C6H3NH+ | 2.7/6 | 45 | 28 |
| Et3NH+ | 0.7/12 | 6 | 0 |
In Table 4, we show how the yield of ammonia depends on the rate of addition of the reducing agent and the pressure. (Only the ammonia yield was measured in these runs.) Addition of all of the reducing agent in 30 s followed by stirring the reaction for 6 h leads to a dramatically reduced yield of ammonia from dinitrogen (24%). Increasing the pressure to 30 psi leads to a small but measurable increase in the yield of ammonia, 71% vs. 63% for a 6-h addition and 55% vs. 45% for a 3-h addition time. The pressure dependence of ammonia formation is consistent with an increase in the amount of Mo(NH3) converted to MoN2 at higher pressures and therefore a greater yield of ammonia. An interesting question is whether the efficiency of conversion of dinitrogen into ammonia can be pushed beyond 75% (1 equiv of dihydrogen per dinitrogen reduced) at several atmospheres N2 pressure.
Table 4.
The yield of ammonia depends on the rate of addition of the reducing agent and the pressure
| Pressure, psi | Catalyst | Add time | Yield, equiv | % from N2 |
|---|---|---|---|---|
| 15 | MoN2 | 6 h | 7.6/12 | 63 |
| 15 |
MoN |
3 h | 5.4/12 | 45 |
| 15 | MoN2 | 30 s | 2.83/12 | 24 |
| 30 | MoN2 | 6 h | 8.55/12 | 71 |
| 30 |
MoN |
3 h | 6.58/12 | 55 |
It occurred to us that the yield of ammonia might depend on the volume above the reaction solution if ammonia diffuses out of solution into that volume relatively quickly during the catalytic reaction and again allows more Mo(NH3) to be converted into MoN2. We now know this hypothesis to not be the case when the volume is increased from 68 to 330 ml (see supporting information). Ammonia produced during catalysis is not efficiently converted to ammonium salts by excess lutidinium, because when a standard reaction is pumped to dryness, the amount of ammonia in the volatiles was found to be 6.3 equiv out of a typical total of 7.5 obtained when the dry residue is also worked up. Therefore, ≈80% of the product is present as ammonia (rather than ammonium) in the headspace and solution, with diffusion of ammonia out of solution and throughout the entire “headspace” being incomplete, relatively slow, or both. In a typical catalytic run, the final solution volume is ≈10 ml and the headspace is 68 ml. If we assume that 8 equiv of ammonia are formed, that the ammonia in solution is in equilibrium with that in the gas phase, and that no ammonium salt forms, then the amount of ammonia in the gas phase is calculated to be ≈75% of the total formed.
To study the longevity of the catalytic reaction, “sequential” catalytic runs were carried out. After the first standard run, the ammonia that was generated and all volatiles (solvent and 2,6-Lut) were removed in vacuo. The solid residue (a mixture of all Mo-species, [NH4]BAr′4, [CrCp2*]BAr′4, and possible products of decomposition such as free ligand) was then reloaded with another 48 equiv of [2,6-LutH]BAr′4 and treated with 36 equiv of CrCp2* as in a normal run. Typically, 6.3 equiv of ammonia are found in the first stage, as noted above, and 1.7 equiv of ammonia in the second stage, for a total of 8.0 equiv. In some standard runs, the total ammonia plus ammonium has totaled 8.0 equiv. Therefore, little or no ammonia appears to be formed in the second stage of a sequential or “double” catalytic run. Use of 2,4,6-pyridinium as the acid source does not alter this result.
We have found that MoH is as competent (65–66% efficiency in electrons) as any other molybdenum species that we have used; apparently at least 1 equiv of dihydrogen is produced under catalytic conditions from MoH (20). Catalytic reactions that employ organometallic derivatives as “precatalysts” also yield ammonia from dinitrogen under standard conditions (Table 5). The most successful are the n-hexyl or n-octyl complexes, which in fact are as successful as MoH. One can imagine that MoR is protonated to yield RH and MoN2 in the presence of reducing agent and dinitrogen (Scheme 3). It is curious that the long chain alkyl complexes are more successful than the shorter-chain ethyl and especially methyl derivatives. The reasons are not yet known.
Table 5.
The yield of ammonia (out of a possible 12 equiv) in a standard reaction using organometallic compounds as catalysts
| Mo compound | NH3, equiv | % from N2 |
|---|---|---|
| [HIPTN3N]Mo(η2-C2H4) | 2.6 | 22 |
| [HIPTN3N]Mo(η2-C2H2) | 2.3 | 19 |
| [HIPTN3N]MoMe | 3.5 | 29 |
| [HIPTN3N]MoEt | 4.9, 5.1 | 42 |
| [HIPTN3N]Mo(n-hexyl) | 6.3 | 53 |
| [HIPTN3N]Mo(n-octyl) | 6.0, 6.4 | 52 |
| [HIPTN3N]MoCH |
1.8 | 15 |
| [HIPTN3N]MoCMe |
4.7 | 39 |
Scheme 3.

Characteristics of MoNNH.
MoNNH (3 in Fig. 2) has been prepared through protonation of {Mo–NN}− with [Et3NH]BAr′4 and has been characterized structurally (16, 17). Although the proton on the β nitrogen atom could not be located in the x-ray study, 15N and proton NMR studies have established conclusively that a proton is present on Nβ (JHNβ = 54 Hz, JHNα = 8 Hz). Upon heating MoNNH in benzene, it slowly decomposes to MoH and dinitrogen in a slow first-order “β-elimination” process (k = 2.2 × 10−6·s−1, t1/2 = 90 h at 61°C) (16). Decomposition of MoNNH is a problem even at room temperature over the long term, a circumstance that complicated obtaining crystals suitable for an x-ray study (17). Decomposition to MoH is accelerated 1 order of magnitude in the presence of 1% [Et3NH]OTf or [Et3NH]BAr′4 to give MoH and dinitrogen (16). A logical proposal is that the metal itself is protonated, and a proton is then lost from the diazenido ligand to generate Mo(H)(N2), which then rapidly loses dinitrogen to leave MoH (Scheme 4). In the presence of [H(Et2O)2]BAr′4, Mo–NNH is protonated at Nβ to yield [MoNNH2]+, but [2,6-LutH]BAr′4 only partially and reversibly protonates Mo–NNH at Nβ. The pKa of MoNNH appears to be approximately the same as that of DBUH+ in THF [pKa = 16.6 vs. 12.5 for Et3N (25)], because MoNNH can be deprotonated to a considerable degree by DBU (1,8-diaza-bicyclo-[5.4.0]-undec-7-ene) in THF (17). It should be noted that the reaction between {MoNN}− and [2,6-LutH]BAr′4 gave largely MoN2, not MoNNH. The relative rates of acid-catalyzed decomposition to MoH vs. protonation at Nβ and further reduction are not known.
Scheme 4.

Alternatives to the [HIPTN3N]3− Ligand.
Three “symmetric” variations of the [HIPTN3N]3− ligand have been explored (20), a hexa-t-butylterphenyl (HTBT)-substituted ([HTBTN3N]3−) ligand, a hexamethylterphenyl-substituted ([HMTN3N]3−) ligand, and a [pBrHIPTN3N]3− ligand in which the para position of the central phenyl ring is substituted with a bromide. IR and electrochemical studies suggest that complexes that contain [HTBTN3N]3− are slightly more electron rich than those that contain the parent [HIPTN3N]3− ligand, whereas those that contain [pBrHIPTN3N]3− are slightly more electron poor (20). An x-ray study of [HTBTN3N]MoCl shows that [HTBTN3N]Mo complexes are significantly more crowded sterically than Mo complexes. [HMTN3N]Mo complexes are likely to be significantly less crowded than Mo complexes, and [pBrHIPTN3N]Mo complexes are likely to have approximately the same steric crowding as Mo complexes. In practice, transformations of [HTBTN3N]3− derivatives that involve electron and proton transfer (e.g., conversion of [HTBTN3N]MoN2 into [HTBTN3N]Mo–NNH) are slower by perhaps an order of magnitude compared with analogous conversions of [HIPTN3N]3− derivatives (20), consistent with a high degree of steric crowding.
[pBrHIPTN3N]Mo°N was found to be a good catalyst for the formation of ammonia, with yields only slightly less than those observed employing [HIPTN3N]3− derivatives (≈65%). On the other hand, [HMBTN3N]MoN was found to be a poor catalyst for the reduction of dinitrogen, with only 1.06 equiv of ammonia being observed. Therefore, only 0.06 equiv are formed from gaseous dinitrogen.
We measured the rates of conversion of the ammonia complex with the dinitrogen complex in the [pBrHIPTN3N]3− and [HTBTN3N]3− systems (Table 2). Conversion of [pBrHIPTN3N]Mo(NH3) into [pBrHIPTN3N]MoN2 under 1 atm of dinitrogen in heptane at 22°C showed that the half-life for formation of [pBrHIPTN3N]MoN2 is ≈2 h, the same as for conversion of Mo(NH3) into MoN2. On the other hand, the half-life for conversion of [HTBTN3N](NH3) into [HTBTN3N]MoN2 under conditions analogous to those used for Mo(NH3) was found to be ≈30 h. This finding is further evidence that the [HTBTN3N]3− system is simply too crowded for what we presume at this stage to be a bimolecular displacement of ammonia by dinitrogen. This problem coupled with the observable (slower) reduction of {[HTBTN3N]Mo(NH3)}+ to [HTBTN3N]Mo(NH3) makes any catalytic reduction of dinitrogen unlikely to be able to compete with “direct” reduction of 2,6-lutidinium to yield dihydrogen.
[HMBTN3N]MoN as a catalyst under standard conditions was also relatively unsuccessful; only 0.47 equiv of ammonia were formed from dinitrogen. One possible problem is the low solubility of [HMBTN3N]MoN, and probably other intermediates (in heptane). However, there may be other problems related to those found for hybrid alternatives below.
It is possible to make the minimum change in a [HIPTN3N]3− ligand, i.e., to reduce the size of the substituent on only one of the arms, as shown in Fig. 5 (W.W.W., R.R.S., A. Hock, and P.M., unpublished results). When these three [(HIPTNCH2CH2)2NCH2CH2N–3,5-R2C6H3]MoN ([RHybrid]3−) species are used in a standard attempted catalytic reaction, no ammonia is produced from dinitrogen using any of them (W.W.W., R.R.S., A. Hock, and P.M., unpublished results). The [CF3Hybrid]3− derivative was chosen to study in more detail.
Fig. 5.
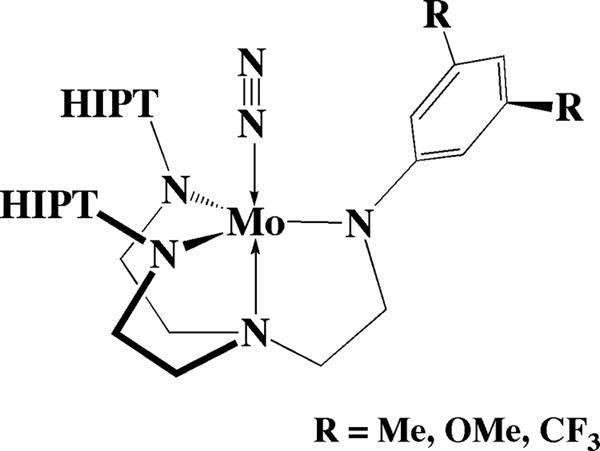
Drawing of a [RHybrid]MoN2 complex.
The half-life for conversion of [CF3Hybrid]Mo(NH3) into [CF3Hybrid]MoN2 was shown to be ≈170 min, which we do not feel is significantly longer than the half-life for conversion of Mo(NH3) into MoN2 (≈120 min) to result in failure of the catalytic reaction in the [CF3Hybrid]3− system. Therefore, reduction must fail in the [CF3Hybrid]3− system for some reason other than a slow conversion of the ammonia complex into the nitrogen complex.
[CF3Hybrid]Mo–NNH can be prepared by treating {[CF3Hybrid]MoN2}Na(THF)2 with H(OEt2)2BAr′4 (0.95 equiv; Scheme 5). It has not been possible to isolate [CF3Hybrid]Mo–NNH, as a consequence of its extreme solubility and instability. Proton NMR spectra revealed a Mo–NNH resonance at 8.6 ppm; in the 15N-labeled compound, JNβH = 54.5 Hz and JNαH = 8 Hz, close to those for the previously characterized Mo–NNH species (15). When prepared in benzene-d6, [CF3Hybrid]Mo–NNH has been observed to decompose to [CF3Hybrid]MoN2 at a rate that is first order in Mo with a rate constant of 6.6 ± 0.7 × 10−4 min−1 (t1/2 = 17 ± 2 h). If we compare this rate with the decomposition of Mo–NNH, two differences are readily apparent. First, Mo–NNH decomposes to MoH, whereas [CF3Hybrid]Mo–NNH decomposes to [CF3Hybrid]MoN2. Second, the rate of decomposition of [CF3Hybrid]Mo–NNH is significantly faster than that of Mo–NNH, which decomposes at a rate where k = 1.3 × 10−4 min−1 (t1/2 = 92 h) at 61°C. It may be important to recognize that [CF3Hybrid]Mo–NNH is prepared in situ, so the product or products of any side reaction (e.g., free ligand) are present. On the basis of the results explained below, the decomposition that we observed therefore may be catalyzed by some side product.
Scheme 5.

Labeling studies are informative. [CF3Hybrid]Mo–NND was found to decompose to [CF3Hybrid]MoN2 with kH/kD = 3.9; therefore, a N–H(D) bond must be cleaved during the rate-limiting step. An interesting observation was made while observing the decomposition of [CF3Hybrid]Mo–15N15NH under 14N2; [CF3Hybrid]Mo–15N15NH was converted into [CF3Hybrid]Mo–14N14NH at a rate that was first order in Mo and with k = 2.6 × 10−2 min−1 (t1/2 = 4.5 h). We have reexamined Mo–15N15NH under similar conditions and found that it also undergoes this exchange reaction, but at a rate at least 100 times slower than that of [CF3Hybrid]Mo–NNH. Whether this difference in 15N exchange rate is important for understanding why catalytic reduction of dinitrogen fails in the [CF3Hybrid]3− system is not yet known.
In an attempt to use catalytically relevant proton sources, [2,6-lutidinium]BAr′4 was used to form [CF3Hybrid]Mo–NNH in situ. The rate of decomposition of [CF3Hybrid]Mo–NNH was found to be 1.05 × 10−2 min−1 (t1/2 = 1.1 h). Use of [Et3NH]BAr′4 as the proton source resulted in a t1/2 of <5 min; only trace amounts of [CF3Hybrid]Mo–NNH were observed 10 min after addition. To confirm that the conjugate base is accelerating the decomposition of [CF3Hybrid]Mo–NNH, 4 equiv of 2,6-Lut, 2,4,6-collidine, or Et3N was added to samples of [CF3Hybrid]Mo–NNH synthesized by using H(OEt2)2BAr′4. In all three cases, all [CF3Hybrid]Mo–NNH decomposed within 5 min to [CF3Hybrid]MoN2.
On the basis of these experiments, we conclude that the less sterically encumbered hybrid systems fail to reduce dinitrogen catalytically as a consequence of a base-catalyzed shunt at the Mo–NNH point in the catalytic cycle that yields dihydrogen (Fig. 6). Because we typically use the nitride as the precatalyst for our catalytic runs, at least 4 equiv of base are present when Mo–NNH is synthesized in catalytic reactions, which is sufficient to reduce the half-life of [CF3Hybrid]Mo–NNH to <5 min. Interestingly, when [CF3Hybrid]MoN2 is used as the precatalyst, 0.7 equiv of NH3 are produced, indicating that the [CF3Hybrid]Mo–NNH step can be (partially) traversed under catalytic conditions when only ≈1 equiv of base is present.
Fig. 6.

A shunt that limits dinitrogen reduction in the less sterically crowded [RHybrid]3− systems.
Conclusions
We have shown that conversion of a Mo(III) ammonia complex (Mo(NH3)) into a Mo(III) dinitrogen complex (MoN2) involves displacement of ammonia by dinitrogen with k estimated to be 2.5 × 10−2 M−1·s−1 and Keq estimated to be 0.1 at room temperature. Therefore, Mo(NH3) should be converted into MoN2 relatively quickly (a half-life of ≈15 min at 1 atm), but conversion is slowed by ≈1 order of magnitude as a consequence of ammonia not being removed efficiently. In contrast, the rate of displacement (it is assumed) of the weaker Brønsted base from Mo(THF) to give MoN2 is 103 times larger. A slower displacement of ammonia by dinitrogen is shown not to be the reason why several catalytic reactions by complexes that contain hybrid ligands fail. Finally, we have found that bimolecular displacement of dinitrogen by a σ bonding ligand, or vice versa, is relatively fast, because three orbitals are available in the transition state, whereas bimolecular displacement of dinitrogen for dinitrogen is slow because four orbitals are required.
We believe the main reason that catalytic reduction of dinitrogen fails for the hybrid systems to be that the Mo–NNH species is decomposed before it can be protonated at the β nitrogen atom (as in 3 → 4 in Fig. 2). It is striking that instability of the Mo–NNH species is the consequence when only one of the three substituents on the amido nitrogens is a “small” 3,5-disubstituted phenyl group. In this situation, there is slightly less steric hindrance, either between the “arms” of the triamidoamine ligand and/or above the apical pocket where the diazenido ligand is located. The most surprising finding is that a base (including 2,6-Lut, the conjugate base of the acid used in a standard reduction) catalyzes decomposition of the Mo–NNH species in the sterically less crowded hybrid species to yield MoN2 and dihydrogen. The rate-limiting step of the lutidine-catalyzed decomposition of Mo–NNH is unimolecular in Mo and appears to involve cleavage of the β NH bond. The precise nature of this apparent base-induced decomposition is unknown at this time.
It clearly will be important to prepare complexes that contain other hybrid ligands that resemble the parent [HIPTN3N]3− ligand system more and more closely. We suspect that the stability of various Mo–NNH species toward 2,6-Lut will correlate with the success of the catalytic reduction of dinitrogen. We also must examine the stability of the parent Mo–NNH species under a variety of conditions, including its stability toward the conjugate bases of the acids shown in Table 3. Nevertheless, it is premature to conclude that failure of any given catalytic reaction can be ascribed to instability of its Mo–NNH intermediate. In any scheme as complex as that shown in Fig. 2, there are many opportunities for failure.
Experimental Methods
All MoX compounds, other than [Mo(THF)][BAr′4], have been reported (14–16). [Mo(THF)][BAr′4] was synthesized by oxidation of MoN2 as described in supporting information. Mo(alkyl) compounds were synthesized as reported (16, 26). [Hybrid]MoX compounds were synthesized in a similar manner to the corresponding MoX system (15, 16). Diazenido compounds (Mo–NNH and [CF3Hybrid]Mo–NNH) were synthesized in situ as described in supporting information.
Electrochemical studies were performed similarly to those reported (11, 16). Catalytic experiments were performed as reported (14). Variations of this procedure are described in supporting information.
MoNH3 and MoN2 exchange studies were performed as described in the supporting information. IR spectroscopy was used to quantify the formation of MoN2.
Hydrogen quantification was performed with a 6890C GC (Hewlett–Packard, Palo Alto, CA) fitted with a 30-m/0.5-mm/25-mm molecular sieve column and a TCD. Briefly, 50 ml of gas was extracted from the experimental system and injected into the GC. The results were compared with a calibration curve to determine the amount of H2 present.
Structural and experimental details and other data are provided in supporting information.
Supplementary Material
Acknowledgments
R.R.S. was supported by National Institutes of Health Grant GM 31978.
Abbreviations
- Ar′
3,5-(CF3)2C6H3
- CV
cyclic voltammogram
- equiv
equivalent(s)
- HIPT
3,5-(2,4,6-i-Pr3C6H2)2C6H3 (hexaisopropylterphenyl)
- HTBT
hexa-t-butylterphenyl
- 2,6-Lut
2,6-lutidine
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
The highest dinitrogen stretch we have found, 2,197 cm−1 in Nujol, is in a bis(imino)pyridine Co(I) cationic complex.
References
- 1.Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Hardy RWF, Bottomley F, Burns RC. A Treatise on Dinitrogen Fixation. New York: Wiley Interscience; 1979. [Google Scholar]
- 3.Veeger C, Newton WE. Advances in Nitrogen Fixation Research. Boston: Dr W Junk/Martinus Nijhoff; 1984. [Google Scholar]
- 4.Coughlan MP, editor. Molybdenum and Molybdenum-Containing Enzymes. New York: Pergamon; 1980. [Google Scholar]
- 5.Eady RR. Chem Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 6.Smith BE. Adv Inorg Chem. 1999;47:159–218. [Google Scholar]
- 7.Rehder D. Coord Chem Rev. 1999;182:297–322. [Google Scholar]
- 8.Rees DC, Howard JB. Curr Opin Chem Biol. 2000;4:559–566. doi: 10.1016/s1367-5931(00)00132-0. [DOI] [PubMed] [Google Scholar]
- 9.Bolin JT, Ronco AE, Morgan TV, Mortenson LE, Xuong LE. Proc Natl Acad Sci USA. 1993;90:1078–1082. doi: 10.1073/pnas.90.3.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Rees DC. Science. 1992;257:1677–1682. doi: 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]
- 11.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 12.Seefeldt LC, Dance IG, Dean DR. Biochemistry. 2004;43:1401–1409. doi: 10.1021/bi036038g. [DOI] [PubMed] [Google Scholar]
- 13.Shilov AE. Russ Chem Bull Int Ed. 2003;52:2555–2562. [Google Scholar]
- 14.Yandulov DV, Schrock RR. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- 15.Yandulov DV, Schrock RR. J Am Chem Soc. 2002;124:6252–6253. doi: 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]
- 16.Yandulov DV, Schrock RR, Rheingold AL, Ceccarelli C, Davis WM. Inorg Chem. 2003;42:796–813. doi: 10.1021/ic020505l. [DOI] [PubMed] [Google Scholar]
- 17.Yandulov D, Schrock RR. Inorg Chem. 2005;44:1103–1117. doi: 10.1021/ic040095w. [DOI] [PubMed] [Google Scholar]
- 18.Humphries MJ, Tellmann KP, Gibson VC, White AJP, Williams DJ. Organometallics. 2005;24:2039–2050. [Google Scholar]
- 19.Greco GE, O'Donoghue MB, Seidel SW, Davis WM, Schrock RR. Organometallics. 2000;19:1132–1149. [Google Scholar]
- 20.Ritleng V, Yandulov DV, Weare WW, Schrock RR, Hock AR, Davis WM. J Am Chem Soc. 2004;126:6150–6163. doi: 10.1021/ja0306415. [DOI] [PubMed] [Google Scholar]
- 21.Hünig S, Wehner I. Synthesis. 1989;1989:552–554. [Google Scholar]
- 22.Kosower EM. Topics Curr Chem. 1983;112:117–162. [Google Scholar]
- 23.Carelli I, Cardinali ME, Casini A, Arnone A. J Org Chem. 1976;41:3967–3969. doi: 10.1021/jo00887a007. [DOI] [PubMed] [Google Scholar]
- 24.Oturan MA. J Electroanal Chem. 1988;242:171–179. [Google Scholar]
- 25.Rodima T, Kaljurand I, Pihl A, Mäemets V, Leito I, Koppel IA. J Org Chem. 2002;67:1873–1881. doi: 10.1021/jo016185p. [DOI] [PubMed] [Google Scholar]
- 26.Byrnes MJ, Dai X, Schrock RR, Hock AS, Müller P. Inorg Chem. 2005;24:4437–4450. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.