

Abstract
The calcineurin inhibitor cyclosporine A (CsA) has emerged as a major cause of secondary hypertension in humans, but the underlying pathogenetic mechanisms have remained enigmatic. Synapsins are a family of synaptic vesicle phosphoproteins that are essential for normal regulation of neurotransmitter release at synapses. In addition to synaptic vesicles, synapsins and other vesicle proteins are found on microvesicles in sensory nerve endings in peripheral tissues. However, the functions of the sensory microvesicles in general, and of synapsins in particular, are unknown. We now demonstrate in a mouse model that CsA raises blood pressure by stimulating renal sensory nerve endings that contain synapsin-positive microvesicles. In knockout mice lacking synapsin I and II, sensory nerve endings are normally developed but not stimulated by CsA whereas a control stimulus, capsaicin, is fully active. The reflex activation of efferent sympathetic nerve activity and the increase in blood pressure by CsA seen in control are greatly attenuated in synapsin-deficient mice. These results provide a mechanistic explanation for CsA-induced acute hypertension and suggest that synapsins could serve as a drug target in this refractory condition. Furthermore, these data establish evidence that synapsin-containing sensory microvesicles perform an essential role in sensory transduction and suggest a role for synapsin phosphorylation in this process.
The immunosuppressive drug cyclosporine A (CsA) has greatly improved both long-term survival after organ transplantation and the treatment of autoimmune diseases. However, CsA also has emerged as a major new cause of secondary hypertension (1). Two syndromes have been described: (i) acute hypertension during initiation of CsA; and (ii) chronic hypertension during long-term administration. In heart transplant recipients, for example, the prevalence of hypertension has increased from 20% in the pre-CsA era to currently 90%. This hypertension typically is difficult to treat and is a major factor contributing to cardiovascular morbidity and mortality in the transplant population (1, 2). Despite the clinical importance of this problem, the underlying pathogenic mechanisms have been enigmatic: renal, vascular, and neuronal mechanisms all have been implicated (1).
In rats, i.v. CsA produces a rapid elevation in blood pressure (BP) that models acute CsA-induced hypertension in patients (3–6). There is abundant evidence, at least in the rat, that the acute hypertensive response to CsA is sympathetically mediated (3, 5–7). The hypertension is accompanied by widespread increases in efferent sympathetic nerve activity (SNA) that persists for hours after a single i.v. dose of CsA, and abrogated by chemical or surgical sympathectomy or ganglionic blockade but unaffected by angiotensin-converting enzyme inhibition or endothelin receptor blockade (3, 4, 6, 8–10). Whereas both central and peripheral neural mechanisms may contribute, this acute neurogenic hypertension is caused largely by CsA acting on renal and other subdiaphragmatic sensory nerves that project centrally via the vagus nerves and dorsal spinal roots to reflexively increase efferent SNA and hypertension (6, 7). When this afferent input is interrupted by dorsal rhizotomy, or cervical or subdiaphragmatic vagotomy (VGX), the pressor response to CsA is greatly attenuated (5).
At the cellular level, CsA forms an immunophilin (cytosolic receptor) complex that is a potent inhibitor of calcineurin, the Ca2+-dependent phosphatase that is ubiquitously expressed in neural tissue (11, 12). We recently implicated a pivotal role for calcineurin inhibition in mediating the stimulation of excitatory renal afferents evoked by i.v. CsA (6, 13). In intact rats, afferent and efferent renal nerve activity and BP also are increased by FK506, another immunophilin ligand inhibitor of calcineurin, but unaffected by rapamycin, a structural analog of FK506 that has no effect on calcineurin (5, 6, 13). We turned our attention to the underlying mechanism linking calcineurin inhibition with renal afferent activation.
If calcineurin inhibition mediates the effects of CsA and FK506 on the neural control of BP, what are the relevant physiological phosphoprotein substrates for calcineurin? The synapsins are a family of vesicle-associated neuronal phosphoproteins that were discovered by Greengard et al. (14) and constitute putative substrates for calcineurin (15–17). There are at least five synapsin proteins (synapsin Ia, Ib, IIa, IIb, and IIIa) which are the alternatively spliced products of three different genes (18, 19). Of these, synapsin I and II account for 98% of all synapsin protein. There is a high degree of structural homology between the synapsins, all of which are phosphorylated at their N terminus by calmodulin kinase I and protein kinase A (20). In addition, synapsin I, but not synapsin II, undergoes phosphorylation at the C terminus by calmodulin kinase II (21). Synapsin I can be dephosphorylated at both sites by calcineurin in vitro (15–17). Although calcineurin inhibitors may enhance vesicle-mediated exocytosis in vitro, it remains to be determined whether synapsins are important neurophysiological substrates for calcineurin in vivo.
Synapsins are best known as universal components of presynaptic vesicles where their primary function is to maintain a stable pool of synaptic vesicles that can be recruited during processes of short-term synaptic plasticity (22). However, synapsins also are expressed and concentrated on small vesicles in numerous somatic sensory nerve endings, although their function in sensory transduction is unknown (23–26). The goal of this study was to determine whether synapsins are present in renal sensory nerve endings and constitute important substrates for calcineurin in mediating the reflex pressor response to CsA. We now demonstrate in a mouse model that CsA raises BP by stimulating renal sensory nerve endings that contain synapsin-positive microvesicles. In double knockout (DKO) mice lacking both synapsin I and II, sensory nerve endings are normally developed but not stimulated by CsA, whereas a control stimulus, capsaicin, is fully active. The reflex activation of efferent SNA and the increase in BP by CsA seen in control mice are greatly attenuated in synapsin DKO mice. These results provide a mechanistic explanation for acute CsA-induced hypertension and suggest that synapsins could serve as a drug target in this refractory condition. Furthermore, these data establish evidence that synapsin-containing sensory microvesicles perform an essential role in sensory transduction and suggest a role for synapsin phosphorylation in this process.
Materials and Methods
Mice.
Male wild-type (WT) mice (Swiss–Webster and C57BL/6 mice; 30–40 g each) were obtained from Harlan Breeders (Indianapolis) or Charles River Breeding Laboratories. Male and female synapsin DKO (27–40 g) or Rab3A knockout (KO) mice (27–40 g) were developed from the same genetic background (C57BL/6) in T.C.S. laboratory (27, 28). Rab3A is a small GTP-binding protein that is abundant in synaptic vesicles. Unlike synapsins, Rab3A does not have Ser-Thr phosphorylation sites that are amenable to dephosphorylation by calcineurin (27). All research protocols were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Research Advisory Committee.
Hemodynamic and Neurophysiological Measurements.
Anesthesia was induced with ketamine (80 mg/kg i.p.) and methohexital (60 mg/kg i.p.), an ultrashort-acting barbiturate. The right jugular vein and left carotid artery were cannulated for drug infusion and BP measurement. The trachea was cannulated for mechanical ventilation and atropine administered (0.5 mg/kg i.p.) to minimize tracheal secretions. Anesthesia was maintained with α-chloralose (60 mg/kg i.v.), which causes less depression of sympathetic reflexes than long-acting barbiturates (29, 30). The left renal nerve was exposed via a retroperitoneal incision. Multifiber efferent SNA was recorded from the intact left renal nerve. In separate experiments, multifiber afferent renal nerve activity was recorded from the cut distal end of the left renal nerve. The nerve fibers were affixed to bipolar platinum electrodes by using dental silicone rubber (Bisico S4i, Bielefeld, Germany) as described (31). The nerve action potentials were detected by a high-impedance probe (Grass Model P511K, Quincy, MA), amplified (gain 5–10 K), and filtered (bandwidth 0.1–1 kHz). All data were simultaneously collected and processed by Maclab System (AD Instruments Ply, Castle Hill, Australia) and stored for offline analysis. Nerve action potentials were counted by using a window discriminator, with the threshold level of detection being set just above the noise level and held constant throughout the experiment. At the end of each experiment, euthanasia was performed (methohexital 100 mg/kg i.v.); any remaining electrical activity was considered noise, which was subtracted from the total activity recorded during the experiment to obtain an estimate of the true neural activity.
Bilateral VGX.
In some WT mice, bilateral VGX was performed by cutting both left and right vagal nerves at lower cervical level (3). In the rat, VGX abrogates the reflex SNA and BP responses to CsA by interrupting the central projections of sensory nerves arising in the kidney, urinary bladder, genitalia, and other subdiaphragmatic (mainly retroperitoneal) viscera (3).
Infusion of Immunophilin Ligands and Other Drugs.
CsA (Novartis, Basel, Switzerland) was infused i.v. over 10 min to a total dose of 10 mg/kg. FK506 (a gift from Fujisawa USA, Deerfield, IL) and rapamycin (Research Biochemicals, Natick, MA) each were infused over 10 min to a total dose of 0.3 mg/kg. All agents were dissolved in the same vehicle (Cremaphor EL, Sigma) as described (5). These produce comparable immunosuppression and hemodynamic responses in rodents (3, 5, 6). Arterial BP and efferent or afferent renal nerve activity were continuously recorded for about 10 min after the end of the infusion.
At the end of each experiment, we assessed: (i) the integrity of the sinoaortic baroreflex by measuring reflex increases in renal SNA induced by lowering BP with nitroprusside (10 μg/kg i.v.); (ii) vascular α-adrenergic sensitivity by measuring the pressor responses to phenylephrine (3 μg/kg i.v.); and (iii) the viability of the renal afferents by measuring the responses of renal afferent discharge to bolus injections of capsaicin (10 μg/kg i.v.).
Immunohistochemistry.
For tissue fixation, synapsin DKO, Rab3A KO, and C57BL/6 mice underwent left ventricular perfusion with 50 ml of 2% paraformaldehyde/0.15% picric acid in PBS (32). The kidneys were removed and fixation continued by overnight immersion at 4°C in the same fixative before transfer to 24–48 h PBS rinse at 4°C. The kidneys were subsequently bisected transversely, embedded in Tissue-TEK OCT matrix, and snap-frozen in supercooled isopentane. The cryoembedments were stored at −80°C. Transverse cryosections were cut at 8 μm by using a Leica CM 3000 Cryostat at −22°C, adhered to Superfrost Plus microscope slides, air dried, and held at −80°C. Cryosections were stained for substance P and synapsin immunoreactivity by an indirect immunofluorescent method. The sections were hydrated in PBS, permeabilized in 0.5% Triton/PBS, washed in PBS, and then incubated in 50 mM ammonium chloride to quench autofluorescence of blood vessel endothelium, and again washed in PBS. Nonspecific immunoreactivity was blocked by incubation of 20 μg/ml goat anti-mouse IgG Fab fragment/3% normal goat serum/PBS before incubation of primary antibodies. Polyclonal rabbit antisera to synapsin I and II (1:1,000), and monoclonal rat anti-substance P (1:60) were diluted in 2% normal goat serum/PBS and applied to sections for overnight incubation at 4°C. Unreacted primary antibodies were washed from the sections in PBS followed by a 40-min incubation with fluorescent secondary antibodies diluted in 2% normal goat serum in PBS. FITC-conjugated goat anti-rabbit IgG (1:100) and Cy 3-conjugated goat anti-rat IgG (1:50) were used, respectively, for synapsin I and II, and substance P labeling. After the incubation, these unreacted secondary antibodies were washed off in PBS (three times for 5 min), and the sections were cover-slipped with Vectashield fluorescence-mounting media. Staining was visualized by using a Leitz Laborlux-S epifluorescence microscope equipped with Endow-GFP and CY3 single band-pass filters. A computer with optronics VI-470 CCD camera and Scion CG7 frame was used for image capture. Layering and composition of obtained images was performed by using SCION IMAGE 1.62 and Adobe PHOTOSHOP 5.0 software.
Substance P Measurements.
Synapsin DKO, Rab3A KO, and C57BL/6 mice underwent left ventricular perfusion with PBS/10 units/ml heparin. The kidneys were immediately removed, weighed, dropped in liquid nitrogen, and held at −80°C until further processing. The frozen kidneys were homogenized in 10 vol of 1 M acetic acid/0.01% mercaptoethanol (33). The homogenate was centrifuged at 14,000 × g for 10 min at 4°C. The supernatant was diluted 1:4 with 4% acetic acid, and passed slowly through the methanol-activated C-18 reverse-phase cartridge (J & W Scientific, Folsom, CA). The reverse-phase cartridge was washed with 10 ml of 4% acetic acid; then the substance P was eluted with ethanol/water/acetic acid (90:10:0.4), and lyophilized by vacuum centrifugation. The samples were reconstituted before the measurement. A substance P enzyme immunoassay kit (Cayman Chemicals, Ann Arbor, MI) was used for the assay (ELISA). The 50% bound/maximum bound of the standard curve was 18.6 pg/ml, and both intra- and interassay coefficients of variation were <10%. The detection limit (80% bound/maximum bound) was 2.9 pg/ml.
Statistics.
Statistical analyses were performed with ANOVA, followed by Newman-Keuls post hoc test. P < 0.05 was considered statistically significant. All values are expressed as means ± SE.
Results
CsA Causes Sympathetic Activation and Acute Hypertension in WT Mice.
To use synapsin DKO for studying the role of synapsin in the neural control of BP in the intact animal, we first miniaturized our experimental preparation to reestablish our rat model in WT mice. In these mice, CsA produced increases in BP and renal SNA (Fig. 1a) that were similar to those reported in anesthetized rats (3, 5–7). Mean arterial pressure (MAP) increased by 14 ± 4 mmHg (P < 0.05), and SNA increased progressively to a value that was 175 ± 52% over baseline (P < 0.05) (Fig. 1b). Furthermore, MAP and SNA were increased by FK506 but unaffected by rapamycin (or vehicle) (12), indicating calcineurin mediation (Fig. 1b). We also demonstrated that the CsA-induced increases in MAP and SNA are almost abrogated by bilateral cervical VGX (Fig. 1c). These mouse data replicate the salient features of our previous rat model, suggesting that the sympathetic activation is a reflex caused by calcineurin inhibition in renal and other subdiaphragmatic visceral chemosensitive nerve ending (6, 13). Thus, we were poised to probe underlying molecular mechanisms of this hypertension by using genetically engineered mouse models.
Figure 1.
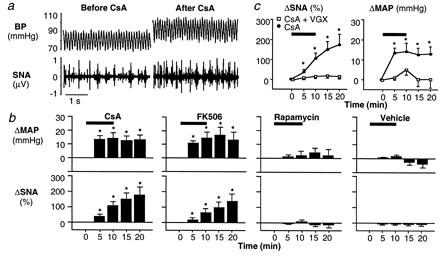
Effects of immunophilin ligands on arterial BP and renal efferent SNA in WT mice. (a) Effects of CsA in a WT mouse. Pulsatile BP and segments of multifiber action potentials of renal efferent SNA were recorded before and after CsA (10 mg/kg i.v.). In this WT mouse, BP and renal efferent SNA increased after CsA. (b) Effects of three immunophilin ligands in WT mice. Changes in MAP and renal efferent SNA were recorded during and after i.v. infusion of CsA (10 mg/kg, n = 7), FK506 (0.3 mg/kg, n = 11), rapamycin (0.3 mg/kg, n = 5), or vehicle (n = 5). MAP and renal efferent SNA increased similarly with CsA or FK506, but were unaffected by rapamycin or vehicle, suggesting calcineurin mediation. (c) Effects of VGX in WT mice. Changes in renal efferent SNA and MAP were recorded during and after CsA in intact WT mice and those with bilateral cervical VGX. CsA-induced increases in renal efferent SNA and MAP were almost abrogated by VGX (n = 7), indicating vagal afferent mediation of these reflex responses. Horizontal bars denote the period of CsA, FK506, rapamycin, or vehicle infusion. *, P < 0.05, compared with baseline (time 0).
CsA-Induced Hypertension and Sympathetic Activation Are Greatly Attenuated in Synapsin DKO Mice.
Synapsin DKO mice are viable, fertile, and survive to adulthood (20, 28). Under anesthesia, basal MAP (mmHg) and heart rate (beats per min) in synapsin DKO (77 ± 5 and 390 ± 24, n = 15) were comparable to those in both WT (85 ± 5 and 413 ± 32, n = 7) and Rab3A KO mice (82 ± 11 and 421 ± 49, n = 5, P = not significant). We found that CsA-induced increases in MAP and renal SNA were greatly attenuated in the DKO mice (Fig. 2 a and b). In contrast, these increases were well preserved in control mice, for example, in mice deficient only in Rab3A (Fig. 2b).
Figure 2.
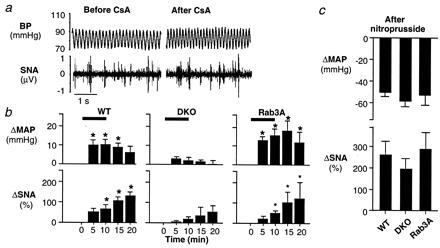
Attenuated effects of CsA on arterial BP and renal efferent SNA in synapsin I and II DKO mice. (a) Attenuated effect of CsA in a DKO mouse. CsA (10 mg/kg i.v.) had little or no effect on BP or renal efferent SNA in this synapsin-deficient mouse. (b) Effects of synapsin deficiency on responses to CsA. Changes in MAP and renal efferent SNA were recorded during and after CsA (10 mg/kg i.v.) in WT (n = 7), Rab3A KO (n = 5), and synapsin DKO (n = 15) mice. In both WT and Rab3A KO mice, MAP and renal efferent SNA increased significantly with CsA. These increases were greatly attenuated in the DKO mice. (c) Effect of synapsin deficiency on the sinoaortic baroreceptor reflexes. Baroreflex function was compared in WT (n = 5), DKO (n = 7), and Rab3A KO (n = 5) mice by measuring the peak reflex increase in renal efferent SNA evoked by decreasing MAP with nitroprusside (5–10 μg/kg i.v.). In synapsin DKO, comparable decreases in MAP to those produced in the two groups of control mice evoked comparable increases in renal efferent SNA, indicating a preserved baroreceptor reflex. Horizontal bars denote the period of CsA infusion. *, P < 0.05, compared with baseline (time 0).
To further investigate the specificity of the attenuated sympathoexcitatory responses to CsA in the DKO mice, we examined the sinoaortic baroreflex by decreasing BP with nitroprusside (Fig. 2c). In the DKO mice, baroreflex-mediated changes in SNA were indistinguishable from those observed in WT and Rab3A KO mice, indicating an intact sinoaortic baroreceptor reflex. Furthermore, the increases in MAP induced by phenylephrine in DKO mice (47 ± 6 mmHg, n = 11) were indistinguishable from those observed in WT (40 ± 6, n = 5), and Rab3A KO mice (45 ± 5, n = 5), indicating preserved vascular α-adrenergic sensitivity to agonist.
Synapsin Immunoreactivity Is Present in WT Mouse Renal Afferent Nerves.
If synapsin is involved in the process that CsA stimulates the renal and other subdiaphragmatic afferents by calcineurin inhibition, which reflexively increases efferent SNA and BP (6, 13), we first must provide evidence that synapsins normally are present in these sensory nerve endings. In WT and Rab3A KO kidney, we demonstrated the colocalization of immunohistochemical staining for synapsin I and II with that for substance P, a specific marker for afferent (rather than efferent) renal nerves (Fig. 3). This colocalization was demonstrated not only in the renal cortex (Fig. 3), but also in renal pelvis and adventitia of arterioles (data not shown). Substance P staining was also evident in synapsin DKO kidney, indicating that synapsins are not essential for the afferent nerve development. Furthermore, substance P concentration in kidney from synapsin DKO was indistinguishable from that in kidney from WT or Rab3A KO. Substance P concentrations (pg/mg renal tissue) were 0.317 ± 0.053 in WT vs. 0.275 ± 0.010 in DKO vs. 0.288 ± 0.046 in Rab3A (n = 3 each, P = not significant).
Figure 3.
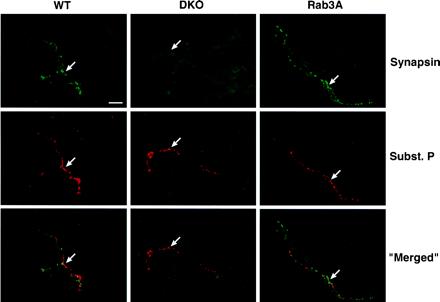
Immunohistochemical staining for synapsin and substance P in mouse kidney. Cross sections of renal cortex from a WT, synapsin DKO, or Rab3A KO mouse were subjected to immunohistochemical staining for synapsin I and II (green, Top) and substance P (red, Middle), a selective marker for afferent nerve endings. (Bottom) The merged images denote areas of double staining (yellow-orange, Bottom). (Horizontal bar = 20 μm.) Within each genotype, arrows denote the same area of interest on all three panels. In renal cortex from both WT and Rab3A KO mice, partial colocalization of synapsin and substance P indicates the presence of synapsin in renal afferent nerve endings. In renal cortex from the DKO mouse, substance P is detectable despite synapsin deficiency, indicating that synapsins are not essential for the development of renal afferents. Similar observations were made in sections of renal pelvis (data not shown).
CsA-Induced Activation of Afferent Renal Nerve Activity Is Impaired in Synapsin DKO Mice.
Having demonstrated the presence of synapsins in WT mouse renal afferent endings, we asked whether the ability of CsA to activate renal afferents in vivo is impaired in synapsin DKO mice. A dose of CsA (10 mg/kg i.v.) that doubled multifiber renal afferent activity in WT mice had little effect in synapsin DKO mice (Fig. 4 a and b). In contrast, in both WT and synapsin DKO mice, afferent renal nerve activity increased comparably (tripled) in response to capsaicin (Fig. 4c), which stimulates nonmyelinated (C-fiber) afferent nerves by enhancing the release of substance P.
Figure 4.
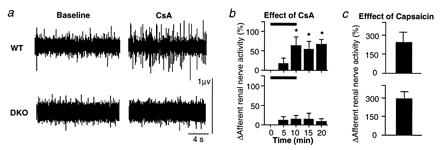
Effects of synapsin deficiency on the activation of renal afferents. (a) Multifiber afferent renal nerve activity was recorded from the cut distal end of the renal nerve at baseline and after CsA. Action potential firing increased with CsA in the WT mouse but was unaffected by CsA in the synapsin DKO mouse. (b) Effects of CsA in WT and synapsin DKO mice. Changes in afferent renal nerve activity in response to CsA (10 mg/kg i.v.) are shown in WT (n = 6) and synapsin DKO (n = 7) mice. Horizontal bars denote the period of CsA infusion. This dose of CsA that doubled afferent renal nerve activity in WT had no effect on the afferent activity in the DKO mice. (c) Effects of capsaicin in WT and synapsin DKO mice. Changes in afferent renal nerve activity in response to capsaicin (5–10 μg/kg i.v.) are shown. Capsaicin, which activates a nonselective cation channel involved in release of substance P, produced comparatively large increases in afferent renal nerve activity in WT and synapsin DKO mice, demonstrating the viability of the afferents. *, P < 0.05, compared with baseline (time = 0).
Discussion
Synapsins are known to be expressed not only on small synaptic vesicles, where they appear to play a major role in processes of short-term synaptic plasticity and neurotransmission, but also on small vesicles in somatic sensory afferents (23–26). The functions of these microvesicles and synapsins in these vesicles are unknown. Here we show that synapsins are present in mouse renal (i.e., visceral) sensory afferents where they are required for the BP-raising effects of calcineurin inhibitors such as CsA. These results suggest a fundamental function of sensory microvesicles in sensory transduction and an involvement of synapsin phosphorylation in acute CsA-induced hypertension.
Synapsin DKO mice provided an opportunity to probe the role of synapsin in the neural control of BP in the intact animal. However, to use this genetically engineered model, we first needed to reestablish the salient features of our rat model of acute CsA-induced hypertension in WT mice. In two different strains, i.v. CsA reproducibly stimulated afferent renal nerve activity and reflexively increased efferent SNA and BP. These increases were of somewhat smaller magnitude than those seen previously in rats (3, 5, 6). The evidence for calcineurin mediation of CsA-induced sympathetic activation in WT mice is that SNA and BP were increased by FK506 as well as CsA but unaffected by rapamycin. The evidence for renal afferent mediation is that the increases in renal SNA were (i) accompanied by parallel increases in afferent renal nerve activity, and (ii) abrogated by VGX, which interrupts the central projections of these and other subdiaphragmatic visceral afferents to the vagal nodosal ganglion.
The major new phenotype in synapsin DKO mice is a marked but selective attenuation of the increases in the afferent and efferent renal nerve activity and BP evoked by i.v. CsA. The attenuated sympathetic response to CsA in the DKO mice is specifically related to synapsin deficiency, because sympathetic activity increased normally when CsA was administered to Rab3A KO mice from the same genetic background (27). The attenuated sympathetic response to CsA in the synapsin DKO mice is not caused by some global defect in sympathetic neural responsiveness, because SNA increased normally in response to baroreceptor deactivation (a non-CsA stimulus). The evidence that synapsin plays a critically important role in the acute BP-raising effect of CsA is that in the synapsin DKO mice, the pressor response to CsA was greatly attenuated even though the pressor response to phenylephrine was well preserved, suggesting normal sensitivity of postjunctional vascular α-adrenergic receptors.
Thus, the primary autonomic defect in the synapsin-deficient mice is that CsA fails to stimulate the renal (and other subdiaphragmatic vagal) afferents that reflexively increase efferent SNA and BP. This defect appears to be functional rather than structural, because in the DKO mice, (i) renal sensory nerve endings develop normally, as evidenced by a normal tissue concentration of substance P; and (ii) renal afferent activity could be readily increased by i.v. capsaicin, which documents the viability of the afferents.
Previous studies have shown that in the rat, i.v. CsA evokes a generalized activation of subdiaphragmatic vagal afferents involving not only renal but also genitofemoral and hypogastric afferents (7). These afferents mainly exert an excitatory effect on SNA (34). In contrast, i.v. CsA did not have an effect on cardiopulmonary vagal afferents (6), which reflexively inhibit SNA (35). The mechanism of the preferential activation of excitatory subdiaphragmatic afferents is unknown, but may be related to differential tissue expression of specific isoforms of immunophilins, calcineurin, or synapsins.
Microvesicles containing synapsin I-like immunoreactivity are present in sensory nerve terminals of the muscle spindle and Golgi tendon organ (23), peripheral vestibular organ (24), lingual taste buds (25), and the mechanosensory neurons of the lyriform slit-sense organ VS-3 and tactile hair sensilla of the Cupiennius salei spider (26). However, the function of synapsin-containing microvesicles in sensory nerve endings is unknown. Our study shows that synapsin is present in renal afferent nerve endings (evidenced by the colocalization of synapsin with substance P in WT mouse kidney) where it plays an important role in mediating the reflex increase in BP induced by CsA.
From the present in vivo mouse experiments, we hypothesize that synapsin phosphorylation-dephosphorylation mediates the ability of calcineurin inhibitors to augment renal afferent discharge. Under physiological conditions, these afferents are thought to signal the central nervous system of changes in the chemical composition of the urine and in the mechanical forces in the renal pelvis, thereby evoking reflex neurocirculatory responses to maintain renal homeostasis (36–38). There is evidence that these afferents are sensitive to activation by uremic or ischemic metabolites (37, 38). Our working hypothesis is that a calcineurin-synapsin pathway is part of a negative feedback mechanism that normally prevents excessive calcium-dependent depolarization of renal afferent endings. During generator potential formation evoked by endogenous stimuli, entry of calcium through voltage-gated channels could activate calcineurin that would then dephosphorylate synapsin, resulting in stabilization of microvesicles in the sensory nerve endings. When CsA inhibits calcineurin, this feedback mechanism would be removed, permitting increased exocytotic release of neurotransmitter-like substances that act locally to augment membrane depolarization. Regardless of the precise molecular mechanisms involved, the present data suggest that synapsins may be a novel drug target for treating refractory hypertension caused by CsA, FK506, or other conditions known to stimulate excitatory renal afferents (e.g., renovascular or renal parenchymal hypertension) (39–42). We should emphasize that the present experiments model the acute CsA-induced hypertension. In the clinical setting, CsA-induced hypertension is undoubtedly multifactorial and mechanisms other than those elucidated here (e.g., increased release of endothelin and expression of endothelin receptors, renal sodium retention) are likely to play a more important role in chronic rather than acute CsA-induced hypertension (1, 43).
Acknowledgments
This work was supported by National Heart Lung and Blood Institute Grant HL 44010.
Abbreviations
- CsA
cyclosporine A
- BP
blood pressure
- SNA
sympathetic nerve activity
- DKO
double knockout
- WT
wild type
- KO
knockout
- MAP
mean arterial pressure
- VGX
vagotomy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170160397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170160397
References
- 1.Sander M, Lyson T, Thomas G D, Victor R G. Am J Hypertens. 1996;9:121S–138S. doi: 10.1016/0895-7061(96)00288-9. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan N M. Clinical Hypertension. Baltimore: Williams & Wilkins; 1998. pp. 1–402. [Google Scholar]
- 3.Morgan B J, Lyson T, Scherrer U, Victor R G. Hypertension. 1991;18:458–466. doi: 10.1161/01.hyp.18.4.458. [DOI] [PubMed] [Google Scholar]
- 4.Chiu P J, Vemulapalli S, Sabin C, Rivelli M, Bernardino V, Sybertz E J. J Pharmacol Exp Ther. 1992;261:994–999. [PubMed] [Google Scholar]
- 5.Lyson T, Ermel L D, Belshaw P J, Alberg D G, Schreiber S L, Victor R G. Circ Res. 1993;73:596–602. doi: 10.1161/01.res.73.3.596. [DOI] [PubMed] [Google Scholar]
- 6.Lyson T, McMullan D M, Ermel L D, Morgan B J, Victor R G. Hypertension. 1994;23:667–675. doi: 10.1161/01.hyp.23.5.667. [DOI] [PubMed] [Google Scholar]
- 7.Moss N G, Powell S L, Falk R J. Proc Natl Acad Sci USA. 1985;82:8222–8226. doi: 10.1073/pnas.82.23.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis L S, Haleen S J, Doherty A M, Cody W L, Keiser J A. J Am Soc Nephrol. 1994;4:1448–1454. doi: 10.1681/ASN.V471448. [DOI] [PubMed] [Google Scholar]
- 9.Brooks D P, Contino L C. Eur J Pharmacol. 1995;294:571–576. doi: 10.1016/0014-2999(95)00591-9. [DOI] [PubMed] [Google Scholar]
- 10.Cavarape A, Endlich K, Feletto F, Parekh N, Bartoli E, Steinhausen M. Kidney Int. 1998;53:963–969. doi: 10.1111/j.1523-1755.1998.00852.x. [DOI] [PubMed] [Google Scholar]
- 11.Snyder S H, Lai M M, Burnett P E. Neuron. 1998;21:283–294. doi: 10.1016/s0896-6273(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 12.Hemenway C S, Heitman J. Cell Biochem Biophys. 1999;30:115–151. doi: 10.1007/BF02737887. [DOI] [PubMed] [Google Scholar]
- 13.Zhang, W. & Victor, R. G. (2000) Am. J. Hypertens., in press. [DOI] [PubMed]
- 14.Johnson E M, Ueda T, Maeno H, Greengard P. J Biol Chem. 1972;274:5650–5652. [PubMed] [Google Scholar]
- 15.King M M, Huang C Y, Chock P B, Nairn A C, Hemmings H C, Jr, Chan K F, Greengard P. J Biol Chem. 1984;259:8080–8083. [PubMed] [Google Scholar]
- 16.Ebihara K, Fukunaga K, Matsumoto K, Shichiri M, Miyamoto E. Endocrinology. 1996;137:5255–5263. doi: 10.1210/endo.137.12.8940343. [DOI] [PubMed] [Google Scholar]
- 17.Steiner J P, Dawson T M, Fotuhi M, Snyder S H. Mol Med. 1996;2:325–333. [PMC free article] [PubMed] [Google Scholar]
- 18.Sudhof T C, Czernik A J, Kao H T, Takei K, Johnston P A, Horiuchi A, Kanazir S D, Wagner M A, Perin M S, De Camilli P, et al. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- 19.Hosaka M, Sudhof T C. J Biol Chem. 1998;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Chacon R, Sudhof T C. Annu Rev Physiol. 1999;61:753–776. doi: 10.1146/annurev.physiol.61.1.753. [DOI] [PubMed] [Google Scholar]
- 21.Czernik A J, Pang D T, Greengard P. Proc Natl Acad Sci USA. 1987;84:7518–7522. doi: 10.1073/pnas.84.21.7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sudhof T C. Nature (London) 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 23.De Camilli P, Vitadello M, Canevini M P, Zanoni R, Jahn R, Gorio A. J Neurosci. 1988;8:1625–1631. doi: 10.1523/JNEUROSCI.08-05-01625.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Favre D, Scarfone E, Di Gioia G, De Camilli P, Dememes D. Brain Res. 1986;384:379–382. doi: 10.1016/0006-8993(86)91176-5. [DOI] [PubMed] [Google Scholar]
- 25.Finger T E, Womble M, Kinnamon J C, Ueda T. J Comp Neurol. 1990;292:283–290. doi: 10.1002/cne.902920210. [DOI] [PubMed] [Google Scholar]
- 26.Fabian-Fine R, Volknandt W, Seyfarth E. Cell Tissue Res. 1999;295:13–19. doi: 10.1007/s004410051208. [DOI] [PubMed] [Google Scholar]
- 27.Geppert M, Bolshakov V Y, Siegelbaum S A, Takei K, De Camilli P, Hammer R E, Sudhof T C. Nature (London) 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- 28.Rosahl T W, Spillane D, Missler M, Herz J, Selig D K, Wolff J R, Hammer R E, Malenka R C, Sudhof T C. Nature (London) 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- 29.Tucker B J, Peterson O W, Ziegler M G, Blantz R C. Am J Physiol. 1982;243:F253–F259. doi: 10.1152/ajprenal.1982.243.3.F253. [DOI] [PubMed] [Google Scholar]
- 30.Bedran-de-Castro M T, Farah V M, Krieger E M. Braz J Med Biol Res. 1990;23:1185–1193. [PubMed] [Google Scholar]
- 31.Zhang W, Thoren P. J Pharmacol Toxicol Methods. 1997;37:117–118. doi: 10.1016/s1056-8719(97)00004-x. [DOI] [PubMed] [Google Scholar]
- 32.Knight D S, Beal J A, Yuan Z P, Fournet T S. J Auton Nerv Syst. 1987;21:145–155. doi: 10.1016/0165-1838(87)90017-8. [DOI] [PubMed] [Google Scholar]
- 33.Reiner A R, Krause J E, Keyser K T, Eldred W D, McKelvy J F. J Comp Neurol. 1984;226:50–75. doi: 10.1002/cne.902260105. [DOI] [PubMed] [Google Scholar]
- 34.Stella A. J Hypertens. 1992;10,Suppl.:S113–S119. [PubMed] [Google Scholar]
- 35.Hainsworth R. Physiol Rev. 1991;71:617–658. doi: 10.1152/physrev.1991.71.3.617. [DOI] [PubMed] [Google Scholar]
- 36.Moss N G. Fed Proc. 1985;44:2828–2833. [PubMed] [Google Scholar]
- 37.Moss N G. Miner Electrolyte Metab. 1989;15:59–65. [PubMed] [Google Scholar]
- 38.Stella A, Zanchetti A. Physiol Rev. 1991;71:659–682. doi: 10.1152/physrev.1991.71.3.659. [DOI] [PubMed] [Google Scholar]
- 39.Abboud F M. Hypertension. 1982;4:208–225. [PubMed] [Google Scholar]
- 40.Wyss J M, Oparil S, Sripairojthikoon W. Can J Physiol Pharmacol. 1992;70:759–770. doi: 10.1139/y92-100. [DOI] [PubMed] [Google Scholar]
- 41.Campese V M. J Nephrol. 1997;10:184–187. [PubMed] [Google Scholar]
- 42.DiBona G F, Kopp U C. Physiol Rev. 1997;77:75–197. doi: 10.1152/physrev.1997.77.1.75. [DOI] [PubMed] [Google Scholar]
- 43.Takeda Y, Miyamori I, Wu P, Yoneda T, Furukawa K, Takeda R. Hypertension. 1995;26:932–936. doi: 10.1161/01.hyp.26.6.932. [DOI] [PubMed] [Google Scholar]