

Abstract
The activity of voltage-gated calcium channels (VGCCs) decreases with time in whole-cell and inside-out patch-clamp recordings. In this study we found that substituting a single amino acid (I1520) at the intracellular end of IIIS6 in the α1 subunit of P/Q-type Ca2+ channels with histidine or aspartate greatly attenuated channel rundown in inside-out patch-clamp recordings. The homologous mutations also slowed rundown of N- and L-type Ca2+ channels, albeit to a lesser degree. In P/Q-type channels, the attenuation of rundown is accompanied by an increased apparent affinity for phosphatidylinositol-4,5-bisphosphate, which has been shown to be critical for maintaining Ca2+ channel activity (Wu et al., 2002). Furthermore, the histidi1ne mutation significantly stabilized the open state, making the channels easier to open, slower to close, harder to inactivate and faster to recover from inactivation. Our finding that mutation of a single amino acid can greatly attenuate rundown provides an easy and efficient way to slow the rundown of VGCCs, facilitating functional studies that require direct access to the cytoplasmic side of the channel.
Keywords: P/Q-type calcium channels, rundown, PIP2, inside-out patch, mutagenesis, S6
1. Introduction
Voltage-gated Ca2+ channels (VGCCs) are subject to extensive regulation in cells [2]. Their activity decreases spontaneously over time in whole-cell or cell-free recordings. This phenomenon, called “rundown”, is a common feature shared by a number of ion channels, including certain types of Na+, K+ and Cl− channels and NMDA and GABAA receptors [3, 4]. Except for the low voltage-activated T-type Ca2+ channels, all high voltage-activated Ca2+ channels, including L-, N-, P/Q- and R-type channels, show rapid rundown in whole-cell and cell-free configurations [5–10]. Extensive efforts have been devoted to studying the mechanisms of rundown of these channels, since a better understanding of this phenomenon could greatly facilitate experimental studies of these channels and help delineate how they are regulated in intact cells. Several mechanisms have been suggested for rundown of VGCCs, including proteolysis [11–15] and dephosphorylation of PKA-mediated phosphorylation [16–19]. Recent work show that phosphatidylinositol-4,5-bisphosphate (PIP2) can greatly stabilize the activity of P/Q-type, and to a lesser degree, N- and L-type Ca2+ channels [1, 20]. This finding has been capitalized to perform functional studies on VGCCs that require direct access to and extensive perfusion of the cytoplasmic side of the channel [21, 22]. In this study, we report another strategy to dramatically attenuate rundown of VGCCs by mutating a single amino acid in the α1 subunit.
2. Materials and Methods
2.1. Molecular biology and oocyte expression
Rabbit brain Cav2.1, Cav2.2, cardiac muscle Cav1.2 and rabbit skeletal muscle α2δ were cloned in an oocyte expression vector, pGEMHE, or one of its variants. Rat brain β4 subunit was cloned in the Bluescript SK vector. Site-directed mutations were generated by PCR mutagenesis and were confirmed by sequencing. cRNA for all constructs (WT α1, mutant α1, α2δ and β4 subunits) were transcribed in vitro using T7 polymerase after linearization. The α1, α2δ and β4 cRNA were injected into Xenopus oocytes with a total concentration of ~0.5μg/μl (ratio α1:α2δ:β2a = 5:5:4) and a volume of ~50nl per oocyte. Oocytes were prepared and maintained as described [23].
2.2. Electrophysiology
Whole-cell currents recorded with two-electrode voltage-clamp with the OC-725C oocyte clamp amplifier (Warner Instruments) were used mainly for checking the functional expression of channels in oocytes. Procedures and protocols were as described [1]. All patch-clamp recordings were performed with the Axopatch 200A amplifier (Axon Instruments, Sunnyvale, CA). Currents were filtered at 2 kHz, digitized at 10–50 kHz. Oocyte was bathed in a control solution containing (in mM) 125 KCl, 4 NaCl, 10 HEPES, 10 EGTA (pH 7.3 with KOH). In all run-down and MARCKS peptide experiments, macroscopic currents were recorded using the inside-out patch configuration. Recording glass pipettes pulled from pyrex glass tubes (Corning, Acton, MA) were filled with a solution of (in mM) 85 BaCl2, 10 HEPES (pH 7.3 with KOH) and had a resistance of 0.2–0.3 MΩ. Currents were evoked from a holding potential of −80 mV every 1.5 s by depolarizations of 6–8 ms from −30 mV to +100 mV in 10-mV increments, followed by a 30-ms repolarization to −30 mV. When some channel properties (activation, inactivation and deactivation) of the WT and I1520H mutant channels were examined, recording pipettes were filled with a solution containing (in mM) 45 BaCl2, 80 KCl and 10 HEPES (pH 7.3 with KOH). In these experiments, the currents were obtained from the cell-attached configuration. To eliminate contamination from the Cl− current mediated by the endogenous Ca2+-activated Cl− channels, 50 nl of 50 mM BAPTA (Sigma, St. Louis, MO) was injected into the oocyte 10–30 min prior to the experiment. MARCKS peptide was synthesized by Global Peptide Services (Ft. Collins, Co). Its sequence is NH2-KKKKKRFS FKKSFKLSGFS-FKKNKK-COOH. 10 mM stock solution of MARCKS peptide was aliquoted and kept at −20°C and was diluted in the control solution before every application. MARCKS peptide was applied until its inhibition could not be distinguished from the intrinsic rundown of the current. Experiments were carried out at 21–23 °C.
2.3. Data analysis
Data acquisition and analysis were performed using pClamp8 (Axon Instruments) on a PC through a Digidata 1200 interface. The time course of decay of the tail currents after patch excision was fitted with a single exponential function. T50 was calculated based on the fitting and defined as the time for the current to decay to 50% of its starting value. Speed of rundown was also quantified as the remaining tail current after 10-minute perfusion for P/Q-type channels and 3-minute perfusion for N-type and L-type channels. The effect of MARCKS peptide was calculated from the tail currents after +100 mV depolarization obtained before application and after washout of the peptide. Data are represented as mean ± s.d. (number of observations). Significance was determined using the Kruskal-Wallis Test with Bonferroni correction. No statistical analysis was performed if the number of observation was less than 5. No corrections were made for leakage current, which was negligible in macropatch recordings.
3. Results
3.1. Mutations of a single residue in IIIS6 reduce rundown of P/Q-type channels
The activity of P/Q-, N- and L-type Ca2+ channels stayed constant in the cell-attached mode but underwent rapid rundown upon inside-out patch excision. In our previous study of P/Q-type channel inner pore [21], we found a cysteine mutation in the CaV2.1 α1 subunit that could greatly reduce rundown in inside-out patches. This amino acid, I1520, is located at the intracellular end of IIIS6 (Fig. 1A), one amino acid below the putative membrane/cytoplasm interface (Fig. 1B). To obtain better mechanistic understanding of why and how the I1520C mutation affected rundown, we systemically mutated this isoleucine to all the other 18 amino acids and examined their effects on channel rundown. All mutant channels, except I1520K and I1520R, were functional.
Figure 1.
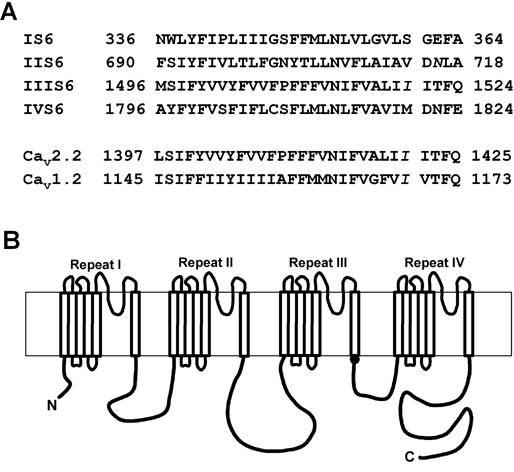
Amino acid sequences and transmembrane topology of the α1 subunit of voltage-gated Ca2+ channels. (A) Amino acid sequences of the S6 segments in the four repeats of Cav2.1 subunit and the S6 segment in the third repeat of Cav2.2 and Cav1.2 subunit. I1520 in Cav2.1 and homologous residues in Cav2.2 and Cav1.2 are shown in italics. (B) Putative transmembrane topology of the α1 subunit of voltage-gated Ca2+ channels. The black dot at the intracellular end of IIIS6 shows the location of I1520.
Using the tail current after a depolarization to +100 mV (Itail(+100 mV)) as a readout of the total number of functional channels, we could faithfully measure the time course of rundown (Fig. 2 A–C), as reported previously [1, 20]. The extent of rundown was quantified as the remaining Itail(+100 mV) after 10-minute perfusion with a control solution. The current of the WT channels decayed quickly, to 38.2% of its starting value (i.e., current obtained immediately after patch excision) 10 minutes after patch-excision (Fig. 2 A and D). In contrast, the current of the I1520H mutant channels decayed much more slowly, to 84.0% of its initial value (Fig. 2 A and D).
Figure 2.

Mutations of I1520 in P/Q-type or homologous positions in N- and L-type channels affect their rundown. (A) Representative time course of rundown of Itail (+100 mV) of the WT P/Q-type channel (•) and I1520H mutant channel (○) in a control solution. Currents are normalized to that obtained immediately after patch excision. (B), (C) Representative current traces for the WT and I1520H mutant channel, taken at the time points indicated in (A). In this and the following figures, channels were activated by depolarization to +100 mV from a holding potential of −80 mV. Tail currents were recorded at −30 mV and were shown on an expanded time scale. (D) Remaining Itail (+100 mV) of the WT and all functional mutant channels after 10-min perfusion in a control solution. Error bar represents S.D. and the number in parenthesis represents number of recordings for each channel type. The histidine, aspartate, alanine and cysteine mutants are significantly different from the WT (P<0.01) but the leucine mutant is not. No statistical analysis was performed on the other mutants due to the small number of observations. (E), (F) Remaining Itail (+100 mV) of the WT and mutant N-type (A) and L-type (C) channels after 3-min perfusion in a control solution. (G), (H) T50 for decay of Itail (+100 mV) of the WT and mutant N-type (B) and L-type (D) channels (** P< 0.001).
Figure 2D shows the relative extent of rundown of all functional mutant channels compared with that of the WT channel. With the exception of the leucine and methionine mutations, all mutations appeared to attenuate rundown by varying degrees. We focused on the histidine and aspartate mutations in later studies because they were among the two most effective mutations and their side-chains carry opposite charges.
We next examined the effect on channel rundown of the histidine mutation at the homologous positions (S360, V714 and M1820, Fig. 1A) in the S6 segment of other three repeats. Interestingly, S360H, V714H and M1820H had no significant effect on rundown of P/Q-type channels (data not shown). Thus, the effect of I1520H appears to be specific for IIIS6.
3.2. Homologous mutations in N- and L-type channels also reduce rundown
We also examined whether histidine and aspartate mutations at the homologous positions of N- and L-type channels also retarded rundown of these channels (Fig. 1A). Because N- and L-type channels rundown much faster than P/Q-type channels do, their rundown was quantified as the remaining Itail(+100 mV) after 3-minute perfusion, or as T50, the time required for Itail(+100 mV) to decay to 50% of its starting value immediately after patch excision. The aspartate mutation, but not the histidine mutation, reduced N-type channel rundown (Fig. 2 E and F). On the other hand, both mutations attenuated L-type channel rundown (Fig. 2 G and H).
3.3. Mutations that reduce rundown increase the apparent affinity for PIP2
We then explored possible mechanisms of attenuation of rundown caused by I1520 mutations in P/Q-type channels. Because phosphatidylinositol-4,5-bisphosphate (PIP2) is essential for maintaining Ca2+ channel activity ([1, 20], we examined the effect of removing PIP2 on the WT and I1520H and I1520D mutant P/Q-type channels by applying a PIP2-sequestering reagent to the intracellular side of the channels. This sequestering reagent is a peptide from the protein “myristoylated alanine-rich protein kinase C substrate” (MARCKS), which has been shown to sequester PIP2, mainly through nonspecific electrostatic interactions between negatively charged PIP2 and a basic region that contains a large number of positively charged amino acids. A peptide corresponding to this basic domain, residue 151 to 175 (NH2-KKKKKRFSFKKSFKLSGFSFKKNKK-COOH), has the same function as the whole protein [24]. We thus used this MARCKS peptide to sequester PIP2 in the membrane. As expected, the MARCKS peptide accelerated the decay of Itail(+100 mV) of the WT channels (Fig. 3A). However, while 3 μM peptide inhibited 74.1 ± 8.8% (n=10) of the WT channel current, 100 μM peptide inhibited only 38.6 ± 12.1% (n=8) of the I1520H channel current. The dose-response curve of the mutant channel was greatly shifted rightward compared with that of the WT channel (Fig. 3B), indicating that the apparent PIP2 binding affinity was markedly increased in the mutant. Similar results were obtained for the I1520D mutant (Fig. 3B). These results indicate that the mutant channels still need PIP2 to maintain their activity, but they have a higher apparent affinity for PIP2.
Figure 3.

Inhibition of WT and I1520 mutant P/Q Ca2+ channels by MARCKS peptide. (A) Inhibition of Itail (+100 mV) of the WT channel by intracellular application of 3 μM MARCKS peptide. (B) Representative current traces for the WT and histidine and aspartate mutant channels. Traces a and b were taken immediately before and after application of MARCKS peptide, respectively, as exemplified in (A). (C) Remaining currents of the WT and I1520H and I1520D mutant channels after steady-state inhibition by different concentrations of MARCKS peptide.
The attenuation effect on rundown of the histidine and aspartate mutations and PIP2 appears non-additive, since application of PIP2 to the mutant channels did not further decrease rundown of N-, L- as well as P/Q-type channels (data not shown).
3.4. I1520H mutation changes channel biophysical properties
I1520 is strategically situated at the intracellular end of the IIIS6 segment, which together with the S6 segment of the other three repeats forms the inner pore [21]. Single mutations at IIIS6 as well as at other S6 segments have been shown to greatly affect inactivation [21, 25–30]. Furthermore, the activation gate is located at or near the membrane/cytoplasm interface [22]. We therefore examined the effect of the I1520H mutation on a host of activation and inactivation properties (Fig. 4). Not surprisingly, there were indeed significant changes. The half activation voltage (V1/2) was shifted from 13.8 ± 2.4 mV (n=10) in the WT channel to 0.6 ± 3.9 mV (n=8) in the mutant channel, while the slope factor (k) remained similar (7.9 ± 0.6 mV (n=10) in WT and 6.1 ± 0.8 mV (n=8) in I1520H, respectively). Although the activation kinetics at a given voltage is the same (Fig. 4B), the deactivation kinetics is slower in the mutant channel (Fig. 4C). The mutant channels also became more resistant to inactivation (Fig. 4D and E). The mid-point voltage and slope factor of steady-state inactivation were −24.1 ± 4.8 mV and −8.5 ± 2.5mV (n=10) for the WT channel and −20.4 ± 4.5 mV and −4.9 ± 1.0 mV (n=10) for the I1520H mutant channel, respectively (Fig. 4D). At +20 mV, the time for 50% inactivation was 0.4 ± 0.1 s (n=10) for WT and 1.4 ± 0.7 s (n=10) for I1520H (Fig. 4E). Correspondingly, the mutant channels recovered more quickly from inactivation than did the WT channels (Fig. 4F).
Figure 4.
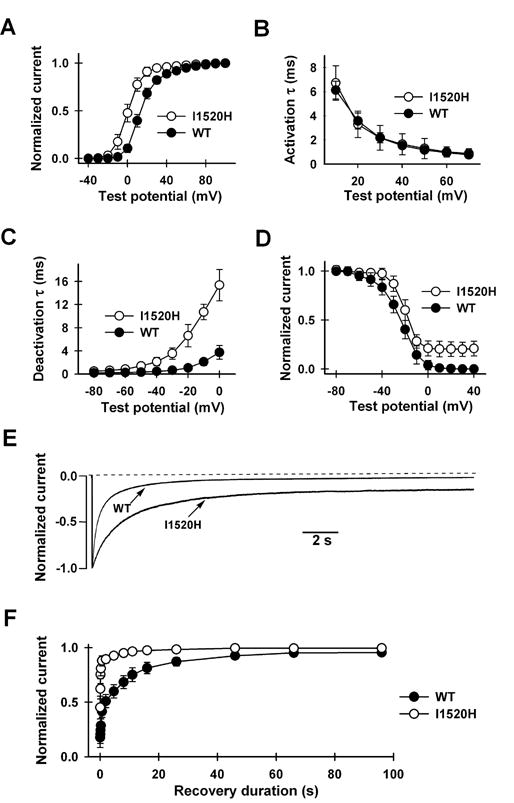
Comparison of biophysical properties of WT and I1520H mutant P/Q Ca2+ channels. (A) Voltage-dependence of activation. Currents were evoked from a holding potential of −80 mV every 3 s by 10-ms depolarizations ranging from −40 mV to +100 mV in 10-mV increment, followed by a 15-ms repolarization to −30 mV. Data points represent normalized tail currents recorded at −30 mV. All activation curves were well fitted by a Boltzmann function. (B) Activation time constants at different test potentials, obtained by fitting the activation phase with a single exponential function (n=10). (C) Deactivation kinetics. Channels were activated by a 20-ms prepulse to +100 mV and deactivation was measured by the decay of the tail current recorded at various repolarizing test potentials, ranging from −80 mV to 0 mV in 10-mV steps. The decay phase was fitted with a single exponential function. Deactivation time constants at different test potentials are plotted. (D) Voltage-dependence of inactivation. Steady-state inactivation was determined by a three-pulse protocol in which a 20-ms normalizing pulse to +20 mV (pulse A) was followed sequentially by a 25-s conditioning pulse (ranging from −60 mV to +40 mV) and a 20-ms test pulse to +20 mV (pulse B). The holding potential was −100 mV and interval between each protocol was 2 min. Peak current evoked by pulse B was normalized by that evoked by pulse A and was plotted against the conditioning potentials. (E) Inactivation kinetics. Current was evoked by a 25-s step depolarization to +20 mV from a holding potential of −100 mV. (F) Kinetics of recovery from inactivation. To obtain the time course of recovery, a control test pulse to +30 mV (pulse #1) was followed by a 4-s conditioning pulse to +30 mV. After varying durations ranging from 10 ms to 96 s at −80 mV, a test pulse to +30 mV (pulse #2) was given again. Recovery was quantified by normalizing the peak current evoked by pulse #2 with that evoked by pulse #1 and plotting it against the recovery duration. The time course was generally fitted well by the sum of two exponential functions. τfast and τslow are 0.11 ± 0.01 s and 7.3 ± 0.8 s (n=10) for the I1520H channel, and 0.47 ± 0.05 s and 12.5 ± 0.7 s (n=10) for the WT.
4. Discussion
4.1. Mechanism of attenuation of rundown
Several mechanisms have been suggested to contribute to Ca2+ channel rundown, including loss of unidentified cytoplasmic factors, reversal of PKA phosphorylation and Ca2+-dependent proteolysis [5]. Studies on cardiac as well as neuronal L-type Ca2+ channels show that exposure of the cytoplasmic side to reagents that promote PKA phosphorylation, such as cAMP, Mg-ATP and the catalytic subunit of PKA, greatly attenuates rundown [5, 11, 16, 31, 32]. In some instances, addition of protease inhibitors such as leupeptin or calpastatin, in combination with Mg-ATP and/or an unknown cytoplasmic factor, further stabilizes Ca2+ channel activity and prevents rundown [5, 11, 13, 33]. However, the stabilizing effect of Mg-ATP and PKA as well as protease inhibitors is highly variable and is usually short-lived in inside-out patches. Indeed, results from a number of studies suggest that neither PKA phosphorylation nor proteolysis plays a critical role in the rundown process [5, 12, 34, 35].
Very little is known about the mechanism of rundown of P/Q- and N-type channels. In general, the molecular and structural mechanisms of Ca2+ channel rundown at the channel level are essentially unclear. Three possibilities can be considered, which are not mutually exclusive. First, the channel enters a permanent but existing inactivated state. Second, a new “rundown” gate is formed along the ion conduction pathway. Third, the voltage sensor and the activation gate are decoupled. Our results do not directly address or distinguish among these possibilities. Nevertheless, based on our recent finding [22] that the activation gate of VGCCs is located at the intracellular end of the S6 segments, at or near the membrane/cytoplasm interface where I1520 is situated, we speculate that some mutations of I1520, such as the histidine mutation, probably help stabilize the open conformation. Furthermore, a previous study has shown that the gating current does not change when the ionic current undergoes rundown, suggesting a disruption of the linkage between the voltage sensor and activation gate after rundown [19]. Thus, strengthening and/or stabilizing the coupling between the voltage sensor and activation gate could be a mechanism for the I1520 mutations to counter rundown. Consistent with this idea, we find that the I1520H mutant channel becomes easier to open (Fig. 4A), slower to close (Fig. 4C), harder to inactivate (Fig. 4 D and E), and faster to recover from inactivation (Fig. 4F). These changes of biophysical properties are not surprising given the strategic location of I1520.
At first glance, there seems no mechanistic picture emerging from the amino acid substitutions at position 1520 since residues that appear able to attenuate rundown can be either large or small, hydrophilic or hydrophobic, negatively charged or positively charged (Fig. 2D). But if one looks at the results from a different perspective, one finds that three residues at this position, isoleucine (WT), leucine and methionine, tend to make the channels rundown faster (Fig. 2D). A common property of these three residues is that they are hydrophobic. It is therefore tempting to speculate that hydrophobic interactions formed between the amino acid at position 1520 and its yet unknown partners play a key role in facilitating channel rundown, and such interactions can only be produced by a hydrophobic residue of certain size at this position.
Another possible mechanism for some I1520 mutations to attenuate rundown is to increase the actual affinity of the mutant channels for PIP2, which is required for maintaining the activity of VGCCs [1, 20]. This possibility, however, seems unlikely since, as a hydrophobic residue, I1520 is unlikely to be directly involved in binding PIP2. Furthermore, substitutions with different types of amino acids, including not only histidine, but also cysteines, alanine and even negatively charged aspartate (which would repel the negatively charged PIP2), were able to reduce channel rundown (Fig. 2D). The apparent enhanced interaction with PIP2 for the I1520H and I1520D mutants is thus most likely an allosteric effect.
4.2. Usefulness of the slow rundown mutant channels
For P/Q-type channels, the attenuation of rundown by the I1520H and I1520D mutations is much stronger and more reliable than that achieved by bath application of PIP2 and Mg-ATP [1]. The effect of analogous mutations in N- and L-type channels is less dramatic, but it is still much more robust than that of PIP2. Furthermore, addition of PIP2 can produce significant shift in the voltage-dependence of activation, especially in the absence of reagents promoting PKA phosphorylation [1]. In practice, introducing a single amino acid mutation is more convenient and reliable than applying Mg-ATP and PIP2 in every patch and is also more economical. Thus, these mutant channels could be useful for functional studies on VGCCs that would benefit from a direct access to the cytoplasmic side of the channels, especially in inside-out membrane patches. Admittedly, the utility of these channels is complicated and limited by the fact that they not only display much slower rundown but also exhibit significantly altered biophysical properties. However, the histidine and aspartate mutant channels still demonstrate more or less proper voltage-dependent gating, so for those studies where the absolute value of the activation and inactivation properties is not the primary concern, such as studies of channel modulation by intracellular proteins, factors and second messengers, these mutant channels could prove to be highly beneficial.
Acknowledgments
We thank Y. Mori for Cav2.1 and Cav2.2 cDNAs, T. Tanabe for Cav1.2 and α2δ cDNAs, and E. Perez-Reyes for β4 cDNA. This work was supported by National Institutes of Health grant NS045819.
References
- 1.Wu L, Bauer CS, Zhen XG, Xie C, Yang J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 2002;419:947–952. doi: 10.1038/nature01118. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 3.Becq F. Ionic channel rundown in excised membrane patches. Biochim Biophys Acta. 1996;1286:53–63. doi: 10.1016/0304-4157(96)00002-0. [DOI] [PubMed] [Google Scholar]
- 4.Hille B. Ion channels of excitable membranes. Sunderland, MA: Sinauer Associates; 2001. [Google Scholar]
- 5.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 6.Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- 7.Williams ME, Brust PF, Feldman DH, Patthi S, Simerson S, Maroufi A, McCue AF, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of an omega-conotoxin-sensitive human N-type calcium channel. Science. 1992;257:389–395. doi: 10.1126/science.1321501. [DOI] [PubMed] [Google Scholar]
- 8.De Waard M, Campbell KP. Subunit regulation of the neuronal alpha 1A Ca2+ channel expressed in Xenopus oocytes. J Physiol. 1995;485(Pt 3):619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mouginot D, Bossu JL, Gahwiler BH. Low-threshold Ca2+ currents in dendritic recordings from Purkinje cells in rat cerebellar slice cultures. J Neurosci. 1997;17:160–170. doi: 10.1523/JNEUROSCI.17-01-00160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martini M, Rossi ML, Rubbini G, Rispoli G. Calcium currents in hair cells isolated from semicircular canals of the frog. Biophys J. 2000;78:1240–1254. doi: 10.1016/S0006-3495(00)76681-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chad JE, Eckert R. An enzymatic mechanism for calcium current inactivation in dialysed Helix neurones. J Physiol. 1986;378:31–51. doi: 10.1113/jphysiol.1986.sp016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belles B, Hescheler J, Trautwein W, Blomgren K, Karlsson JO. A possible physiological role of the Ca-dependent protease calpain, its inhibitor calpastatin on the Ca current in guinea pig myocytes. Pflügers Arch. 1988;412:554–6. doi: 10.1007/BF00582548. [DOI] [PubMed] [Google Scholar]
- 13.Romanin C, Grosswagen P, Schindler H. Calpastatin and nucleotides stabilize cardiac calcium channel activity in excised patches. Pflügers Arch. 1991;418:86–92. doi: 10.1007/BF00370456. [DOI] [PubMed] [Google Scholar]
- 14.Kameyama M, Kameyama A, Nakayama T, Kaibara M. Tissue extract recovers cardiac calcium channels from ‘run-down’. Pflügers Arch. 1988;412:328–30. doi: 10.1007/BF00582516. [DOI] [PubMed] [Google Scholar]
- 15.Kameyama M, Kameyama A, Takano E, Maki M. Run-down of the cardiac L-type Ca2+ channel, partial restoration of channel activity in cell-free patches by calpastatin. Pflügers Arch. 1998;435:344–349. doi: 10.1007/s004240050521. [DOI] [PubMed] [Google Scholar]
- 16.Armstrong D, Eckert R. Voltage-activated calcium channels that must be phosphorylated to respond to membrane depolarization. Proc Natl Acad Sci USA. 1987;84:2518–22. doi: 10.1073/pnas.84.8.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ono K, Fozzard HA. Phosphorylation restores activity of L-type calcium channels after rundown in inside-out patches from rabbit cardiac cells. J Physiol. 1992;454:673–688. doi: 10.1113/jphysiol.1992.sp019286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yazawa K, Kameyama A, Yasui K, Li JM, Kameyama M. ATP regulates cardiac Ca2+ channel activity via a mechanism independent of protein phosphorylation. Pflügers Arch. 1997;433:557–62. doi: 10.1007/s004240050314. [DOI] [PubMed] [Google Scholar]
- 19.Costantin JL, Qin N, Waxham MN, Birnbaumer L, Stefani E. Complete reversal of run-down in rabbit cardiac Ca2+ channels by patch-cramming in Xenopus oocytes; partial reversal by protein kinase A. Pflügers Arch. 1999;437:888–894. doi: 10.1007/s004240050859. [DOI] [PubMed] [Google Scholar]
- 20.Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol [correction] 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhen XG, Xie C, Fitzmaurice A, Schoonover CE, Orenstein E, Yang J. Functional architecture of the inner pore of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:193–204. doi: 10.1085/jgp.200509292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie C, Zhen XG, Yang J. Localization of the activation gate of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:205–212. doi: 10.1085/jgp.200509293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu T, Nguyen B, Zhang XM, Yang J. Architecture of a K+ channel inner pore revealed by stoichiometric covalent modification. Neuron. 1999;22:571–580. doi: 10.1016/s0896-6273(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 24.McLaughlin S, Wang JY, Gambhir A, Murray D. PIP(2) and proteins, interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 25.Hering S, Aczel S, Grabner M, Doring F, Berjukow S, Mitterdorfer J, Sinnegger MJ, Striessnig J, Degtiar VE, Wang Z, Glossmann H. Transfer of high sensitivity for benzothiazepines from 1-type to class A (BI) calcium channels. J Biol Chem. 1996;271:24471–24475. doi: 10.1074/jbc.271.40.24471. [DOI] [PubMed] [Google Scholar]
- 26.Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J. Familial hemiplegic migraine mutations change alpha1A Ca2+ channel kinetics. J Biol Chem. 1998;273:5586–5590. doi: 10.1074/jbc.273.10.5586. [DOI] [PubMed] [Google Scholar]
- 27.Kraus RL, Sinnegger MJ, Koschak A, Glossmann H, Stenirri S, Carrera P, Striessnig J. Three new familial hemiplegic migraine mutants affect P/Q-type Ca(2+) channel kinetics. J Biol Chem. 2000;275:9239–9243. doi: 10.1074/jbc.275.13.9239. [DOI] [PubMed] [Google Scholar]
- 28.Berjukow S, Marksteiner R, Sokolov S, Weiss RG, Margreiter E, Hering S. Amino acids in segment IVS6 and beta–subunit interaction support distinct conformational changes during Ca(v)2.1 inactivation. J Biol Chem. 2001;276:17076–17082. doi: 10.1074/jbc.M010491200. [DOI] [PubMed] [Google Scholar]
- 29.Hering S, Berjukow S, Aczel S, Timin EN. Ca2+ channel block and inactivation: common molecular determinants. Trends Pharmacol Sci. 1998;19:439–443. doi: 10.1016/s0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- 30.Stotz SC, Zamponi GW. Identification of inactivation determinants in the domain IIS6 region of high voltage-activated calcium channels. J Biol Chem. 2001;276:33001–33010. doi: 10.1074/jbc.M104387200. [DOI] [PubMed] [Google Scholar]
- 31.Kostyuk PG, Veselovsky NS, Fedulova SA. Ionic currents in the somatic membrane of rat dorsal root ganglion neurons – II. Calcium currents Neurosci. 1981;6:2431–2437. doi: 10.1016/0306-4522(81)90089-0. [DOI] [PubMed] [Google Scholar]
- 32.Doroshenko PA, Kostyuk PG, Martynyuk AE. Intracellular metabolism of adenosin 3′,5′-cyclic monophosphate and calcium inward current in perfused neurons of Helix Pomatia. Neurosci. 1982;7:2125–2134. doi: 10.1016/0306-4522(82)90124-5. [DOI] [PubMed] [Google Scholar]
- 33.Hao LY, Kameyama A, Kameyama M. A cytoplasmic factor, calpastatin and ATP together reverse run-down of Ca2+ channel activity in guinea-pig heart. J Physiol. 1999;514:687–699. doi: 10.1111/j.1469-7793.1999.687ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byerly L, Yazejian B. Intracellular factors for the maintenance of calcium currents in perfused neurons from the snail, Lymnaea stagnalis. J Physiol. 1986;370:631–650. doi: 10.1113/jphysiol.1986.sp015955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kameyama A, Yazawa K, Kaibara M, Ozono K, Kameyama M. Run-down of the cardiac Ca2+ channel: characterization and restoration of channel activity by cytoplasmic factors. Pflügers Arch. 1997;433:547–556. doi: 10.1007/s004240050313. [DOI] [PubMed] [Google Scholar]