

Abstract
Background & Aims:
Tumor necrosis factor (TNF) plays critical roles in intestinal disease. In intestinal epithelia, TNF causes tight junction disruption and epithelial barrier loss by upregulating myosin light chain kinase (MLCK) activity and expression. The aim of this study was to determine the signaling pathways by which TNF causes intestinal epithelial barrier loss.
Methods:
Caco-2 cells that were either non-transfected or stably-transfected with human TNF receptor 1 (TNFR1) or TNFR2 and mouse colonocytes were used for physiological, morphological, and biochemical analyses.
Results:
Colitis induced in vivo by adoptive transfer of CD4+CD45RBhi T cells was associated with increased epithelial myosin light chain kinase (MLCK) expression and myosin II regulatory light chain (MLC) phosphorylation as well as morphological tight junction disruption. In vitro studies showed that TNF caused similar increases in MLCK expression and MLC phosphorylation, as well as barrier dysfunction, in Caco-2 monolayers only after IFN-γ pretreatment. This reductionist model was therefore used to determine the molecular mechanism by which IFN-γ and TNF synergize to cause intestinal epithelial barrier loss. IFN-γ priming increased TNFR1 and TNFR2 expression and blocking antibody studies showed that TNFR2, but not TNFR1, was required for TNF-induced barrier dysfunction. Transgenic TNFR2, but not TNFR1, expression allowed IFN-γ-independent TNF responses.
Conclusions:
IFN-γ primes intestinal epithelia to respond to TNF by inducing TNFR2 upregulation, which in turn mediates the TNF-induced MLCK-dependent barrier dysfunction. The data further suggest that epithelial TNFR2 blockade may be a novel approach to restore barrier function in intestinal disease.
Keywords: tight junction, interferon-γ, tumor necrosis factor, cytoskeleton, myosin, inflammatory bowel disease
Introduction
The intestinal epithelial barrier is frequently compromised in intestinal disease. Although the precise mechanisms of intestinal barrier dysfunction are incompletely understood in human disease, recent data suggest that interferon-γ (IFN-γ) and tumor necrosis factor (TNF) may be important mediators of barrier dysfunction in vitro and in vivo 1-4. Consistent with this, intestinal barrier function is restored by anti-TNF therapy in Crohn’s disease patients 1.
We have recently shown that TNF treatment of cultured intestinal epithelial cells induces barrier dysfunction by a mechanism that requires myosin II regulatory light chain (MLC) phosphorylation 2. We went on to show that this TNF-induced barrier dysfunction only occurred in cultured monolayers that had been primed with IFN-γ 5. In IFN-γ-primed, but not control, monolayers, TNF induced increased MLC kinase (MLCK) expression and increased MLC phosphorylation 5. This led to disruption of the tight junction, the primary determinant of epithelial barrier function 5. To determine the relevance of these in vitro observations to human disease, we studied intestinal epithelia of Crohn’s disease and ulcerative colitis patients. Both MLCK protein expression and MLC phosphorylation were increased and the extent of these increases correlated with disease activity 6. Together with data showing that immune activation correlates with barrier dysfunction in Crohn’s disease patients 7, this suggests that, in human inflammatory bowel disease, intestinal epithelial MLCK expression, MLC phosphorylation, and barrier function are intimately related.
These data also suggest that the mechanisms by which anti-TNF therapy restores the barrier in Crohn’s disease patients may include blockade of TNF-induced epithelial MLCK upregulation. However, it is clear that anti-TNF therapy targets many other aspects of the immune response. For example, anti-TNF therapy has been reported to cause lymphocyte apoptosis 8 and downregulate IFN-γ production by colonic T cells from Crohn’s disease patients 9. While these and other effects of anti-TNF therapy that do not directly target epithelia are likely important in anti-TNF therapy, the global immunomodulation caused by anti-TNF puts patients at increased risk for potentially fatal infectious complications 10-13. It is possible that the risk of these complications could be decreased through more specific therapy using antibodies that selectively block epithelial responses to TNF. Unfortunately, the roles of TNF receptor 1 (TNFR1) or TNFR2 in TNF-induced epithelial barrier dysfunction have not been defined. Our data show that TNFR2, but not TNFR1, is required for TNF-induced barrier dysfunction to occur and that induction of TNFR2 expression is the primary mechanism by which IFN-γ primes intestinal epithelia to respond to TNF with MLCK-dependent tight junction disruption and barrier dysfunction.
Materials and Methods
CD4+CD45RBhi-adoptive transfer.
C57BL/6 and RAG1-/- mice were obtained from Jackson Laboratories (Bar Harbor, ME) and maintained in a specific pathogen-free facility. All experiments were carried out in accordance with National Institutes of Health guidelines under protocols approved by the Institutional Animal Care and Use Committee at the University of Chicago. CD4+CD45RBhi lymphocytes were isolated from C57BL/6 splenocytes using a MO-FLO sorter (Dako, Carpinteria, CA) after staining with allophycocyanin-conjugated anti-CD4 and phycoerythrin-conjugated anti-CD45RB (BD Pharmingen, San Jose, CA). After adoptive transfer of 5x105 CD4+CD45RBhi cells, RAG1-/- mice were monitored for weight loss and clinical evidence of colitis and sacrificed after 10 weeks. RAG1-/- littermates injected with buffer instead of CD4+CD45RBhi cells were used as controls. Colonocytes were isolated as previously described 4 and MLCK expression and MLC phosphorylation were assessed by SDS-PAGE immunoblot. Sections of colon were snap-frozen in OCT and stored at -80°C.
Cell culture and measurement of transepithelial electrical resistance.
Caco-2BBE cell 14, 15 cultures were grown as monolayers on collagen-coated polycarbonate membrane Transwell supports (Corning, Cambridge, MA) with 0.4 μm pores for 17-20 days after confluence, as described previously 5, 14. Transwell supports with 0.33- and 5-cm2 surface areas were used for electrophysiological and biochemical studies, respectively. Transepithelial resistance (TER) was measured with an epithelial voltohmmeter under open circuit conditions (World Precision Instruments, Sarasota, FL) as described previously 5.
Cytokine and blocking antibody treatment.
Recombinant human TNF and IFN-γ (R&D Systems, Minneapolis, MN) were added to the basal chamber, without manipulating the apical media, at 2.5ng/ml and 10ng/ml, respectively, unless indicated otherwise. TNFR1 and TNFR2 blocking antibodies (R&D Systems) were similarly added to the basal chamber at 2 μg/ml.
Semi-quantitative RT-PCR analysis of TNFR1 and TNFR2 mRNA.
Total RNA was isolated from TRIzol (Invitrogen, Carlsbad, CA) extracts of Caco-2BBE monolayers.CDNA was generated from polyA+ mRNA by RT-PCR (Invitrogen). Primers used for analysis of TNFR1 and TNFR2 were: TNFR1 forward beginning at nucleotide 542 5’-TCC TTC ACC GCT TCA GAA AA-3’, TNFR1 reverse ending at nucleotide 960 5’-GGG ATA AAA GGC AAA GAC CAA-3; TNFR2 forward beginning at nucleotide 350 5’-AAC TGG GTT CCC GAG TGC TTG-3’, TNFR2 reverse ending at nucleotide 794 5’-AGT GCT GGG TTC TGG AGT TGG -3’. After 35 cycles of PCR, which preliminary studies showed was within the linear range for these primers and samples, amplified fragments were separated by electrophoresis, stained with ethidium bromide, and visualized under UV light.
Stable transfection of human TNFR1 and TNFR2.
Human TNFR1 cDNA (TNFRSF1A, IMAGE clone 4131360, American Type Culture Collection, Manassas, VA) was subcloned into pCNDA3.1/Zeo+ (Invitrogen) using BamHI and XhoI restriction sites and subsequently verified by sequencing. Human TNFR2 cDNA (TNFRSF1B, IMAGE clone 6198614, Open Biosystems, Huntsville, AL) was subcloned into pcDNA3.1/Zeo+ using KpnI and XhoI restriction sites and verified by sequencing. The pcDNA3.1/Zeo+ expression constructs were transfected separately using 24 μg DNA and 120 μl lipofectamine 2000 (Invitrogen) to produce clones stably expressing TNFR1 or TNFR2. Selection medium containing 300 μg/ml zeocin (Invitrogen) was added 48 hours after transfection to select stably expressing clones. Individual clones were isolated and expanded in media with 300 μg/ml zeocin 16. Expression of TNFR1 or TNFR2 was assessed by SDS-PAGE and immunoblot. After selection stable transfectants were maintained in media with 50 μg/ml zeocin. For experimental use, monolayers were allowed to differentiate in media without zeocin.
SDS-PAGE and immunoblot.
Monolayers grown on 5-cm2 Transwell supports were used for the determination of TNFR1, TNFR2, MLCK, and MLC protein expression as well as MLC phosphorylation. Monolayers were scraped directly into 0.5 ml SDS-PAGE sample buffer, sonicated, separated on SDS-PAGE gels (Bio-Rad, Hercules, CA), and transferred to polyvinylidene difluoride membranes (Bio-Rad). Lysates of isolated colonocytes were processed similarly 4. Immunoblots were performed using antibodies specific for TNFR1, TNFR2, MLCK (clone K36, Sigma, St. Louis, MO), total MLC 4, and phosphorylated MLC 17 followed by HRP-conjugated secondary antibodies (Cell Signaling Technology, Beverly, MA). These were detected by chemiluminescence using Super Signal West Pico Reagents (Pierce Biotechnology Inc, Rockford, IL). Quantitative analysis was performed using Metamorph 6.2 (Universal Imaging Corp., Downingtown, PA).
Immunofluorescent staining, microscopy, and image analysis.
Frozen tissue sections (5 μm) were collected on coated slides, fixed in 1% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, washed thrice with PBS, and nonspecific binding blocked with 1% normal goat serum in PBS. Adjacent frozen sections were stained with hematoxylin and eosin 6. Caco-2BBE cell monolayers grown on 0.33-cm2 Transwell supports were fixed with 1% paraformaldehyde in PBS for 30 min, washed, quenched with 50 mM NH4Cl in PBS, and permeabilized in 0.1% Triton X-100 in PBS. Specimens were then incubated with monoclonal antibody against ZO-1 (Invitrogen) or polyclonal antisera against occludin (Invitrogen), TNFR1, or TNFR2. After washing, tissues or monolayers were incubated with Alexa 488- or Alexa 594-conjugated secondary antisera (Invitrogen) and Hoechst 33342 (Invitrogen), washed, and mounted in Slowfade (Invitrogen). Monolayers were imaged using a Leica DM4000 epifluorescence microscope equipped 63X and 100X PL-APO objectives, DAPI, EGFP, and Texas Red zero pixel shift filter sets (Chroma Technology, Brattleboro, VT), and Coolsnap HQ camera (Roper Scientific, Tucson, AZ) controlled by MetaMorph 6.2. For monolayers, postacquisition deconvolution used Autodeblur × (AutoQuant Imaging, Troy, NY) with standard settings for 10 iterations.
Results
Experimental immune-mediated inflammatory bowel disease is associated with increased MLCK expression, increased MLC phosphorylation, and tight junction disruption in vivo.
Several studies using in vitro 2, 5, 18 and acute in vivo 4 models have shown that MLCK-mediated MLC phosphorylation is central to TNF-induced epithelial tight junction disruption and barrier dysfunction. In the setting of chronic disease, it has recently been reported that intestinal epithelial MLCK expression and MLC phosphorylation are increased in patients with active inflammatory bowel disease and that the magnitude of these changes correlate with the grade of disease activity 6.However, only patients with longstanding disease were evaluated, making it impossible to distinguish between primary and secondary changes. We therefore asked if epithelial MLCK expression and MLC phosphorylation were increased in experimental chronic inflammatory bowel disease. We used the CD4+CD45RBhi T cell transfer into RAG1-/- as an established model of chronic immune-mediated colitis that can be limited by neutralization of either IFN-γ or TNF signaling 19. In this model, weight loss can be used to indicate disease severity, and we too observed significant weight loss after CD4+CD45RBhi adoptive transfer (Fig. 1A). Immunoblot analysis of purified colonocytes showed MLCK expression and MLC phosphorylation were markedly increased in diseased animals (Fig. 1B), suggesting that processes similar to those seen in human inflammatory bowel disease patients 6 are activated in CD4+CD45RBhi adoptive transfer-induced colitis. Histological evidence of active mucosal damage was present (Fig. 1C) and the distribution of tight junction proteins occludin (Fig. 1D) and ZO-1 (Fig. 1E) were disrupted in colonocytes of animals with CD4+CD45RBhi adoptive transfer-induced disease. These data show that increased MLCK expression and activity as well as tight junction disruption occur in an in vivo model of chronic immune-mediated inflammatory bowel disease that depends on both IFN-γ and TNF.
Figure 1.
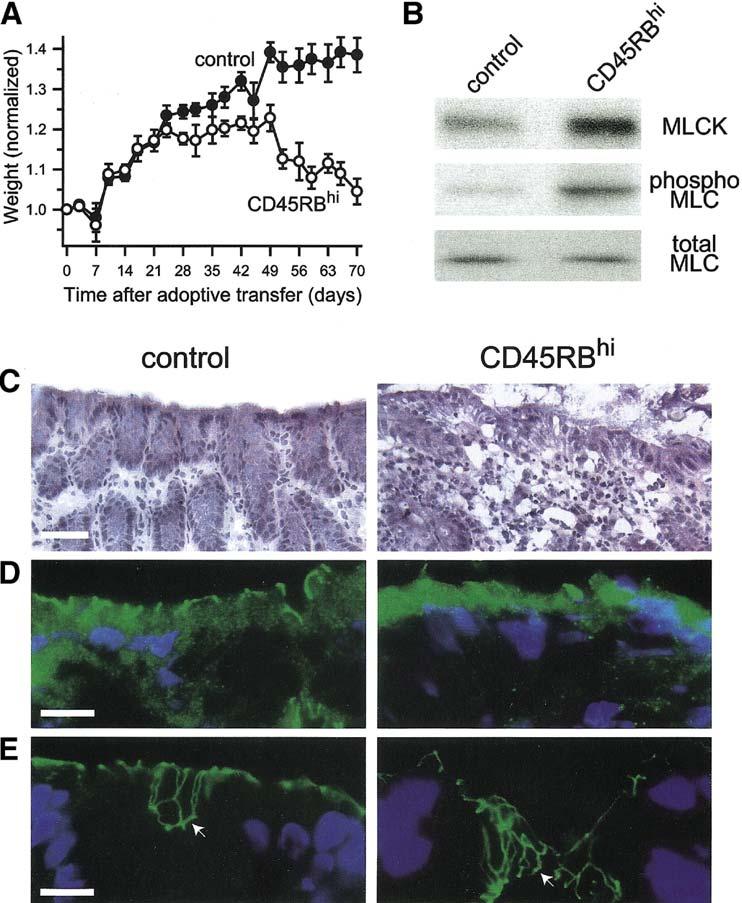
In vivo immune-mediated colitis is associated with increased epithelial MLCK expression, increased MLC phosphorylation, and tight junction disruption.
A. Adoptive transfer of CD4+CD45RBhi T cells (CD45RBhi) causes weight loss. Data are mean ± SE of four mice for each condition and are representative of 2 independent experiments. B. Colonocytes were isolated from control mice or after CD4+CD45RBhi adoptive transfer and analyzed by SDS-PAGE immunoblot for MLCK expression, MLC phosphorylation, and total MLC as a loading control. CD4+CD45RBhi adoptive transfer caused a 3.3±0.5-fold increase in MLCK expression and a 2.7±0.3-fold increase in MLC phosphorylation (p<0.03 for both). Data are mean ± SE of two samples from separate mice for each condition and are representative of 2 independent experiments. C.Histological examination shows that CD4+CD45RBhi adoptive transfer caused chronic inflammatory bowel disease with a moderate activity. Bar = 40 μm. D.Immunofluorescent analysis of occludin (green) localization in colon sections shows marked loss from the epithelial tight junction and appearance in intracellular vesicles after CD4+CD45RBhi adoptive transfer. Nuclei (blue) are shown for orientation. Bar = 10 μm. E. Immunofluorescent analysis of ZO-1 (green) localization in colon sections shows the development of an undulating network at the tight junction (compare areas marked by arrows) after CD4+CD45RBhi adoptive transfer. Nuclei (blue) are shown for orientation. Bar = 10 μm.
IFN-γ primes model epithelia to regulate MLCK and tight junctions in response to TNF
Like the in vivo disease model presented above, we have recently shown that, in terms of MLCK expression and activity as well as tight junction structure, Caco-2 monolayers require both IFN-γ and TNF 5. While TNF causes MLCK upregulation, MLC phosphorylation, tight junction disruption, and barrier dysfunction in monolayers pre-treated with IFN-γ, neither IFN-γ nor TNF alone caused any of these changes (Figs. 2A, 2B). We have previously shown that TNF pre-treatment does not sensitize monolayers to IFN-γ 5. The response to TNF was highly polarized, as IFN-γ-primed Caco-2 monolayers responded to basal, but not apical, TNF (Fig. 2C). Thus, like the in vivo model 19, the requirement for IFN-γ appears to precede that of TNF in this in vitro model of barrier dysfunction.
Figure 2.

IFN -γ primes Caco-2 monolayers for TNF-induced barrier dysfunction.
A. Caco-2 cells were pre-incubated with IFN-γ, 10 ng/ml, for 24 hours prior to transfer to fresh media, without IFN-γ, either with or without TNF, 2.5 ng/ml, as indicated. Monolayers were then harvested and analyzed by SDS-PAGE immunoblot for MLCK expression, MLC phosphorylation, and total MLC as a loading control. When IFN-γ-primed cells were treated with TNF, MLCK expression increased 2.1±0.1-fold and MLC phosphorylation increased 3.0±0.6-fold (p < 0.05 for both). Other treatments did not significantly alter MLCK expression or MLC phosphorylation. B. TER of control monolayers (black triangles) was measured for 8 hours after transfer to fresh media. Addition of TNF, 2.5 ng/ml, to the basal media had no effect on TER (black circles). Pre-incubation with IFN-γ, 10 ng/ml, for 24 hours prior to transfer to fresh media without IFN-γ, also had no effect on TER (white triangles). In contrast, the TER of monolayers pre-incubated with IFN-γ prior dropped within 3 hours after transfer to fresh media with TNF (white circles, p<0.01). Data are mean ± SE of triplicate monolayers. Data are representative of more than 10 independent experiments. C. All monolayers were pre-incubated with IFN-γ, 10 ng/ml, for 24 hours. Transfer to fresh media without cytokines (white bar) had no effect on TER, while transfer to media with 2.5 ng/ml TNF in the basal chamber induced marked TER loss (black bar, p<0.02). Transfer to media with 2.5 ng/ml TNF in the apical chamber had no effect on TER. Data are mean ± SE of triplicate monolayers 8 hours after transfer and were normalized to the overall mean TER of monolayers pre-incubated with IFN-γ. Data are representative of 4 independent experiments.
IFN-γ upregulates both TNFR1 and TNFR2 expression
Based on published reports showing that IFN-γ can upregulate TNF receptor expression 3, 20, we hypothesized that this might be the mechanism by which IFN-γ primes epithelial monolayers to respond to TNF. To determine if IFN-γ did increase expression of TNF receptors, Caco-2 monolayers were treated with IFN-γ for 24 hrs and then harvested for analysis. Semi-quantitative RT-PCR showed that both TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2) mRNA were upregulated after IFN-γ treatment (Fig. 3A). Similarly, SDS-PAGE immunoblot showed that corresponding protein levels of TNFR1 and TNFR2 were increased by IFN-γ treatment (Fig. 3A). Morphometric analyses confirmed this upregulation of TNFR1 and TNFR2 and showed that both proteins were localized to the basolateral membranes of polarized Caco-2 cells (Fig. 3B). This localization is consistent with the response of IFN-γ-primed monolayers to basolateral, but not apical, TNF.
Figure 3.

IFN-γ upregulates both TNFR1 and TNFR2 in Caco-2 monolayers.
A. RT-PCR and SDS-PAGE immunoblot analyses show that both TNFR1 and TNFR2 mRNA and protein expression are upregulated after 24 hours of culture with IFN-γ added to the basal chamber (p<0.01 for both). Data are representative of 4 independent experiments, each with duplicate samples. B. Immunofluorescence of monolayers before or after 24 hours of culture with IFN-γ confirms increased surface expression of TNFR1 and TNFR2. The images shown were taken at focal planes basal to the tight junction and TNF receptors were only detected on lateral membranes. Exposures and post-processing were identical for images of each TNF receptor. Bar = 10 μm.
TNFR2 mediates TNF-induced barrier dysfunction
Of the two TNF receptors described, many more biological roles have been reported for TNFR1. These include systemic responses to bacterial products 21, 22, innate immunity 21, 22, inhibition of intestinal epithelial wound closure 23, and induction of intestinal epithelial apoptosis 24. In contrast, TNFR2 expressed on lymphocytes has been reported to be necessary for the development of intestinal graft vs. host disease 25 and TNFR2 expressed on intestinal epithelial cells seems to mediate the increased epithelial migration induced by TNF 23. To determine whether one or both TNF receptors mediated TNF-induced barrier dysfunction, IFN-γ-primed Caco-2 monolayers were pre-incubated with functional blocking antibodies directed against either TNFR1 or TNFR2. In the absence of TNF, neither antibody had any effect on barrier function (Fig. 4). Moreover, TNFR1 blocking antibodies did not prevent or reduce TNF-induced barrier dysfunction. In contrast, TNFR2 blocking antibodies completely prevented TNF-induced barrier dysfunction (Fig. 4). Thus, the effect of TNF on barrier function in IFN-γ-primed intestinal epithelia requires TNFR2, but not TNFR1.
Figure 4.

Blockade of TNFR2, but not TNFR1, prevents TNF-induced barrier dysfunction.
All monolayers used in this experiment were pre-incubated with IFN-γ, 10 ng/ml, for 24 hours prior to experimental use. These IFN-γ-primed monolayers were then incubated in media with indicated TNFR1 or TNFR2 blocking antibodies, 2 μg/ml, in the basal compartment for 30 minutes prior to addition of TNF, 2.5 ng/ml, to the basal chamber. Only TNFR2 blocking antibodies prevented TNF-induced TER loss (p<0.02). Data are mean ± SE of triplicate monolayers 8 hours after TNF addition. Data are representative of 3 independent experiments.
IFN-γ primes Caco-2 monolayers to respond to TNF by upregulating TNFR2 expression
Treatment of intestinal epithelial cells with IFN-γ has been reported to modify expression of many proteins, including class II HLA 3, 26-28, transcellular transporters 29, and tight junction components 30. Thus, while the data above suggest that IFN-γ-induced TNFR2 upregulation is required for TNF-induced barrier dysfunction to occur, they do not exclude the possibility that IFN-γ-priming also modifies expression of other essential mediators. To determine if the primary role of IFN-γ in priming Caco-2 monolayers to regulate barrier function in response to TNF is induction of TNFR2 expression, Caco-2 cell lines were stably transfected with human TNFR1 or TNFR2. Immunoblot analysis confirmed that these stable transfectants expressed TNF receptors at levels comparable to those induced by IFN-γ priming of nontransfected monolayers (Figs. 5A, 6A). Transfected monolayers were then assessed for their ability to respond to TNF without IFN-γ priming. As shown in Fig. 5B, Caco-2 monolayers stably expressing TNFR1 did not respond to TNF under these conditions. However, they were able to regulate barrier function in response to TNF after IFN-γ-priming (Fig. 5C). Similar results were obtained with two additional TNFR1-transfected clones. Thus, stable expression of TNFR1 did not eliminate the need for IFN-γ priming. When analyzed in an identical manner, monolayers of cells stably expressing TNFR2 responded to TNF despite the absence of IFN-γ priming (Fig. 6B). This occurred in a dose-dependent manner and was quantitatively similar to the response to TNF after IFN-γ pretreatment (Fig. 6C). Similar results were obtained with three different TNFR2-transfected clones. Thus, the requirement for IFN-γ priming is abrogated by transgenic TNFR2 expression. These data show that the primary role of IFN-γ in priming intestinal epithelial cells to regulate barrier function in response to TNF is induction of TNFR2 expression.
Figure 5.

Transgenic TNFR1 expression does not confer TER responses to TNF in the absence of IFN-γ-priming.
A. SDS-PAGE immunoblot of TNFR1 comparing two independently-generated TNFR1-transfected clones and nontransfected control and IFN-γ-primed Caco-2 monolayers. Data are representative of 3 independent experiments.
B. Treatment of TNFR1-transfected monolayers with TNF at doses from 0.1 ng/ml to 10 ng/ml does not induce significant TER loss. Data are mean ± SE of triplicate monolayers normalized to TER of monolayers not treated with TNF. Data are representative of 3 independent experiments.
C. Although TNFR1-transfected monolayers are not responsive to TNF without IFN-γ, they are able to regulate TER in response to TNF, 2.5 ng/ml, after IFN-γ-priming (p<0.02). Data are mean ± SE of triplicate monolayers 8 hours after transfer to TNF. Data were normalized to TER prior to TNF treatments and are representative of 4 independent experiments.
Figure 6.

Transgenic TNFR2 expression abrogates the requirement for IFN-γ-priming.
A. SDS-PAGE immunoblot of TNFR2 comparing two independently-generated TNFR2-transfected clones and nontransfected control and IFN-γ-primed Caco-2 monolayers. Data are representative of 3 independent experiments.
B. Treatment of TNFR2-transfected monolayers with TNF at doses from 0.1 ng/ml to 10 ng/ml shows dose-dependent TER loss (p<0.02). Data are mean ± SE of triplicate monolayers normalized to TER of monolayers not treated with TNF. Data are representative of 3 independent experiments.
C. TNFR2-transfected monolayers are similarly responsive to 2.5 ng/ml TNF in the presence or absence of IFN-γ-priming. Data are mean ± SE of triplicate monolayers 8 hours after transfer and were normalized to TER prior to transfer. Data are representative of 4 independent experiments.
TNFR2-mediated barrier dysfunction is associated with tight junction disruption
The data above show that expression of both TNFR1 and TNFR2 are upregulated by IFN-γ priming, but only TNFR2 is required for TNF to disrupt barrier function. Epithelial barrier function is dependent on both the integrity of the epithelium and the permeability characteristics of the tight junction. To determine which of these was affected by TNFR2-mediated TNF signaling, TNFR2-expressing monolayers were assessed before and after TNF treatment. Although some have reported that TNF-dependent effects on barrier function are due to apoptosis 31, 32, we and others have reported that TNF-dependent barrier dysfunction can occur in the absence of significant epithelial apoptosis, both in vitro and in vivo 2, 4, 5, 33. TNF-treatment of TNFR2-expressing monolayers, without IFN-γ priming, did not increase epithelial apoptosis, as determined by assessment of caspase-3 and caspase-9 cleavage (Fig. 7A) and nuclear fragmentation (Fig. 7B). Thus, overall epithelial integrity was not compromised and apoptosis was not induced by TNF treatment of TNFR2-expressing monolayers. Together with the absence of induction of short circuit current by TNF treatment (data not shown), these observations also suggest that the effect of TNF on epithelial barrier function is primarily due to increases in paracellular, rather than transcellular, permeability.
Figure 7.

Treatment with TNF at 2.5 ng/ml does not induce apoptosis of TNFR-transfected Caco-2 cells.
A. Increased cleavage of caspase-3 and caspase-9 cleavage are not detected in TNFR1- or TNFR2-transfected 8 hours monolayers after TNF treatment. IFN-γ-primed non-transfected monolayers, without TNF treatment, are shown for reference. Data are representative of 3 independent experiments.
B. Nuclear fragmentation is induced by 100 ng/ml TNF, but not 2.5 ng/ml TNF, in IFN-γ primed monolayers. TNF, 2.5 ng/ml, does not induce nuclear fragmentation in TNFR1-nor TNFR2-transfected monolayers. Data are representative of 3 independent experiments. Bar = 10 μm.
Having excluded gross TNF-induced damage to the monolayer, we considered the possibility that tight junctions are disrupted by TNF via TNFR2-mediated signaling. TNF treatment of TNFR2-expressing monolayers induced marked reorganization of occludin from smooth arcs into irregularly undulated profiles as well as internalization into cytoplasmic vesicles (Fig. 8). Similar changes were induced in nontransfected monolayers after, but not before, IFN-γ-priming (Fig. 8). Like occludin, ZO-1 distribution in TNFR2-expressing monolayers was transformed into irregularly undulating profiles after TNF treatment (Fig. 9). These morphological changes were also similar to those induced by TNF in IFN-γ-primed monolayers (Fig. 9). Thus, Caco-2 monolayers transgenically expressing TNFR2 respond to TNF by reorganizing tight junction proteins in a manner indistinguishable from that of IFN-γ-primed monolayers. Remarkably, the changes in both occludin and ZO-1 distribution were similar to those seen in colonocytes in vivo after CD4+CD45RBhi transfer-induced colitis.
Figure 8.

TNF induces occludin redistribution in IFN-γ-primed and TNFR2-expressing Caco-2 cells.
Monolayers of control, IFN-γ-primed, TNFR1-transfected, and TNFR2-transfected Caco-2 cells were treated with TNF, 2.5 ng/ml, as indicated, for 8 hours. Occludin distribution was detected by deconvolution immunofluorescence microscopy. TNF caused irregular occludin undulations at the tight junction and the appearance of intracellular occludin-containing vesicles in IFN-γ-primed and TNFR2-transfected, but not control or TNFR1-transfected monolayers. Bar = 10 μm.
Figure 9.

TNF induces ZO-1 redistribution in IFN-γ-primed and TNFR2-expressing Caco-2 cells.
Monolayers of control, IFN-γ-primed, TNFR1-transfected, and TNFR2-transfected Caco-2 cells were treated with TNF, 2.5 ng/ml, as indicated, for 8 hours. ZO-1 distribution was detected by deconvolution immunofluorescence microscopy. TNF caused irregular ZO-1 undulations at the tight junction in IFN-γ-primed and TNFR2-transfected, but not control or TNFR1-transfected monolayers. Bar = 10 μm.
TNFR2-dependent tight junction disruption is accompanied by increases in MLC phosphorylation and MLCK expression
Tight junction disruption by TNF in IFN-γ-primed monolayers is due to MLCK protein upregulation and the resulting increases in MLC phosphorylation 5. It has also been reported that MLCK phosphorylation alone is sufficient to induce irregularly undulating ZO-1 profiles similar to those induced by TNF 34. To determine if TNFR2-mediated tight junction disruption occurs through increases in MLCK expression and activity, TNFR2-expressing monolayers were treated with TNF and both MLC phosphorylation and MLCK expression assessed by SDS-PAGE immunoblot. TNF treatment induced increased MLC phosphorylation and MLCK expression in TNFR2-transfected monolayers (Fig. 10). TNF induced similar changes in non-transfected monolayers after, but not before, IFN-γ-priming (Fig. 10). The data therefore show that TNFR2-dependent signaling causes increases in intestinal epithelial MLCK expression and activity.
Figure 10.

TNFR2-dependent tight junction disruption is accompanied by increases in MLC phosphorylation and MLCK expression.
IFN-γ-primed non-transfected monolayers and TNFR2-transfected monolayers were harvested 8 hours after treatment with 2.5 ng/ml TNF, as indicated. SDS-PAGE and immunoblot shows 2.8±0.4-fold and 2.9±0.1-fold induction of MLCK expression after TNF treatment of IFN-γ-primed non-transfected monolayers and TNFR2-transfected monolayers, respectively (p<0.02 for both). MLC phosphorylation increased 2.0±0.1-fold and 1.9±0.2-fold after TNF treatment of IFN-γ-primed non-transfected monolayers and TNFR2-transfected monolayers, respectively (p<0.01 for both). Total MLC is shown as a loading control. Data are representative of 3 independent experiments, each with triplicate samples.
Discussion
Compromised intestinal barrier function is a common feature of inflammatory, infectious, ischemic, and immune-mediated intestinal disease 35, and abundant data suggest that compromised intestinal barrier function may be a critical pathogenic event in these processes 36-40. For example, in patients with inactive Crohn’s disease, barrier loss predicts disease reactivation 41, 42. The degree of barrier dysfunction also parallels expression of activation markers on circulating lymphocytes 7. Thus, many have proposed that compromised barrier function may be critical to both initial pathogenesis and reactivation of chronic intestinal disease. We have suggested that this reflects the linkage of barrier dysfunction and immune activation in a self-amplifying loop where barrier dysfunction allows increased interaction between luminal contents and mucosal immune cells that, in turn, leads to cytokine release, further barrier dysfunction, and further immune activation 35. Consistent with this hypothesis, we 2, 5 and others 3, 20, 33, 43 have shown that epithelial barrier dysfunction can be induced in vitro by synergistic signaling between IFN-γ and TNF. Moreover, we have shown that this barrier dysfunction requires MLCK activity and expression 2, 5. To better understand the mechanisms by which this occurs we focused on understanding the mechanisms by which IFN-γ and TNF signal to epithelia to reduce barrier function.
We began using an in vivo immune-mediated model of inflammatory bowel disease in which both IFN-γ and TNF are important to disease pathogenesis 19. These data showed that intestinal epithelial MLCK expression and MLC phosphorylation are increased during disease progression and that this is associated with tight junction disruption.
To better characterize the mechanism of cytokine signaling to the epithelium, we used a well-characterized in vitro intestinal epithelial model in which TNF regulates barrier function via MLCK 2, 5, 18. Under the conditions studied here, IFN-γ synergizes with TNF by priming intestinal epithelia to respond to TNF; there is no response to either cytokine alone. As our primary measure of barrier function was TER, a measure of tight junction ion conductance, it is conceivable that IFN-γ could change size selectivity without altering TER. However, our previous work using the same experimental model showed that, while the combination of IFN-γ and TNF did increase transepithelial 3kD dextran flux, this did not occur with either cytokine alone 5. One might also ask if TNF changes charge selectivity of tight junctions. Although this has not been investigated in detail, a recent study did compare two cytokine-induced pathways of in vitro barrier loss and found that while IL-13 dramatically increased claudin-2 expression at the tight junction, combined IFN-γ and TNF treatment reduced expression of claudin-2, claudin-3, and claudin-4 44. As available data suggest that it is the pattern of claudin isoforms expressed that defines tight junction charge selectivity 45, 46, TNF may act by globally removing claudins from the tight junction but, in contrast to IL-13 44, 47, not by altering expression of specific claudin isoforms.
We next turned our attention to proteins induced by IFN-γ that might increase sensitivity to TNF signaling. Our data show that intestinal epithelial expression of both TNFR1 and TNFR2 are upregulated by IFN-γ-priming. These TNF receptors are expressed on lymphoid and non-lymphoid cells, and roles for each have been implicated in inflammatory disease 21-23, 25, 48-50. In addition, several studies have suggested a cooperative interaction between TNFR1 and TNFR2 51, 52. We therefore sought to determine whether one or both TNF receptors were necessary for TNF-induced barrier dysfunction to occur.
Use of specific TNFR1 and TNFR2 blocking antibodies in vitro clearly showed that epithelial TNFR2 is required for TNF-induced barrier dysfunction to occur. Moreover, the lack of an effect of TNFR1 blocking antibodies suggests that epithelial TNFR1 is not necessary for TNF-induced barrier dysfunction. Thus, TNFR2, but not TNFR1, mediates the effects of TNF on epithelial barrier function. These data indicate that a critical role of IFN-γ priming is TNFR2 upregulation. We therefore sought to determine whether TNFR2 expression alone was sufficient to prepare intestinal epithelia to respond to TNF. Transgenic TNFR2 expression allowed intestinal epithelial monolayers to regulate barrier function in response to TNF without IFN-γ-priming. This suggests that the primary function of IFN-γ-priming is TNFR2 upregulation. Based on our experience with MLCK-dependent in vivo barrier dysfunction in acute T cell-mediated disease 4, we performed similar studies using TNFR1 or TNFR2 knockout mice. These studies were unsuccessful, likely due to the ubiquitous expression of both TNF receptors on many cell types and the presence of redundant signaling pathways in vivo.
Critical downstream mediators of TNF-induced barrier dysfunction include increased MLCK expression, MLC phosphorylation, and tight junction protein reorganization 5. We therefore assessed these to determine if signaling pathways activated by TNF in monolayers transgenically expressing TNFR2 are the same as those activated by TNF in IFN-γ-primed monolayers. Increased MLCK expression, MLC phosphorylation, and tight junction protein redistribution were all similarly induced by TNF in TNFR2-transfected monolayers and IFN-γ-primed nontransfected monolayers. Thus, like TNF-induced barrier dysfunction in IFN-γ-primed monolayers, TNF disrupts barrier function in TNFR2-transfected monolayers via MLCK upregulation and its downstream sequelae.
Although this is the first study showing the critical role of epithelial TNFR2 in mediating TNF effects on barrier function, previous data from diverse sources are consistent with this important role for TNFR2. For example, TNFR2 has been suggested to mediate inflammation-induced epithelial hyperplasia 48 and TNFR2 polymorphisms have been reported in Crohn’s disease patients 49. Moreover, TNFΔARE mice, in which deletion of TNF AU-rich elements results in TNF mRNA stabilization and TNF protein overexpression, develop joint and intestinal disease 53. Remarkably, only joint disease occurs when TNFΔARE bone marrow is transplanted into TNFR2 knockout mice 53. This result is consistent with our conclusion that TNFR2 signals epithelial barrier dysfunction and suggests that this may be critical for the development of chronic immune-mediated intestinal disease, although an important role for TNFR2 expressed on nonepithelial radioresistant stromal cells cannot be excluded. Consistent with a role for TNFR2 in disease, transgenic mice expressing human TNFR2 develop severe systemic inflammatory disease 54. However, once again, the fact that TNFR2 is normally expressed on multiple cell types complicates interpretation of these data. For example, the systemic disease induced by transgenic expression of human TNFR2 is characterized by increased NF-κB activity in peripheral blood mononuclear cells 54. Tissue-specific TNFR2 knockout or transgenic animals would help to resolve these issues, but such animals have not been reported. Thus, in vitro models represent the best available opportunity to understand the mechanisms by which TNF signals intestinal epithelia to regulate barrier function and the means by which IFN-γ prepares epithelia to receive this signal. Moreover, a central role of IFN-γ in this process is to upregulate TNFR2 expression, consistent with reports of intestinal epithelial TNFR2 upregulation in ulcerative colitis and Crohn’s disease 48. Together with in vivo reports, our data are consistent with the hypothesis that TNFR2-mediated signaling in intestinal epithelial cells, including MLCK upregulation, is critical to the pathogenesis of inflammatory disease. The data also support the hypothesis that specific blockade of epithelial TNFR2 may be effective in inflammatory bowel disease. This is worthy of future investigation, as more precisely targeted approaches may have fewer systemic complications than current anti-TNF-based therapies 10, 11, 13, 55, 56.
In summary, our data show that TNFR2-mediated TNF signaling to epithelia triggers increased MLCK expression, increased MLC phosphorylation, tight junction reorganization, and barrier dysfunction. Moreover, TNFR2 upregulation is the primary role of the IFN-γ priming that is necessary before TNF can cause intestinal epithelial barrier dysfunction in response to TNF. Finally, while the in vivo situation is certainly far more complex, these data also suggest that development of agents that block specific aspects of TNF signaling may lead to improved therapies for inflammatory intestinal disease.
Acknowledgements
We are pleased to acknowledge Drs. Sean Colgan, Yang-Xin Fu, Le Shen, Cormac Taylor, and Judy Turner for their insightful comments and Harn Shiue for technical assistance. This work was supported by the National Institutes of Health (DK61931 and DK68271), the Crohn’s Colitis Foundation of America, The University of Chicago Digestive Disease Center (DK42086), and The University of Chicago Cancer Center (CA14599).
Footnotes
- IFN-γ
- interferon-γ
- MLC
- myosin II regulatory light chain
- MLCK
- myosin light chain kinase
- TER
- transepithelial resistance
- TNFR1
- tumor necrosis factor receptor 1 or TNFRSF1A
- TNFR2
- tumor necrosis factor receptor 2 or TNFRSF1B
- TNF
- tumor necrosis factor
References
- 1.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, Rutgeerts P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol. 2002;97:2000–4. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 2.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 3.Taylor CT, Dzus AL, Colgan SP. Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology. 1998;114:657–68. doi: 10.1016/s0016-5085(98)70579-7. [DOI] [PubMed] [Google Scholar]
- 4.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–19. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 7.Yacyshyn BR, Meddings JB. CD45RO expression on circulating CD19+ B cells in Crohn’s disease correlates with intestinal permeability. Gastroenterology. 1995;108:132–7. doi: 10.1016/0016-5085(95)90017-9. [DOI] [PubMed] [Google Scholar]
- 8.Shen C, Van Assche G, Rutgeerts P, Ceuppens JL. Caspase activation and apoptosis induction by adalimumab: demonstration in vitro and in vivo in a chimeric mouse model. Inflamm Bowel Dis. 2006;12:22–8. doi: 10.1097/01.mib.0000194185.69800.07. [DOI] [PubMed] [Google Scholar]
- 9.Agnholt J, Kaltoft K. Infliximab downregulates interferon-gamma production in activated gut T-lymphocytes from patients with Crohn’s disease. Cytokine. 2001;15:212–22. doi: 10.1006/cyto.2001.0919. [DOI] [PubMed] [Google Scholar]
- 10.Dimakou K, Papaioannides D, Latsi P, Katsimboula S, Korantzopoulos P, Orphanidou D. Disseminated tuberculosis complicating anti-TNF-alpha treatment. Int J Clin Pract. 2004;58:1052–5. doi: 10.1111/j.1742-1241.2004.00061.x. [DOI] [PubMed] [Google Scholar]
- 11.Molenaar ET, Bultink IE, Dijkmans BA, Lems WF. Development of fatal tuberculosis in a patient with rheumatoid arthritis after three years of treatment with infliximab: comment on the article by Wolfe et al. Arthritis Rheum. 2005;52:1334–6. doi: 10.1002/art.20973. [DOI] [PubMed] [Google Scholar]
- 12.Jarnerot G, Hertervig E, Friis-Liby I, Blomquist L, Karlen P, Granno C, Vilien M, Strom M, Danielsson A, Verbaan H, Hellstrom PM, Magnuson A, Curman B. Infliximab as rescue therapy in severe to moderately severe ulcerative colitis: a randomized, placebo-controlled study. Gastroenterology. 2005;128:1805–11. doi: 10.1053/j.gastro.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Lichtenstein GR, Feagan BG, Cohen RD, Salzberg BA, Diamond RH, Chen DM, Pritchard ML, Sandborn WJ. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol. 2006;4:621–30. doi: 10.1016/j.cgh.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–85. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 15.Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- 16.Zhao H, Shiue H, Palkon S, Wang Y, Cullinan P, Burkhardt JK, Musch MW, Chang EB, Turner JR. Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc Natl Acad Sci U S A. 2004;101:9485–90. doi: 10.1073/pnas.0308400101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berglund JJ, Riegler M, Zolotarevsky Y, Wenzl E, Turner JR. Regulation of human jejunal transmucosal resistance and MLC phosphorylation by Na+-glucose cotransport. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1487–93. doi: 10.1152/ajpgi.2001.281.6.G1487. [DOI] [PubMed] [Google Scholar]
- 18.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–30. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 19.Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–62. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 20.Fish SM, Proujansky R, Reenstra WW. Synergistic effects of interferon gamma and tumour necrosis factor alpha on T84 cell function. Gut. 1999;45:191–8. doi: 10.1136/gut.45.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 22.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–67. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 23.Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, Whitehead R, Polk DB. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Physiol Cell Physiol. 2003;284:C953–61. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 24.Piguet PF, Vesin C, Guo J, Donati Y, Barazzone C. TNF-induced enterocyte apoptosis in mice is mediated by the TNF receptor 1 and does not require p53. Eur J Immunol. 1998;28:3499–505. doi: 10.1002/(SICI)1521-4141(199811)28:11<3499::AID-IMMU3499>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 25.Brown GR, Lee E, Thiele DL. TNF-TNFR2 interactions are critical for the development of intestinal graft-versus-host disease in MHC class II-disparate (C57BL/6J-->C57BL/6J × bm12)F1 mice. J Immunol. 2002;168:3065–71. doi: 10.4049/jimmunol.168.6.3065. [DOI] [PubMed] [Google Scholar]
- 26.Colgan SP, Parkos CA, Matthews JB, D’Andrea L, Awtrey CS, Lichtman AH, Delp-Archer C, Madara JL. Interferon-gamma induces a cell surface phenotype switch on T84 intestinal epithelial cells. Am J Physiol. 1994;267:C402–10. doi: 10.1152/ajpcell.1994.267.2.C402. [DOI] [PubMed] [Google Scholar]
- 27.Salomon P, Pizzimenti A, Panja A, Reisman A, Mayer L. The expression and regulation of class II antigens in normal and inflammatory bowel disease peripheral blood monocytes and intestinal epithelium. Autoimmunity. 1991;9:141–9. doi: 10.3109/08916939109006750. [DOI] [PubMed] [Google Scholar]
- 28.Hughes A, Bloch KJ, Bhan AK, Gillen D, Giovino VC, Harmatz PR. Expression of MHC class II (Ia) antigen by the neonatal enterocyte: the effect of treatment with interferon-gamma. Immunology. 1991;72:491–6. [PMC free article] [PubMed] [Google Scholar]
- 29.Rocha F, Musch MW, Lishanskiy L, Bookstein C, Sugi K, Xie Y, Chang EB. IFN-gamma downregulates expression of Na(+)/H(+) exchangers NHE2 and NHE3 in rat intestine and human Caco-2/bbe cells. Am J Physiol Cell Physiol. 2001;280:C1224–32. doi: 10.1152/ajpcell.2001.280.5.C1224. [DOI] [PubMed] [Google Scholar]
- 30.Youakim A, Ahdieh M. Interferon-gamma decreases barrier function in T84 cells by reducing ZO-1 levels and disrupting apical actin. Am J Physiol. 1999;276:G1279–88. doi: 10.1152/ajpgi.1999.276.5.G1279. [DOI] [PubMed] [Google Scholar]
- 31.Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–302. doi: 10.1136/gut.2003.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gitter AH, Bendfeldt K, Schulzke JD, Fromm M. Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. Faseb J. 2000;14:1749–53. doi: 10.1096/fj.99-0898com. [DOI] [PubMed] [Google Scholar]
- 33.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–72. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 34.Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119:2095–106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- 35.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–91. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 36.Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000;95:2754–9. [PubMed] [Google Scholar]
- 37.Hollander D. Is Crohn’s a tight junction disease? Gut. 1989;21:315–19. [Google Scholar]
- 38.Hollander D. Permeability in Crohn’s disease: Altered barrier functions in healthy relatives? Gastroenterology. 1993;104:1848–1851. doi: 10.1016/0016-5085(93)90668-3. [DOI] [PubMed] [Google Scholar]
- 39.Meddings JB, Sutherland LR, May GR. Intestinal permeability in patients with Crohn’s disease. Gut. 1994;35:1675–6. doi: 10.1136/gut.35.11.1675-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suenaert P, Bulteel V, Vermeire S, Noman M, Van Assche G, Rutgeerts P. Hyperresponsiveness of the mucosal barrier in Crohn’s disease is not tumor necrosis factor-dependent. Inflamm Bowel Dis. 2005;11:667–73. doi: 10.1097/01.mib.0000168371.87283.4b. [DOI] [PubMed] [Google Scholar]
- 41.Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol. 2000;35:1163–9. doi: 10.1080/003655200750056637. [DOI] [PubMed] [Google Scholar]
- 42.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–9. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 43.Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T, Okawa K, Iwamatsu A, Kita T. Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. J Immunol. 1999;163:553–7. [PubMed] [Google Scholar]
- 44.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on Claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85:1139–62. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 45.Van Itallie C, Rahner C, Anderson JM. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest. 2001;107:1319–27. doi: 10.1172/JCI12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–6. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 47.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, Zeitz M, Fuss I, Strober W, Schulzke JD. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–64. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 48.Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, de Jong YP, Ooi CJ, Xavier RJ, Terhorst C, Podolsky DK, Bhan AK. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology. 2002;122:134–44. doi: 10.1053/gast.2002.30347. [DOI] [PubMed] [Google Scholar]
- 49.Sashio H, Tamura K, Ito R, Yamamoto Y, Bamba H, Kosaka T, Fukui S, Sawada K, Fukuda Y, Satomi M, Shimoyama T, Furuyama J. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and Crohn’s disease, respectively. Immunogenetics. 2002;53:1020–7. doi: 10.1007/s00251-001-0423-7. [DOI] [PubMed] [Google Scholar]
- 50.Musch MW, Clarke LL, Mamah D, Gawenis LR, Zhang Z, Ellsworth W, Shalowitz D, Mittal N, Efthimiou P, Alnadjim Z, Hurst SD, Chang EB, Barrett TA. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J Clin Invest. 2002;110:1739–47. doi: 10.1172/JCI15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexopoulou L, Pasparakis M, Kollias G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur J Immunol. 1997;27:2588–92. doi: 10.1002/eji.1830271018. [DOI] [PubMed] [Google Scholar]
- 52.Pinckard JK, Sheehan KC, Schreiber RD. Ligand-induced formation of p55 and p75 tumor necrosis factor receptor heterocomplexes on intact cells. J Biol Chem. 1997;272:10784–9. doi: 10.1074/jbc.272.16.10784. [DOI] [PubMed] [Google Scholar]
- 53.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–98. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 54.Douni E, Kollias G. A critical role of the p75 tumor necrosis factor receptor (p75TNF-R) in organ inflammation independent of TNF, lymphotoxin alpha, or the p55TNF-R. J Exp Med. 1998;188:1343–1352. doi: 10.1084/jem.188.7.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kollias G, Kontoyiannis D. Role of TNF/TNFR in autoimmunity: specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine & Growth Factor Reviews. 2002;13:315–321. doi: 10.1016/s1359-6101(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 56.van der Klooster JM, Bosman RJ, Oudemans-van Straaten HM, van der Spoel JI, Wester JP, Zandstra DF. Disseminated tuberculosis, pulmonary aspergillosis and cutaneous herpes simplex infection in a patient with infliximab and methotrexate. Intensive Care Med. 2003;29:2327–9. doi: 10.1007/s00134-003-1867-z. [DOI] [PubMed] [Google Scholar]