

Abstract
G protein-coupled inwardly rectifying potassium (GIRK) channels can be activated or inhibited by different classes of receptors, suggesting a role for G proteins in determining signaling specificity. Because G protein βγ subunits containing either β1 or β2 with multiple Gγ subunits activate GIRK channels, we hypothesized that specificity might be imparted by β3, β4, or β5 subunits. We used a transfection assay in cell lines expressing GIRK channels to examine effects of dimers containing these Gβ subunits. Inwardly rectifying K+ currents were increased in cells expressing β3 or β4, with either γ2 or γ11. Purified, recombinant β3γ2 and β4γ2 bound directly to glutathione-S-transferase fusion proteins containing N- or C-terminal cytoplasmic domains of GIRK1 and GIRK4, indicating that β3 and β4, like β1, form dimers that bind to and activate GIRK channels. By contrast, β5-containing dimers inhibited GIRK channel currents. This inhibitory effect was obtained with either β5γ2 or β5γ11, was observed with either GIRK1,4 or GIRK1,2 channels, and was evident in the context of either basal or agonist-induced currents, both of which were mediated by endogenous Gβγ subunits. In cotransfection assays, β5γ2 suppressed β1γ2-activated GIRK currents in a dose-dependent manner consistent with competitive inhibition. Moreover, we found that β5γ2 could bind to the same GIRK channel cytoplasmic domains as other, activating Gβγ subunits. Thus, β5-containing dimers inhibit Gβγ-stimulated GIRK channels, perhaps by directly binding to the channels. This suggests that β5-containing dimers could act as competitive antagonists of other Gβγ dimers on GIRK channels.
Potassium channels that are active near resting membrane potentials are key determinants of cellular excitability. The G protein-coupled inwardly rectifying K+ (GIRK; Kir3.x) channels are particularly interesting in that they are differentially regulated by receptors that couple to different classes of heterotrimeric G proteins: GIRK channels are activated by receptors that couple to Gαi/o and inhibited by receptors that couple to Gαq (1, 2). This dual up- and down-regulation of GIRK channels by different receptor classes has been described in atrial cells (3), aminergic brainstem neurons (4, 5), and enteric neurons of the peripheral nervous system (6).
Mechanisms underlying inhibition of GIRK channels are not well understood. By contrast, the characteristics of receptor-mediated activation of GIRK channels have been worked out in detail. It is now clear that Gβγ subunits liberated from G protein heterotrimers bind directly to GIRK channels to enhance channel activity (reviewed in refs. 1 and 2). This mechanism raises an interesting conundrum: If all G protein-coupled receptors release Gβγ subunits when activated and all Gβγ subunits tested to date activate GIRK channels (7), how is signaling specificity obtained such that different classes of receptor can activate or inhibit GIRK channels?
One possibility is that specificity derives from associations of different receptors with particular combinations of G protein subunits, which either activate or inhibit GIRK channels. Indeed, exquisite specificity in receptor–G protein subunit interactions has been demonstrated by using antisense approaches in a number of test systems (reviewed in ref. 8), but there is currently no direct evidence for specificity of Gβγ effects on GIRK channels (1, 2, 7). However, of the five Gβ subunits identified to date by molecular cloning, only β1 and β2 have been systematically tested for effects on GIRK channels (7).
Limited functional evidence indicates that dimers containing β3 and β4 can activate GIRK channels (9, 10), but yeast two-hybrid assays suggested that β3 and β4 do not interact with GIRK channels (11). The most structurally divergent Gβ subunit is β5 (12). It is expressed in a number of tissues (e.g., brain, heart, and kidney; refs. 12–14), and forms Gβγ dimers that interact preferentially with Gαq-coupled receptors (15, 16), the same receptors that mediate GIRK channel inhibition (1, 3–6). Effects on GIRK channels of β5-containing Gβγ dimers have not been previously examined.
Here, by using transfection and glutathione-S-transferase (GST) pull-down assays, we show that β3 and β4 are like β1, inasmuch as they form Gβγ dimers that bind directly to GIRK channel proteins and activate GIRK currents in mammalian cells. By contrast, the β5 subunit is distinctly different in its effects on GIRK channels; it forms dimers that inhibit Gβγ-activated GIRK channel currents. Because β5-containing dimers bind to the same cytoplasmic GIRK channel domains as the activating Gβγ subunits, we suggest that β5-mediated GIRK channel inhibition results from competitive interactions between β5 and other subunits for active sites on the channels.
Methods
Stable Cell Lines Expressing GIRK Channels.
We obtained GIRK1 in pBSII (KS−) and GIRK4 in pcDNA3 from N. Davidson (California Institute of Technology, Pasadena, CA) and subcloned GIRK1 into pcDNA3 (Invitrogen). HEK 293 cells were transfected with pcDNA3-GIRK1 and pcDNA3-GIRK4, maintained under G418 selection (400 μg/ml; GIBCO/BRL), and a G418-resistant cell line (G1,4 cell line) was chosen for study based on robust expression of inwardly rectifying K+ currents. Western blots of crude cell lysates and immunocytochemistry with GIRK1 antisera (Alomone Labs, Jerusalem, Israel) verified GIRK expression in G1,4 cells that was absent in HEK 293 cells and diminished by antigenic peptide (data not shown). An additional HEK 293 cell line that stably expresses GIRK1 and GIRK2 together with the m4-muscarinic receptor (G1,2m4) was provided by L. Y. Jan (University of California, San Francisco) (17).
Expression and Purification of GST-GIRK Fusion Constructs and G Protein βγ Subunits.
Cytoplasmic C-terminal regions of GIRK1(182–501), GIRK4(187–419), and the N-terminal region of GIRK4(1–92) were amplified by PCR, inserted in-frame with the GST-coding sequence in pGEX-2T (Pharmacia), and sequenced. The N-terminal region of GIRK1(1–84) was obtained in pGEX-2T from E. Peralta (Harvard University, Cambridge, MA) (18). GST-GIRK fusion proteins were expressed in BL21 cells (Stratagene) and purified with glutathione-conjugated agarose beads (18).
Gβγ dimers containing β1, β3, β4, and β5, together with γ2 or a modified γ2 carrying a 5′-hexahistidine (His6) sequence and a FLAG epitope (γ2HF), were expressed in Sf9 cells. Baculoviruses encoding β1, β5, γ2, and γ2HF subunits have been described (16); those encoding β3 and β4 were prepared after excision of the cognate cDNAs from β3-pCI (S. R. Ikeda, Guthrie Institute, Sayre, PA) and β4-pcDNA3 (W. F. Simonds, National Institutes of Health, Bethesda, MD), essentially as described (16).
Recombinant β1γ2, β1γ2HF, β3γ2, and β4γ2 were extracted from Sf9 cells and purified by DEAE and a Gαi1-affinity chromatography (16). To facilitate purification of β5-containing dimers, which bind poorly to Gαi columns (15), Sf9 cells were infected with β5 and γ2HF; the expressed Gβ5γ2HF was purified by using anti-FLAG and nickel column chromatography (16).
Binding of Purified G Proteins and GST-GIRK Fusion Constructs.
Purified Gβγ subunits (200 nM) were incubated with GST-GIRK fusion proteins (200 nM) at 4°C for 2 h in binding buffer (20 mM Hepes, pH 7.5/3 mM MgCl2/150 mM NaCl/1 mM β-mercaptoethanol/0.1% Genapol C-100). The bead complex was pelleted in a microfuge, washed, boiled in sample loading buffer, separated by SDS/PAGE, and immunoblotted with Gβ and Gγ antisera (see below). To decrease background binding observed with affinity-tagged β1γ2HF and β5γ2HF, the protocol was modified by adding 1% fraction V BSA (Boehringer Mannheim) throughout the incubation and wash, including a 30-min preincubation with the beads, and by eluting proteins from bead pellets using glutathione [0.5 mM Tris, pH 8.0/15 mM reduced glutathione (Sigma)/0.1% Genapol C-100] for 30 min at room temperature. Aliquots of the eluate were separated by SDS/PAGE and immunoblotted.
Silver Staining, Protein Quantification, Immunoprecipitation, and Immunoblotting.
Protein samples were resolved by SDS/PAGE and protein concentrations determined by comparison with ovalbumin standards on silver-stained gels by using a Molecular Dynamics densitometer and wholeband software (BioImage, Ann Arbor, MI) (16, 19). Immunoprecipitation of His6-FLAG-tagged G protein subunits from lysates of transfected G1,4 cells was achieved by using M2 anti-FLAG antisera (27 μg; Sigma) and protein A-Sepharose beads (Pharmacia). For immunoblots, proteins were transferred to nitrocellulose and Gβ subunits were identified by using an anti-Gβ common subunit antibody (β1, β3, and β4 subunits; NEN 808, 1:1,000) or the SGS anti-Gβ5 subunit antisera (W. F. Simonds, NIH; 1:1,000). The γ2 and Gαq/11 subunits were identified with antibodies from Santa Cruz (1:100), GIRK1 with an antibody from Alomone Labs (1:500), and green fluorescent protein (GFP) with a mAb from Chemicon (1:1,000). Primary antibodies were detected by enhanced chemiluminescence using secondary antisera coupled to horseradish peroxidase.
Transfection of Stable Cell Lines Expressing GIRK Channels.
Stable cell lines (G1,4 and G1,2m4) were transiently transfected with plasmids that express G protein α, β, or γ subunits under the control of a cytomegalovirus promoter. We obtained cDNAs for: Gαs in pCW1 and Gαi2 in pCMV5 (G. L. Johnson, National Jewish Medical and Research Center, Denver); Gαt in pcDNAI (F. L. Kolakowski, University of Texas, San Antonio); Gαq in pcDNAI and β1, β4, β5, and γ2 in pcDNA3 (W. F. Simonds; National Institutes of Health); and β3 in pCI (S. R. Ikeda, Guthrie Research Institute). The γ11 cDNA was excised from pVL1393 (19) and subcloned into pcDNA3. γ2 and β5 were modified by PCR to add XbaI and ApaI sites and subcloned into cognate sites in frame with a 5′ His6-FLAG sequence that was inserted into pcDNA3. The βARK construct (W. J. Koch, Duke University, Durham, NC) was subcloned in-frame with a myristic acid attachment signal in pGTM, a pcDNA3 derivative (E. A. Golemis, Fox Chase Cancer Center, Philadelphia, PA). The 5-HT1A receptor cDNA (D. K. Grandy, Vollum Institute, Portland, OR) was subcloned into pcDNA3. These expression plasmids were transfected in 35-mm culture dishes by CaPO4 precipitation together with a plasmid (pGreenLantern; GIBCO) that directs expression of an enhanced GFP in a ratio (μg) of 6:1 (test plasmids:GFP plasmid), except where noted.
Electrophysiological Recordings from Transfected Cells.
Cells were plated onto glass coverslips 48–72 h after transfection to obtain single cells for electrical recording. Coverslips were submerged in a recording chamber at room temperature on microscopes equipped with epifluorescent optics (Zeiss Axioskop FS or Nikon TE300). Individual cells that expressed GFP were identified by using standard fluorescein isothiocyanate filter sets, and targeted for recording with an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA). Recording pipettes (2–4 MΩ) were filled with an internal solution containing 120 mM KCH3O3S/4 mM NaCl/1 mM MgCl2/0.5 mM CaCl2/10 mM Hepes/10 mM EGTA/3 mM MgATP/0.3 mM GTP⋅Tris, pH 7.2. External solution contained 137 mM NaCl/6 mM KCl/10 mM Hepes/2 mM CaCl2/2 mM MgCl2/10 mM glucose, pH 7.3. To enable recording of larger currents from G1,2 m4 cells, extracellular K+ was increased to 25 mM by equimolar substitution for Na+.
Electrophysiological data were acquired and analyzed by using pCLAMP (Axon Instruments). Currents were filtered at 1–2 kHz and digitized at 2–10 kHz; series resistance was typically <10 MΩ and compensated by ≈70–80%. Membrane voltages were corrected for a 10-mV liquid junction potential. Whole cell currents were evoked by using a voltage ramp command (Δ −90 mV; 0.1 V/s) applied at 0.1 Hz from −50 mV. Slope conductance was obtained from a linear fit to current-voltage data from −100 to −120 mV. Receptor-activated GIRK currents were elicited by adding agonists to the superfusate for 1–3 min. Conductance was determined at the peak of the agonist-induced response (within ≈40–50 s); effects decreased by 20–25% over the ensuing 30 s, but this desensitization was not different under any conditions examined (data not shown). All data are presented as mean ± SEM.
Results
Endogenous Gβγ-Mediated GIRK1,4 Channel Currents in a Stable HEK 293 Cell Line.
To study G protein modulation of GIRK channels in a mammalian cell system, we prepared a stable HEK 293 cell line that expresses Kir3.1/3.4 (GIRK1,4) channels. Hyperpolarizing ramp voltage commands evoked inwardly rectifying currents in these G1,4 cells that were not apparent in the control HEK 293 cells (Fig. 1), indicating that a substantial amount of GIRK current is present in G1,4 cells under nonstimulated conditions. This basal current was significantly decreased by proteins that sequester Gβγ; each of four different classes of Gα subunit (Gαq, Gαs, Gαi2, and Gαt), as well as the C-terminal fragment of βARK (βARK-ct), decreased basal conductance in G1,4 cells (Fig. 1, Inset; refs. 9, 20, and 21). In two cases (i.e., cells transfected with Gαq and Gαi2), the basal conductance appeared to be completely blocked because it was reduced to the same level as was seen in HEK 293 cells. These data indicate that channel activation by endogenous Gβγ subunits accounts in large part for basal GIRK current in G1,4 cells, as reported for other native and heterologous expression systems (4, 5, 9).
Figure 1.
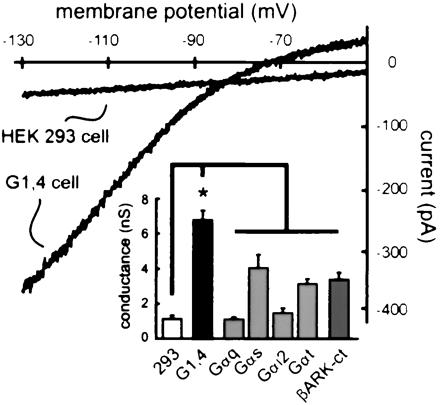
Endogenous Gβγ subunits activate GIRK currents in G1,4 cells. Sample current traces show a large, inwardly rectifying current response to ramp hyperpolarizing voltage commands in G1,4 cells that was absent in HEK 293 cells. (Inset) Basal GIRK currents in G1,4 cells are inhibited by proteins that sequester Gβγ subunits. Conductance (i.e., the slope of I–V curve between −100 and −120 mV; mean ± SEM) was determined in HEK 293 cells (293), in G1,4 cells transfected with GFP alone (G1,4) and in G1,4 cells transfected with the indicated constructs. Note that Gα subunits of diverse families, as well as the C-terminal fragment of βARK, inhibited basal conductance in G1,4 cells. *, Significantly different from G1,4 (P < 0.05 by ANOVA with post hoc Dunnett's test).
Activation and binding of GIRK channels by Gβ3- and Gβ4-containing Gβγ dimers.
It is well accepted that activation of GIRK channels requires binding of Gβγ. Because β3 and β4 reportedly did not interact with GIRK1 in yeast two-hybrid assays (11), we determined if β3- and β4-containing dimers support GIRK channel activation. Cotransfection of either β1, β3 or β4, together with γ2, caused a marked enhancement of basal GIRK channel currents relative to control G1,4 cells transfected with GFP alone (Fig. 2A, Table 1). There was no effect on GIRK channel currents of any Gβ or Gγ subunits expressed alone (Table 1). β3 and β4 also activated GIRK currents when combined with γ11 (Table 1), a Gγ subunit that is distantly related to γ2 and differentially isoprenylated (22). In light of these results, we assessed directly the ability of β3γ2 and β4γ2 to bind to GIRK channel proteins. Purified, recombinant β1γ2, β3γ2, and β4γ2 (Fig. 2B, Left) were incubated with equimolar concentrations of GST-fusion proteins of the N- and C-termini of both GIRK1 and GIRK4 immobilized on glutathione agarose beads (Fig. 2B, Right). The beads were pelleted and proteins that coprecipitated were immunoblotted by using pan-Gβ and γ2 antisera. All three Gβγ dimers bound to all of the cytoplasmic domains of both GIRK1 and GIRK4 (Fig. 2C) and none bound to beads containing GST alone. Thus, β3- and β4-containing dimers bind directly to cytoplasmic domains of GIRK channel proteins, and activate the channels.
Figure 2.
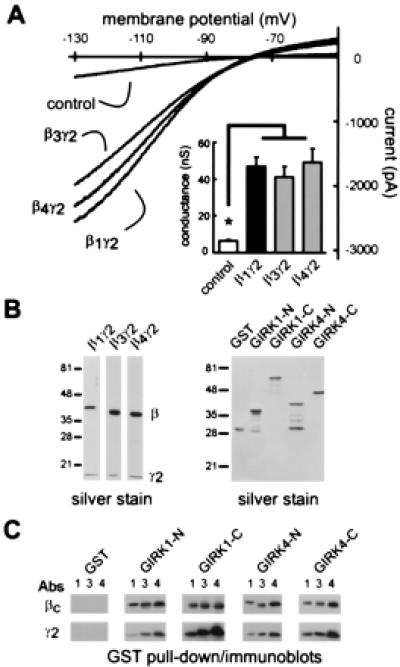
β3γ2 and β4γ2 activate GIRK channels and bind to GIRK1,4 cytoplasmic domains. (A) Sample current traces from control cells (i.e., G1,4 cells transfected with GFP alone) and G1,4 cells expressing β1γ2, β3γ2, or β4γ2; expression of these Gβγ subunits is associated with a large increase in inwardly rectifying current. (Inset) Averaged data (± SEM) show that conductance in cells expressing β3γ2 and β4γ2 is significantly higher than in control cells and comparable to that induced by β1γ2. *, Significantly different from control (P < 0.05; ANOVA with Dunnett's test). (B) Silver stain of recombinant β3γ2 and β4γ2 subunits purified from Sf9 cells (Left) and of GST fusion proteins of N- and C-termini of GIRK1 and GIRK4 isolated from bacterial cells (Right). (C) Immunoblots from GST pull down assays with Gβcommon (βc) and γ2 antisera demonstrate binding of β1 (1), β3 (3), and β4 (4), as well as γ2, to each of the GIRK-GST fusion proteins; Gβγ subunits did not bind to GST alone. Binding data are representative of two replicate experiments.
Table 1.
Conductance was determined in HEK 293 cells and in G1,4 cells transfected with the indicated G protein subunits
| Condition | Conductance, nS ± SEM (n) |
|---|---|
| HEK293 | 1.1 ± 0.2 (8)* |
| G1,4 + GFP | 6.8 ± 0.5 (68) |
| + Gγ2 | 6.3 ± 0.6 (16) |
| + Gβ1 | 7.2 ± 1.3 (8) |
| + Gβ1γ2 | 47.5 ± 4.3 (51)* |
| + Gβ1γ2HF | 79.1 ± 9.5 (8)* |
| + Gβ1γ11 | 38.8 ± 4.2 (27)* |
| + Gβ3 | 6.3 ± 1.0 (8) |
| + Gβ3γ2 | 43.3 ± 6.5 (22)* |
| + Gβ3γ11 | 20.8 ± 1.7 (38)* |
| + Gβ4 | 8.8 ± 2.8 (8) |
| + Gβ4γ2 | 49.1 ± 7.2 (20)* |
| + Gβ4γ11 | 22.8 ± 3.1 (14)* |
| + Gβ5 | 7.1 ± 1.6 (7) |
| + Gβ5γ2 | 3.8 ± 0.2 (65)* |
| + Gβ5γ2HF | 2.3 ± 0.4 (8)* |
| + Gβ5γ11 | 2.5 ± 0.3 (25)* |
Data from experiments in which GFP was transfected at 1 μg, with test constructs at 6 μg each.
Significantly different from control G1,4 cells transfected with GFP alone, by ANOVA with post hoc Dunnett's test (P < 0.05).
Gβ5-containing Gβγ dimers inhibit basal GIRK channel currents.
Of the Gβ subunits identified to date, β5 is the most divergent in primary structure (12). The β5 subunit also was distinctly different in its effects on GIRK channel currents. In G1,4 cells expressing β5γ2, GIRK currents were not increased as in β1γ2-expressing cells but actually diminished relative to control (Fig. 3A). GIRK currents were diminished by 44%–63% in cells transfected with β5γ2 or β5γ11 (Fig. 3A, Inset; Table 1). Because it occurred with two Gγ subunits that are distantly related by homology and differentially isoprenylated (22), the inhibitory effect of Gβ5-containing dimers was probably not a function of the type of associated Gγ subunit.
Figure 3.
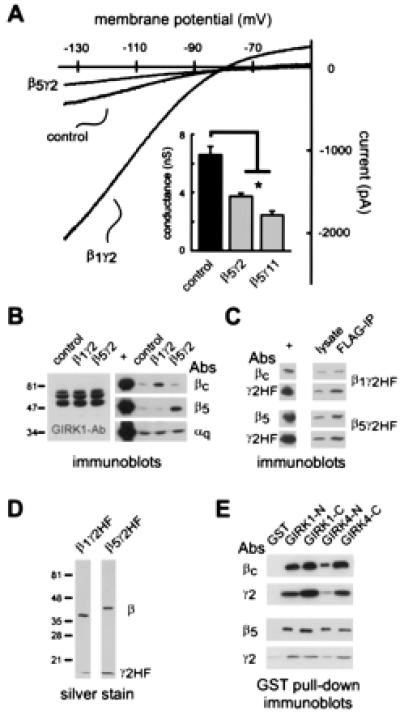
Gβ5-containing dimers inhibit basal GIRK1,4 currents and bind to GIRK1,4 channel cytoplasmic domains. (A) Sample current traces from control cells and G1,4 cells expressing either β1γ2 or β5γ2. Inwardly rectifying current was enhanced by β1γ2 but inhibited by β5γ2. (Inset) Conductance (±SEM) is decreased in cells transfected with β5γ2 or β5γ11. *, Statistically significant differences from control. *, Significantly different from control (P < 0.05; ANOVA with Dunnett's test). (B) Immunoblots of cell lysates from control cells, and cells transfected with β1γ2 or β5γ2. Overexpression of β1 (Right, Top) and β5 (Right, Middle) is apparent in the relevant transfected cells. Note, however, that there is no compensatory change in GIRK1 expression in transfected cells (Left), nor is there any change in Gαq expression (Right, Bottom). Positive controls (+) are lanes loaded with the cognate purified protein. (C) Immunoblots from G1,4 cells transfected with β1γ2HF (Top) or β5γ2HF (Bottom) demonstrate that both β1 and β5 were coprecipitated from cell lysates by using anti-FLAG antibodies, indicating that these Gβ subunits associate with γ2HF in our test system. Positive controls (+) are lanes loaded with the cognate-purified protein. (D) Silver stain of recombinant β1γ2HF and β5γ2HF subunits purified from Sf9 cells. (E) Gβ and Gγ2 immunoblots obtained after incubation of purified βγHF dimers with GIRK1,4-GST fusion proteins demonstrate that both β1γ2HF (Top) and β5γ2HF (Bottom) bind to each of the GIRK-GST fusion proteins, but not to GST alone. Binding data are representative of four replicate experiments.
Inhibition of GIRK channels in β5γ2-transfected cells was not caused by compensatory changes in Gαq expression (Fig. 3B), the Gα subunit with which β5 preferentially associates (15, 16), and which decreased basal GIRK currents when overexpressed (see Fig. 1C). In addition, down-regulation of GIRK channel expression could not account for decreased GIRK currents in β5γ2-transfected cells (Fig. 3B). Among Gβ subunits, only β5 interacts with regulator of G protein signaling (RGS) proteins that contain a G-gamma-like (GGL) domain (i.e., RGS 6, 7, 9, and 11; reviewed in ref. 23). Our functional data indicate that β5-mediated inhibition of GIRK currents was mediated by Gβγ dimers, and not due to interactions with endogenous GGL-containing RGS proteins, because the inhibition required cotransfection of a Gγ subunit. Moreover, we found that β5 interacts with Gγ subunits in our expression system because β5 could be coimmunoprecipitated with epitope-tagged γ2HF (Fig. 3C). The epitope tag did not interfere with effects of Gβγ dimers in control experiments; when transfected into G1,4 cells, β1γ2HF activated and β5γ2HF inhibited GIRK channel currents as effectively as the nontagged counterparts (see Table 1). Thus, these data indicate that β5 indeed forms Gβγ dimers in G1,4 cells but, unlike all other Gβγ subunits tested to date, β5-containing dimers inhibit rather than activate GIRK channels.
Gβ5γ2 Binds N- and C-Termini of GIRK Channels.
To determine whether inhibitory β5-containing dimers bind to GIRK channel proteins in a manner similar to that of activating Gβγ subunits, we performed binding assays using GST-fusion constructs of the cytoplasmic domains of GIRK1 and GIRK4. Because β5γ2 does not bind well to Gαi columns (15), β5 was expressed in Sf9 cells together with γ2HF and purified by epitope affinity chromatography (16); purified β1γ2HF was used as a control for these experiments (Fig. 3D). As illustrated in immunoblots obtained from GST pull down assays (Fig. 3E), both β1γ2HF and β5γ2HF bound to N- and C-termini of GIRK1 and GIRK4, and neither bound to the GST control. These results indicate that inhibitory β5γ2 dimers bind directly to GIRK channel cytoplasmic domains.
Gβ5γ2 Inhibits Gβ1γ2-Mediated Activation of GIRK Channels.
These binding data suggested a mechanism for GIRK inhibition: β5-containing dimers could inhibit GIRK channel activity by competing with other, activating Gβγ dimers. This possibility was tested by functional experiments in G1,4 cells.
In transient transfection assays, increasing the amount of β1 cDNA in the context of constant, excess γ2 cDNA resulted in a dose-dependent enhancement of GIRK currents (Fig. 4A, ○). Addition of β5 cDNA (10 μg) caused a clear rightward shift in the β1 dose-response relationship (Fig. 4A, ●) that was overcome at high levels of β1 (≥6 μg), consistent with a competitive mode of inhibition. Likewise, GIRK currents activated by β1 cDNA (4 μg) were inhibited in a dose-dependent manner by increasing concentrations of β5 cDNA (Fig. 4B, filled symbols); again, increasing β1 concentration to 6 μg overcame the β5-mediated inhibition (Fig. 4B, □). GIRK inhibition could not be explained by a β5-induced decrease in β1 expression (Fig. 4B, Inset) and was apparently not due to competition between β1 and β5 for the γ2 subunit because the β5-mediated inhibition was most prominent at low β1 concentrations when γ2 was in the greatest excess (Fig. 4A) and was identical at three different γ2 concentrations (Fig. 4B). It is noteworthy, however, that levels of β5 that were ≈2.5-fold higher than β1 (i.e., 10 μg vs. 4 μg) were required for GIRK current inhibition. This could be due to differences in expression of the β subunits but might also reflect a lower affinity of β5 for γ2 (14, 24) or of β5γ2 for the GIRK channels. In any case, these functional data are consistent with the interpretation that β5-containing dimers inhibit βγ-activated GIRK channel currents by a competitive mechanism.
Figure 4.
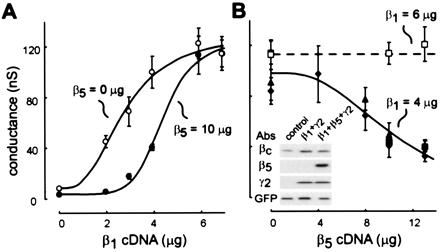
β5γ2 inhibits GIRK currents activated by β1γ2. (A) Concentration-response curve depicting the averaged conductance (±SEM) measured in G1,4 cells with increasing amounts of β1 included in transient transfections (○); the curve was shifted to the right when β5 was included at 10 μg (●). Note that when β1 was not included (β1 = 0 μg), β5 caused ≈50% inhibition of basal currents (8.3 ± 1.3 vs. 3.6 ± 0.6 nS) whereas β5 was without effect at the highest concentrations of β1 (≥6 μg). In all experiments, γ2 was included at 17 μg, and total DNA adjusted to 35 μg by addition of empty pcDNA3 vector. (B) Concentration-response curve depicting the averaged conductance (±SEM) measured in G1,4 cells transfected with increasing amounts of β5. When β1 was included at 4 μg, conductance was decreased by β5 at levels >8 μg (filled symbols). Note that a similar level of GIRK inhibition was observed with three different levels of γ2 (12 μg: ⧫, 17 μg: ▴, 20 μg: ▪; total DNA adjusted to 30, 35 and 40 μg, respectively). GIRK conductance was unaffected when β1 was included at 6 μg (□; γ2, 20 μg). Data points represent averages from ≥6 cells, and were fitted with equations of the form: G = a − d/(1 + ([β cDNA]/b)c), where a and d represent maximum and minimum conductance, b is the half maximal β cDNA concentration, and c is a slope factor. (Inset) Immunoblot of cell lysates from G1,4 cells transfected with: GFP alone (control); GFP, β1 and γ2 (β1 + γ2); or GFP, β1, β5, and γ2 (β1 + β5 + γ2) in μg amounts of 1:4:13:17 (GFP:β1:β5:γ2, with total DNA adjusted to 35 μg with empty vector). Blot is representative of two replicate transfections.
Gβ5γ2 Inhibits Agonist-Induced GIRK Channel Currents.
A hallmark of GIRK channels is their activation by receptors that couple to PTx-sensitive G proteins (Gαi/o), an effect that is mediated by Gβγ(1, 2). We therefore tested if receptor-activated currents also were inhibited by β5-containing Gβγ dimers. GIRK channel currents were activated by 5-HT in G1,4 cells transfected with a 5-HT1A receptor (Fig. 5A), an effect that was completely blocked by pertussis toxin (data not shown). 5-HT also increased GIRK currents in 5-HT1A-transfected cells that were cotransfected with either β5γ2 or β5γ11, but in these β5-expressing cells the 5-HT-induced conductance was ≈50% smaller than it was in control cells (1.4 ± 0.3 nS vs. 2.9 ± 0.7 nS; Fig. 5A, Inset). Thus, β5-containing dimers inhibit agonist-induced GIRK channel currents to approximately the same extent as they inhibit basal currents in G1,4 cells (≈40–60%; see Table 1 and Fig. 3A, Inset).
Figure 5.
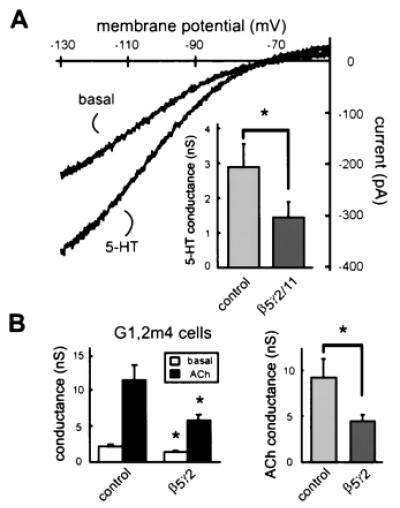
Agonist-induced currents are inhibited by Gβ5-containing dimers. (A) Sample current traces from G1,4 cell line transfected with 5-HT1A receptor under control conditions (basal) and after agonist treatment (5-HT; 50 μM); 5-HT activates an inwardly rectifying current. (Inset) As compared to control cells, the 5-HT-induced conductance is significantly reduced in cells that coexpress β5-containing dimers (data from cells expressing β5γ2 and β5γ11 are combined). (B, Left) Basal conductance is significantly lower in G1,2m4 cells expressing β5γ2, as compared to control cells. Peak conductance obtained during exposure to 10 μM ACh is reduced in β5γ2-expressing cells. (Right) As compared to control cells, the ACh-induced conductance is significantly reduced in cells that express β5γ2. *, Statistically significant differences from control (P < 0.05 by unpaired t test).
Gβ5γ2 also Inhibits Basal and Agonist-Stimulated GIRK1,2 Channel Currents.
The mammalian GIRK channel family includes at least four members (1): GIRK1,4 heterotetramers represent the native atrial GIRK channel (25), whereas GIRK1,2 heterotetramers have been identified in neuronal cells (26). We tested whether inhibitory effects of β5 observed in the context of atrial-type, GIRK1,4 channels also might be manifest in neuronal-type, GIRK1,2 channels. As illustrated in Fig. 5B, β5γ2 inhibited both basal and agonist-induced GIRK1,2 channel currents in the G1,2m4 cell line, a stable cell line that expresses GIRK1,2 channels and the m4-muscarinic receptor (17). The agonist-independent basal conductance was reduced by ≈36% in cells transfected with β5γ2 (from 2.2 ± 0.3 nS to 1.4 ± 0.2 nS) and the ACh-induced conductance was decreased by ≈53% (from 9.2 ± 2.0 nS to 4.4 ± 0.7 nS). These data indicate that inhibitory effects of β5-containing dimers on GIRK channels are not limited to GIRK1,4 heterodimers and suggest that inhibition may be a general outcome of GIRK channel interactions with Gβγ subunits that include β5.
Discussion
In the present study, we used a transfection assay in mammalian cells expressing GIRK channels to identify heretofore unrecognized specificity, conferred by the Gβ subunit, in effects of Gβγ on GIRK channels. We find that Gβγ subunits containing either β3 or β4 behave much like previously characterized β1 and β2 subunits inasmuch as they bind to GIRK channel cytoplasmic domains and activate the channels. Given the 85–95% sequence identity among these Gβ subunits (12, 22), their similarity in effects on GIRK channels is perhaps not surprising. When formed with the more divergent β5 subunit, however, we found that Gβγ dimers actually inhibit GIRK channel currents; this inhibition was apparent under basal conditions as well as during agonist stimulation of cells expressing either the 5-HT1A or m4-muscarinic receptor, conditions in which channels were activated by endogenous Gβγ. In cotransfection assays, β5γ2 inhibited β1γ2-stimulated GIRK channel activity in a manner consistent with competitive antagonism and moreover, β5-containing dimers were capable of binding to the same N- and C-terminal fragments of the GIRK channels as activating Gβγ dimers. These data support burgeoning evidence that β5 imparts unique effector specificity on Gβγ (16, 27, 28), and are particularly provocative from the standpoint of understanding receptor signaling to GIRK channels, because β5-containing dimers couple preferentially to Gαq-linked receptors (15, 16), which inhibit rather than activate GIRK channels (1, 3–6, 29).
Gβ3- and Gβ4-Containing Dimers Bind and Activate GIRK Channels.
We found that β3 and β4 were capable of activating GIRK channels, and, consistent with the accepted mechanism of GIRK activation, we showed that they could bind to GIRK channel N- and C-terminal cytoplasmic domains. Together with earlier data indicating that multiple Gβγ dimers of defined composition (including β1 and β2 with different Gγ subunits) all activated GIRK channels (7), these studies provide no evidence for specificity in signaling to GIRK channels by β1-β4 subunits. Likewise, although not extensively studied, there is only sparse evidence for specificity among β1-β4 subunits with other effectors (e.g., in Gβγ modulation of calcium channels; ref. 30, but see ref. 10). Perhaps systematic exploration of these four highly homologous Gβ subunits in the context of multiple Gγs and distinct effector systems may yet reveal differences in their functional properties.
Unique Activities of Gβ5-Containing Dimers.
The β5 subunit is a clear outlier in the Gβ gene family, and it has become increasingly clear that it possesses characteristics distinctly different from other Gβ subunits. For example, only the β5 subunit associates with the GGL domain-containing RGS proteins (reviewed in ref. 23), a property that predicts novel and specific roles for β5-RGS dimeric complexes. In this respect, heterologous coexpression of β5 with either RGS7 or RGS9 accelerates receptor-mediated GIRK channel activation and deactivation (31). Our experiments did not specifically address these issues but the present data do not suggest any role for β5 interactions with endogenous RGS proteins in modulation of GIRK current amplitude in G1,4 cells (i.e., expressing β5 without a Gγ subunit was without effect).
Together with Gγ subunits in more traditional dimeric complexes, β5 also imparts unique functional properties. Receptor interactions of β5-containing Gβγ pairs are determined by a strong selectivity for Gαq subunits because β5-containing dimers associate preferentially with Gαq subunits (15) and reconstitute agonist-stimulated G protein activation only in the context of Gαq-coupled receptors (16). Furthermore, studies of known Gβγ effectors indicate that whereas β5-containing dimers activate PLC-β (12, 16, 28) and inhibit N-type calcium channels (10, 30), they activate adenylyl cyclase II weakly or not at all (16, 27) and do not stimulate the mitogen-activated protein kinase-signaling pathway (28). Our results with GIRK channels extend the evidence for unique effector specificity of β5-containing dimers. However, β5-containing dimers are not merely inactive at this effector; rather, they bind directly to GIRK channels and cause inhibition of Gβγ-mediated channel activity.
Proposed Mechanism of GIRK Channel Inhibition by Gβ5-Containing Dimers.
The mechanism of GIRK channel activation by Gβγ has been studied extensively, and it is now clear that Gβγ binds directly to the channels to cause activation (reviewed in refs. 1 and 2). The electrophysiological and binding data we present suggest a mechanism by which β5-containing dimers mediate GIRK channel inhibition. Inhibition was evident when GIRK channels were activated by endogenous free Gβγ, either present under basal conditions or following receptor stimulation, and β5 inhibited GIRK channel activation by cotransfected β1 in a competitive manner. Binding data showed that inhibitory β5-containing dimers interact with GIRK channel cytoplasmic domains. Taken together, it seems reasonable to propose that inhibition of GIRK channels by β5-containing dimers could proceed via their displacement of activating Gβγ subunits (i.e., competitive antagonism). It remains to be determined which specific region(s) are critical for inhibition by β5-containing dimers. Given current evidence that favors an important, if not crucial, role for the proximal C-termini of GIRK channels in channel binding and activation by Gβγ subunits (9, 18, 32), it seems likely that competition for those domains may be important for inhibition by Gβ5.
Receptor-Mediated Inhibition of GIRK Channels.
Dual-regulation of GIRK channels by different receptor classes has been demonstrated in various cell contexts, where Gαq-coupled receptors can inhibit channels preactivated by Gαi/o-coupled receptors (3–6); this dual regulation can be recapitulated in G1,4 cells transfected with TRH (Gαq-coupled) and 5-HT1A (Gαi/o-coupled) receptors (Q.L. and D.A.B., unpublished observations). We have shown here that β5-containing dimers, which display a clear specificity for Gα subunits of the Gαq family (15, 16), can interfere with channel activation by Gαi/o-coupled receptors coexpressed in a heterologous cell system. This finding is certainly consistent with the possibility that β5 plays a role in the inhibition of GIRK channels by Gαq-coupled receptors. However, it is important to point out that we have not shown that β5 is directly responsible for receptor-mediated GIRK inhibition, and it remains to be determined whether Gαq or its associated Gβγ subunit is primarily responsible for downstream signaling that culminates in GIRK channel inhibition by those receptors. Nevertheless, given their preferential association with Gαq, our current data indicate that the β5-containing dimers could contribute directly to GIRK channel inhibition—or at the least, serve to exclude other Gβγ subunits that would provide a signal at crossed purposes with that of the associated Gαq subunit.
Acknowledgments
We thank William F. Simonds, who generously supplied most of the Gβ and Gγ subunit cDNAs used in these studies and the Gβ5-specific antibody. We also thank other investigators (see text) who kindly provided additional reagents that were necessary for these studies. This work was supported by grants from the National Institutes of Health [NS33583 and NS39553 (to D.A.B.) and DK19952 (to J.C.G.)] and from the Council for Tobacco Research [4421 (to D.A.B.)].
Abbreviations
- GIRK
G protein-coupled inwardly rectifying potassium
- GST
glutathione-S-transferase
- GFP
green fluorescent protein
- RGS
regulator of G protein signaling
- GGL
G-gamma-like domain
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Dascal N. Cell Signalling. 1997;9:551–573. doi: 10.1016/s0898-6568(97)00095-8. [DOI] [PubMed] [Google Scholar]
- 2.Wickman K, Clapham D E. Physiol Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 3.Braun A P, Fedida D, Giles W R. Pflugers Arch. 1992;421:431–439. doi: 10.1007/BF00370253. [DOI] [PubMed] [Google Scholar]
- 4.Farkas R H, Chien P Y, Nakajima S, Nakajima Y. Neurosci Lett. 1997;231:21–24. doi: 10.1016/s0304-3940(97)00530-2. [DOI] [PubMed] [Google Scholar]
- 5.Velimirovic B M, Koyano K, Nakajima S, Nakajima Y. Proc Natl Acad Sci USA. 1995;92:1590–1594. doi: 10.1073/pnas.92.5.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanner S, Evans R J, Matsumoto S G, Surprenant A. J Neurophysiol. 1993;69:1632–1644. doi: 10.1152/jn.1993.69.5.1632. [DOI] [PubMed] [Google Scholar]
- 7.Wickman K D, Iniguez-Lluhl J A, Davenport P A, Taussig R, Krapivinsky G B, Linder M E, Gilman A G, Clapham D E. Nature (London) 1994;368:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 8.Gudermann T, Kalkbrenner F, Schultz G. Annu Rev Pharmacol Toxicol. 1996;36:429–459. doi: 10.1146/annurev.pa.36.040196.002241. [DOI] [PubMed] [Google Scholar]
- 9.He C, Zhang H L, Mirshahi T, Logothetis D E. J Biol Chem. 1999;274:12517–12524. doi: 10.1074/jbc.274.18.12517. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Velasco V, Ikeda S R. J Neurosci. 2000;20:2183–2191. doi: 10.1523/JNEUROSCI.20-06-02183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan K, Gautam N. J Biol Chem. 1996;271:17597–17600. doi: 10.1074/jbc.271.30.17597. [DOI] [PubMed] [Google Scholar]
- 12.Watson A J, Katz A, Simon M I. J Biol Chem. 1994;269:22150–22156. [PubMed] [Google Scholar]
- 13.Jones P G, Lombardi S J, Cockett M I. Biochim Biophys Acta. 1998;1402:288–291. doi: 10.1016/s0167-4889(98)00017-2. [DOI] [PubMed] [Google Scholar]
- 14.Snow B E, Betts L, Mangion J, Sondek J, Siderovski D P. Proc Natl Acad Sci USA. 1999;96:6489–6494. doi: 10.1073/pnas.96.11.6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher J E, Lindorfer M A, DeFilippo J M, Yasuda H, Guilmard M, Garrison J C. J Biol Chem. 1998;273:636–644. doi: 10.1074/jbc.273.1.636. [DOI] [PubMed] [Google Scholar]
- 16.Lindorfer M A, Myung C S, Savino Y, Yasuda H, Khazan R, Garrison J C. J Biol Chem. 1998;273:34429–34436. doi: 10.1074/jbc.273.51.34429. [DOI] [PubMed] [Google Scholar]
- 17.Chuang H H, Yu M, Jan Y N, Jan L Y. Proc Natl Acad Sci USA. 1998;95:11727–11732. doi: 10.1073/pnas.95.20.11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunkel M T, Peralta E G. Cell. 1995;83:443–449. doi: 10.1016/0092-8674(95)90122-1. [DOI] [PubMed] [Google Scholar]
- 19.Myung C S, Yasuda H, Liu W W, Harden T K, Garrison J C. J Biol Chem. 1999;274:16595–16603. doi: 10.1074/jbc.274.23.16595. [DOI] [PubMed] [Google Scholar]
- 20.Jeong S W, Ikeda S R. J Neurosci. 1999;19:4755–4761. doi: 10.1523/JNEUROSCI.19-12-04755.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kammermeier P J, Ikeda S R. Neuron. 1999;22:819–829. doi: 10.1016/s0896-6273(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 22.Gautam N, Downes G B, Yan K, Kisselev O. Cell Signalling. 1998;10:447–455. doi: 10.1016/s0898-6568(98)00006-0. [DOI] [PubMed] [Google Scholar]
- 23.Hepler J R. Trends Pharmacol Sci. 1999;20:376–382. doi: 10.1016/s0165-6147(99)01369-3. [DOI] [PubMed] [Google Scholar]
- 24.Jones M B, Garrison J C. Anal Biochem. 1999;268:126–133. doi: 10.1006/abio.1998.3064. [DOI] [PubMed] [Google Scholar]
- 25.Krapivinsky G, Gordon E A, Wickman K, Velimirovic B, Krapivinsky L, Clapham D E. Nature (London) 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 26.Inanobe A, Yoshimoto Y, Horio Y, Morishige K, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, et al. J Neurosci. 1999;19:1006–1017. doi: 10.1523/JNEUROSCI.19-03-01006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayewitch M L, Avidor-Reiss T, Levy R, Pfeuffer T, Nevo I, Simonds W F, Vogel Z. J Biol Chem. 1998;273:2273–2276. doi: 10.1074/jbc.273.4.2273. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S, Coso O A, Lee C, Gutkind J S, Simonds W F. J Biol Chem. 1996;271:33575–33579. doi: 10.1074/jbc.271.52.33575. [DOI] [PubMed] [Google Scholar]
- 29.Sharon D, Vorobiov D, Dascal N. J Gen Physiol. 1997;109:477–490. doi: 10.1085/jgp.109.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.García D E, Li B, García-Ferreiro R E, Hernández-Ochoa E O, Yan K, Gautam N, Catterall W A, Mackie K, Hille B. J Neurosci. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovoor A, Chen C K, He W, Wensel T G, Simon M I, Lester H A. J Biol Chem. 2000;275:3397–3402. doi: 10.1074/jbc.275.5.3397. [DOI] [PubMed] [Google Scholar]
- 32.Krapivinsky G, Kennedy M E, Nemec J, Medina I, Krapivinsky L, Clapham D E. J Biol Chem. 1998;273:16946–16952. doi: 10.1074/jbc.273.27.16946. [DOI] [PubMed] [Google Scholar]