


Abstract
Among the three mammalian genes encoding DNA ligases, only the LIG3 gene does not have a homolog in lower eukaryotes. In somatic mammalian cells, the nuclear form of DNA ligase IIIα forms a stable complex with the DNA repair protein XRCC1 that is also found only in higher eukaryotes. Recent studies have shown that XRCC1 participates in S phase-specific DNA repair pathways independently of DNA ligase IIIα and is constitutively phosphorylated by casein kinase II. In this study we demonstrate that DNA ligase IIIα, unlike XRCC1, is phosphorylated in a cell cycle-dependent manner. Specifically, DNA ligase IIIα is phosphorylated on Ser123 by the cell division cycle kinase Cdk2 beginning early in S phase and continuing into M phase. Interestingly, treatment of S phase cells with agents that cause oxygen free radicals induces the dephosphorylation of DNA ligase IIIα. This oxidative stress-induced dephosphorylation of DNA ligase IIIα is dependent upon the ATM (ataxia-telangiectasia mutated) kinase and appears to involve inhibition of Cdk2 and probably activation of a phosphatase.
INTRODUCTION
DNA ligases are critical enzymes in DNA metabolism because of their role in the completion of DNA replication, repair and recombination. In mammals, three genes encoding ATP-dependent DNA ligases, designated LIG1, LIG3 and LIG4, have been identified (1). The LIG3 gene is unusual for the following reasons. Firstly, unlike LIG1 and LIG4, there are no homologs of LIG3 in lower eukaryotes such as Saccharomyces cerevisiae. Secondly, as described below, the LIG3 gene generates several distinct gene products. In somatic cells, the use of alternative translation initiation start sites generates nuclear and mitochondrial forms of DNA ligase IIIα (2) whereas an alternative germ cell-specific splicing mechanism generates DNA ligase IIIβ, which has a different C-terminal amino acid sequence than DNA ligase IIIα (3).
The discovery that DNA ligase IIIα forms a stable complex with the DNA repair protein, XRCC1, which is involved in base excision repair (BER) and the repair of DNA single-strand breaks, provided the first insights into the cellular functions of the LIG3 gene products (4). In subsequent studies it was shown that complex formation occurs via interactions between the C-terminal BRCT motifs of XRCC1 and DNA ligase IIIα (3,5,6) and that XRCC1 is necessary for the stability and activity of DNA ligase IIIα in vivo (7). Since xrcc1 mutant cells are deficient in DNA ligase IIIα activity, the phenotype of these cells is likely to be, at least in part, a consequence of reduced DNA joining.
Although XRCC1 has no catalytic activity, it appears to co-ordinate the activities of proteins involved in BER and the repair of DNA single-strand breaks via protein–protein interactions (4,8–14). More recently, it has been found that XRCC1 also interacts with aprataxin (15–17), a protein which is required to prevent ataxia ocular apraxia (18). Intriguingly, both XRCC1 and DNA ligase IIIα appear to be components of two different complexes, one of which contains known DNA repair protein partners of XRCC1 whereas the other complex contains aprataxin (19). Since reduced cellular levels of aprataxin result in reduced levels of XRCC1 and increased DNA damage sensitivity (19), it appears that aprataxin stabilizes XRCC1 (and presumably DNA ligase IIIα), thereby indirectly regulating the base excision and single strand break repair pathways.
XRCC1 functions independently of DNA ligase IIIα in a DNA repair pathway that plays a major role in the repair of DNA damage induced by DNA alkylating agents in replicating cells (20). Moreover, XRCC1 does not appear to be involved in the function of DNA ligase IIIα in mitochondrial DNA metabolism (21). Thus, the role of DNA ligase IIIα in maintaining genome stability and cell viability cannot be deduced from the phenotype of xrcc1 mutant cells. Here we show that, in contrast to the constitutive phosphorylation of XRCC1 by casein kinase II (22), DNA ligase IIIα is specifically phosphorylated in replicating cells by the cell cycle kinase Cdk2. However, in response to oxidative DNA damage, DNA ligase IIIα is dephosphorylated in a pathway that is dependent upon the DNA damage-activated, phosphatidylinositol 3-phosphate (PI3)1-related kinase ATM.
MATERIALS AND METHODS
Cell culture
The human cancer cell lines, T24 and HeLa were obtained from America Type Culture Collection and cultured in DMEM with low glucose (1 mg/ml) plus 10% fetal bovine serum. Cultures of T24 cells were synchronized by density arrest and then replated at a lower density to allow the cells to synchronously re-enter the cell cycle. To obtain M phase cells, nocodazole was added to the medium 26 h after replating and incubation continued for 5–8 h (23). The progression of T24 cells through the cell cycle was monitored by the phosphorylation status of the retinoblastoma protein (23) and confirmed by FACS analysis (data not shown). HeLa cells were synchronized in S phase by double thymidine treatment as described (24). A population of cells enriched for the G1 phase was prepared by releasing the nocodazole-arrested G2/M cells and allowing them to progress into G1.
The cell lines, FT/pEBS7 and FT/pEBS7-YZ5 were derived from immortalized human fibroblast AT22IJE-T, which contains homozygous frameshift mutations in the ATM gene, by transfection with the empty vector (pEBS7) and a recombinant vector expressing wild type ATM, respectively (25). The cells lines were cultured in DMEM with low glucose (1 mg/ml) plus 10% fetal bovine serum.
Cell cultures were treated with various genotoxic agents. For γ irradiation, cells were exposed at a rate of 2.5 Gray/min in an Irradiator (Mark I, model 68A, JL Shepherd & Assoc.) and then harvested 1 h post-irradiation. The DNA alkylating agent ethyl methanesulfonate (EMS) and the oxidizing agents menadione and hydrogen peroxide (30% w/w solution) were from Sigma. Cells were treated with either EMS, hydrogen peroxide or menadione for 1 h. To induce oxidative stress, glucose oxidase was added at various concentrations to cells cultured in DMEM with high glucose (4.5 mg/ml). After 1 h of incubation, the cells were harvested. Wortmannin (Sigma) was added 30 min before exposure to the damaging agent to inhibit PI3 kinases (26).
Immunoblotting and antibodies
Cells were harvested and lysed in lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.1% NP-40, 1 mM phenylmethylsulfonyl fluoride and 1 mM NaF]. After centrifugation at 14 000 g for 5 min, aliquots of the cleared lysates were resolved by sodium dodecyl-polyacrylamide gel (8.5% polyacrylamide, 17 × 22 cm gel) electrophoresis (SDS–PAGE) and then transferred to a nitrocellulose membrane (Optitran, Schleicher & Schuell) prior to incubation with the indicated antibody. Antigen-antibody complexes were detected by either enhanced chemiluminescence (ECL, Pierce) or an alkaline phosphatase/colorimetric assay (AP, Bio-Rad). Antibodies against DNA ligase III (1F3, monoclonal) and XRCC1 (rabbit polyclonal) antibodies were from GeneTex. Antibodies against Cdk6, Cdk2 and Cdc2 were from Santa Cruz Biotechnology. Antibody against human Rb protein (RBmAb11D7), a gift from Dr Phang-Lang Chen, was used to verify cell cycle status. Enzyme-linked secondary antibodies were from Bio-Rad. Antibodies against cyclin A and B are from Santa Cruz Biotechnology. α-phos-Ser123, antiserum against a DNA ligase III peptide (amino acid 116–130) encompassing phosphorylated Ser123 was also generated and affinity purified (Bethyl Laboratories).
Immunoprecipitations
Cleared cell lysates, prepared as described above, were mixed with the indicated antibody and protein G (or A) Sepharose beads equilibrated with lysis buffer. After incubation at 4°C with constant rolling, the beads were collected and washed extensively with lysis buffer. Proteins were eluted from the beads by incubation in SDS sample buffer for 5 min at 100°C and then separated by SDS–PAGE.
Phosphatase assays
Cleared cell lysates were prepared as described above except that NaF was omitted from the lysis buffer. The lysates were incubated with or without the indicated phosphatase (lambda phosphatase or calf intestinal phosphatase, NEB) at 30°C for 30 min. Where indicated, the nonspecific phosphatase inhibitor NaF (1 mM) was also included in this assay to demonstrate that changes in mobility were caused by phosphatase action. Similarly, DNA ligase IIIα was co-immunoprecipitated with XRCC1 antibodies and protein G beads from cell lysates as described above. Proteins bound to the protein G beads were incubated with calf intestine phosphatase (NEB ) at 37°C for 60 min. After separation by SDS–PAGE, DNA ligase III was detected by immunoblotting.
Kinase assays
Cells were harvested and lysed in TGN buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Tween-20 (v/v), 0.2% NP-40 (v/v), 1 mM NaF, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 2 μg/ml pepstatin]. Cleared lysates (2 mg protein) were incubated with 30 μl protein G Sepharose beads for 1 h with constant rolling at 4°C. After removing the beads, antibodies against Cdk2, Cdk6 and Cdc2 (Santa Cruz Biotechnology) were added and incubation continued. After 1 h, protein G Sepharose beads (20 μl) were added and incubation continued for 2 h. Beads were collected, washed with TGN buffer and then with Cdk buffer (50 mM HEPES (pH 7.5), 30 mM NaCl, 4 mM MnCl2, 6 mM MgCl2, 10% glycerol, 1 mM DTT and 100 μM sodium orthovanadate). For each kinase assay, the beads were incubated with 1 μg of substrate protein, 20 μM ATP and 10 μCi [γ-32P]ATP in Cdk buffer for 15 min at 30°C. After separation by SDS–PAGE (10% polyacrylamide, 7 × 8 cm gel), labeled proteins were detected in the dried gel by phosphorimager analysis. Cdk6 substrate, an Rb fragment corresponding to amino acids 769–921 of mouse Rb, was from Santa Cruz Biotechnology. Cdk2 and Cdc2 substrate, histone H1 was from Sigma.
Plasmids
Wild type and dominant negative Cdk2 cDNAs (27) were subcloned in-frame with an HA-tag into the mammalian expression vector pRC/RSV (Invitrogen) to generate pRCH-cdk2(wt) and pRCH-cdk2(dn). The plasmid pQE32 (Qiagen) was used to express all versions of his-tagged DNA ligase IIIβ. The pQE32 plasmid expressing full length DNA ligase IIIβ was described previously (28,29). Various truncated versions of DNA ligase IIIβ cDNA were subcloned into pGEX-4T to generate plamids expressing GST fusion proteins.
PCR-based site directed mutagenesis was performed using DNA ligase IIIβ cDNA as a template. Three primers were designed for mutation of each of the three putative Cdk sites: Thr104/Ala, Ser123/Ala and Ser826/Ala. The sequences of these primers are listed. Primer pair I, 5′-GTCTTCTAAGGCA(T)GCAGGTA(G)CACCAAAGAAGAAAGC-3′; 5′-GCTTTCTTCTTTGGTGCACCTGCAGCCTTAGAAGAC-3′. To facilitate identification of mutants, a PstI site was introduced by replacing GCA with GCT. This mutation does not affect amino acid sequence. Similarly, a HindIII site was introduced into the second and third primers without changing the amino acid sequence. Primer pair II 5′-CCAGGCTAAGT(C)TG(T)ACAACCACTGGCCAGGTGACTT(G)CTCCAGTGAAAG-3′; 5′-CTTTCACTGGAGCAGTCACCTGGCCAGTGGTTGTAAGCTTAGCCTGG-3′. Primer pair III, 5′-GGGGAGAAGCTG(T)GCCACAAAGTCTT(G)CTCCAGTGAAAGTAG-3′; 5′-CTACTTTCACTGGAGCAGACTTTGTGGCAAGCTTCTCCCC-3′. Amplification was carried out in a 50 μl volume containing 50 ng of plasmid DNA, 200 μM dNTPs, 125 nM primers, 1 μl Pfu buffer (Stratagene), and 2.5 U of Pfu DNA polymerase. A pre-incubation at 95°C for 30 s was performed, followed by 16 cycles of 95 C for 30 s, 55 C for 1 min, and 68 C for 15 min. The PCR products were verified by agarose gel electrophoresis and treated with the restriction endonuclease DpnI at 37°C for 1 h to remove template plasmids. An aliquot (1 μl) from each reaction was used for transformation of competent bacterial cells. LB plates containing isopropyl-β-d-thiogalactopyranoside (0.1 mM) and 5-bromo-4-chloro-3-indolyl-β-d-galactoside (0.1 mg/ml) were used for selection. Plasmids bearing mutations were selected and verified by restriction enzyme digestion and DNA sequencing.
Expression and purification of his-tagged and GST fusion proteins
Wild type and mutation versions of his-tagged DNA ligase IIIβ were overexpressed in Escherichia coli and purified as described previously (28,29). The plasmid pGEX-4T encoding truncated versions of DNA ligase III as GST fusions were transformed into E.coli BL-21 competent cells. Expression of GST fusion proteins was induced by IPTG and verified by Coomassie blue staining after SDS–PAGE. The purification of tagged proteins was carried out as described previously (28,29).
RESULTS
DNA ligase IIIα is phosphorylated during S phase
The recent observations that the contributions of XRCC1 and DNA ligase IIIα to the repair of DNA damage differ depending on cell cycle stage (20) and that XRCC1 is constitutively phosphorylated by casein kinase II (22) prompted us to examine whether DNA ligase IIIα is subject to post translational modification during cell cycle progression. In initial studies, density-arrested human T24 cells derived from a bladder carcinoma were stimulated to synchronously re-enter the cell cycle by replating at a lower density (23). The cell cycle regulatory protein, Rb, which becomes hyper-phosphorylated during the cell cycle progression, has been used as an indicator of cell cycle stage (23). As expected, Rb is increasingly phosphorylated as the T24 cell population proceeds through the cell cycle (Figure 1A, left panel). The cellular levels of XRCC1 (Figure 1A, right panel) and DNA ligase IIIα (Figure 1A, left panel) did not change significantly as the cell population progressed through the cell cycle stages. However, a form of DNA ligase IIIα with reduced mobility was initially detected ∼6–12 h after replating, which corresponds to when the cells are transitioning from G1 to S phase (23). This form accumulated as the cell population progressed through the cell cycle with a concomitant reduction of the major form of DNA ligase IIIα detected in early G1 cells (Figure 1A).
Figure 1 .

DNA ligase IIIα is phosphorylated during cell cycle progression. (A) Human T24 cells were synchronized by density arrest. Aliquots of the culture were removed at the times indicated after replating at a lower density. The addition of nocodazole (Noc) blocked cells at G2/M. DNA ligase IIIα, Rb, (left panel) β-actin and XRCC1 (right panel) were detected in cell extracts by immunoblotting. The phosphorylation status of Rb serves as an indicator of cell cycle progression (23). (B) HeLa cell populations were enriched for the G1 (G1), G2/M (Noc) and S phases (DT) of the cell cycle as described in Materials and Methods section. DNA ligase IIIα was detected in the extracts by immunoblotting. (C) Cell extracts were prepared from asynchronous CHO cells (As) and CHO cells treated with hydroxyurea (Hu) and nocodazole (Noc). DNA ligase III was detected by immunoblotting. (D) Aliquots of an extract from an asynchronous population of T24 cells were incubated as indicated with λ phosphatase (λ) either in the presence (+) or absence (−) of the phosphatase inhibitor, NaF for 30 min at 37°C. DNA ligase IIIα was detected by immunoblotting. (E) DNA ligase IIIα was co-immunoprecipitated with XRCC1 antibody from an S phase HeLa cell extract. The immunoprecipitate was incubated with (+) or without (−) calf intestinal phosphatase (CIP) prior to the detection of DNA ligase IIIα by immunoblotting.
To demonstrate that the change in mobility of DNA ligase IIIα is not cell-type specific, we examined extracts of human HeLa cells enriched for different cell cycle stages by treatment with chemical inhibitors of cell cycle progression (Figure 1B). In accord with the studies described above (Figure 1A), a DNA ligase IIIα doublet was detected in extracts of HeLa cells enriched for early S phase by double thymidine block (Figure 1B). In extracts from cell populations enriched for M phase by nocodazole treatment, the upper band of the doublet was the major species whereas the lower band of the doublet was the major species in extracts from cell populations enriched for G1 phase by releasing cells arrested in M phase by nocodazole (Figure 1B). Similar cell cycle-dependent changes in the mobility of DNA ligase IIIα were observed in experiments with Chinese Hamster Ovary cells (Figure 1C).
Incubation of extracts from cell populations enriched for S phase with λ phosphatase resulted in a reduction in the upper band of the DNA ligase IIIα doublet and a comcomitant increase in the lower band (Figure 1D). A similar result was obtained when DNA ligase IIIα was immunoprecipitated from S phase cell extracts with an XRCC1 antibody and then incubated with calf intestinal phosphatase (Figure 1E). Together these results demonstrate that DNA ligase IIIα is phosphorylated when cells enter S phase with an increasing fraction of DNA ligase IIIα complexed with XRCC1 becoming phosphorylated as the cells progress through the S and G2 phases of the cell cycle. During the transition from mitosis into the G1 phase of the next cell cycle, DNA ligase IIIα is dephosphorylated.
Cdk2 phosphorylates DNA ligase III in vitro and in vivo
The pattern of DNA ligase IIIα phosphorylation suggests that it may be a substrate for one of the cell cycle-regulated cyclin-dependent kinases (CDKs). Indeed, a search of the mammalian DNA ligase IIIα amino acid sequence for regions that are homologous to the consensus CDK site (Figure 2A) revealed three putative CDK sites that are conserved in the human and mouse enzymes (Figure 2A). To determine which of the CDK(s) phosphorylates DNA ligase III, Cdk6 and Cdk2 were immunoprecipitated from extracts of an asynchronous HeLa cell population. Although both these kinases phosphorylated their native substrate, Rb protein fragment and histone H1, respectively (Figure 2B), only Cdk2 phosphorylated DNA ligase III in vitro. This result is consistent with the observed phosphorylation of DNA ligase IIIα in S phase (Figure 1) because Cdk2 is active in both the S and G2 phases of the cell cycle whereas Cdk6 is a G1 phase kinase. Since DNA ligase IIIα is also phosphorylated in M phase (Figure 1), we asked whether Cdc2, a cyclin-dependent kinase active in G2/M, can also phosphorylate DNA ligase III. As expected, activity Cdk2 was co-immunoprecipitated with cyclin A from S and G2/M extracts whereas robust Cdc2 activity was only co-immunoprecipitated with cyclin B from a G2/M extract of synchronized human T24 cells (Figure 2C). The immunoprecipitated Cdk2 and Cdc2 kinases phosphorylated both histone H1 and DNA ligase III (Figure 2D).
Figure 2 .

Cdk2 phosphorylates DNA ligase III in vitro. (A) The consensus motif recognized by CDKs is shown with conserved putative CDK phosphorylation sites in mouse and human DNA ligase III below. (B) In the upper panel, Cdk6 and Cdk2 were immunoprecipitated from asynchronous HeLa cell extracts. In vitro kinase assays were performed with [γ-32P]ATP using an Rb fragment (Santa Cruz) or histone H1 (Sigma) as phosphorylation substrates. The protein kinase substrates were detected by Coomassie blue staining and phosphorimaging. In the lower panel, in vitro phosphorylation of DNA ligase III by Cdk6 and Cdk2 immunoprecipitated from asynchronous HeLa cell extracts was detected by phosphorimaging. (C) T24 cells density-arrested in G0 were released into the cell cycle and harvested at 6 h (G1) or 24 h (S). Cells were treated with nocodazole as described in Materials and Methods section to obtain G2/M enriched cells. In the upper and middle panels, Cdk2, Cdc2, cyclin A and cyclin B were detected in the cell extracts by immunoblotting. In the lower left panel, Cdk2 co-immunoprecipitated with cyclin A antibody (IP) from the indicated cell extract was detected by immunoblotting (IB). In the lower right panel, Cdc2 co-immunoprecipitated with cyclin B antibody (IP) from the indicated cell extract was detected by immunoblotting (IB). (D) Phosphorylation of histone H1 and DNA ligase III by CDK kinases immunoprecipitated with cyclin A (left panel) and cyclin B (right panel) antibodies from the indicated extracts. The protein kinase substrates were detected by Coomassie blue staining and phosphorimaging.
Since DNA ligase IIIα is fully phosphorylated during cell progression prior to the the presence of robust Cdc2 activity, we focused on mapping the DNA ligase III sites phosphorylated by Cdk2. Several truncated versions of DNA ligase III were expressed as GST fusion proteins and purified by affinity chromatography (Figure 3A). When these fragments were incubated with immunoprecipitated Cdk2 in kinase assays, an N-terminal fragment containing two of the three putative CDK sites (Thr104 and Ser123) was efficiently phosphorylated by Cdk2, whereas a fragment containing the third putative CDK site (Ser826) was phosphorylated to a much lesser extent (Figure 3B). To determine whether Cdk2 was indeed phosphorylating the putative CDK sites in vitro, we used site-directed mutagenesis to change the key serine or threonine residues to alanine. Replacement of Ser123 greatly reduced DNA ligase III phosphorylation whereas replacement of both Thr104 and Ser826 had only a minor effect on DNA ligase III phosphorylation. Together these results show that DNA ligase III is specifically phosphorylated by Cdk2 and Cdc2 in vitro and that Ser123 is the major site of phosphorylation by Cdk2.
Figure 3 .

Mapping the DNA ligase III sites phosphorylated in vitro by Cdk2. (A) The indicated fragments of DNA ligase III were expressed as GST fusion proteins. The grey and black boxes mark the positions of the N-terminal zinc finger and C-terminal BRCT domain, respectively. (B) Purified GST and the GST fusion proteins were incubated with immunoprecipitated Cdk2 and [γ-32P]ATP. (C) Wild type (wt) and mutant versions of DNA ligase III (S123/A and T104/A, S286/A), in which putative amino acid targets for Cdk2 phosphorylation were replaced with alanine, were incubated with immunoprecipitated Cdk2 and [γ-32P]ATP. After separation by SDS–PAGE, unlabeled proteins were visualized by Coomassie blue staining [upper panel in both (B) and C)] and labeleled proteins by phosphorimager analysis [lower panel in both (B) and (C)].
To demonstrate that Ser123 of DNA ligase III is phosphorylated in vivo, we generated an antibody against a DNA ligase III peptide (residues 116–130) encompassing phosphorylated Ser123. As expected, the phosphopeptide antibody preferentially recognized DNA ligase III that had been phosphorylated in vitro by immunoprecipitated Cdk2 (Figure 4A). In this experiment, we did not detect phosphorylation of DNA ligase III by Cdk2 immunoprecipitated by cyclin B antibody, presumably because of the low levels of the G2/M kinase in an extract from asynchronous cells. Next we examined the reactivity of DNA ligase in extracts in G1 and S phase cell extracts with the phosphopeptide antibody by immunoblotting. As expected, the DNA ligase III monoclonal antibody that recognizes both phosphorylated and unphosphorylated DNA ligase III, detected a DNA ligase III doublet in the S phase extract but similar total levels of DNA ligase III protein were present in both cell extracts (Figure 4B). In contrast, only the S phase extract contained versions of DNA ligase III recognized by the phosphopeptide antibody under these conditions (Figure 4B), indicating that Ser123 is phosphorylated in vivo.
Figure 4 .

Cdk2 phosphorylates DNA ligase III in vivo. (A) Proteins were immunoprecipitated with Cdk2, cyclin A or cyclin B antibodies from extracts of asynchronous HeLa cells. The immunoprecipitates were incubated with purified DNA ligase III and histone H1 in the presence of [γ-32P]ATP. DNA ligase III was detected by immunblotting with the DNA ligase III monoclonal antibody IF3 that recognizes unmodified and phosphorylated DNA ligase III and the antibody (α-phos-Ser123) raised against a DNA ligase III peptide (residues 116–130) encompassing phosphorylated Ser123. Purified histone H1 (Sigma) was detected by Coomassie blue staining. Labeled DNA ligase III and histone H1 were detected by phosphorimaging. (B) DNA ligase III from G1 (G1) and S (S) phase T24 cells was detected by immunoblotting with either 1F3 or α-phos-Ser123. (C) HeLa cells were transfected with expression vectors expressing HA-tagged versions of wild type (Cdk2-wt) and dominant negative cdk2(dn) (Cdk2-dn), and the empty expression vector (vector). HA-tagged Cdk2 and DNA ligase III were detected in cell extracts by immunoblotting.
To confirm that Cdk2 phosphorylates DNA ligase IIIα in vivo, we transiently overexpressed wild type Cdk2 and a dominant negative version (Cdk2 dn), which interacts with cyclins but lacks kinase activity (27). In cells overexpressing HA-tagged Cdk2 dn, there were markedly reduced levels of phosphorylated DNA ligase III compared with cells transfected with the empty expression vector (Figure 4C). By contrast, overexpression of HA-tagged wild type Cdk2 did not significantly change the levels of the phosphorylated and dephosphorylated DNA ligase IIIα (Figure 4C). Thus, we conclude that Cdk2 phosphorylates DNA ligase IIIα in vivo.
Dephosphorylation of DNA ligase IIIα is induced by oxidative stress
Having established the pattern of DNA ligase IIIα phosphorylation as a function of cell cycle stage, we next examined whether DNA damage influences the post-translational modification of this enzyme. In experiments with G1 cells, we were unable to detect a change in the mobility of the dephosphorylated form of DNA ligase IIIα following treatment with various DNA damaging agents (data not shown). However, treatment of an S phase-enriched population of HeLa cells with ionizing radiation (IR) but not the DNA alkylating agent EMS resulted in the dephosphorylation of DNA ligase IIIα (Figure 5A). Using the phosphopeptide antibody, we failed to detect EMS-induced dephosphorylation of DNA ligase III at concentrations ranging from 0.01 to 0.1% EMS whereas exposure to 1 Gray of IR induced DNA ligase III dephosphorylation (Figure 5B). DNA ligase IIIα molecules immunoprecipitated by XRCC1 antibody were dephosphorylated in response to IR demonstrating that, as shown previously (Figure 1), DNA ligase IIIα complexed with XRCC1 is being modified and that complex formation with XRCC1 is not influenced by the phosphorylation status of DNA ligase IIIα (Figure 5C).
Figure 5 .

Oxidative damage induces dephosphorylation of DNA ligase IIIα. (A) DNA ligase III was detected in extracts from undamaged (con) S phase-enriched HeLa cells and from cells that had been either exposed to 10 Gy of ionizing radiation (IR) or 0.5% EMS (EMS) by immunoblotting. (B) Asynchronous HeLa cells were treated with the indicated concentrations of EMS and doses of IR. DNA ligase III in extracts from the damaged and undamaged (con) cell extracts was detected by immunoblotting with the antibody (α-phos-Ser123) raised against a DNA ligase III peptide (residues 116–130) encompassing phosphorylated Ser123. To control for protein loading, β-actin in the extracts was detected by immunoblotting. (C) Populations of HeLa cells enriched for S phase as described in Materials and Methods section were either mock treated or treated with 12 Gy of ionizing radiation (IR). DNA ligase III was detected in the cell extracts either directly by immunoblotting (upper panel) or by co-immunoprecipitation with an XRCC1 antibody followed by immunoblotting (lower panel). (D) S phase-enriched HeLa cells were treated with either hydrogen peroxide (H2O2, upper panel), menadione (MN, middle panel) or glucose oxidase (GOX, lower panel) as described in Material and Methods section. DNA ligase III was detected in cell extracts by immunoblotting.
The ability of IR but not EMS to induce DNA ligase III dephosphorylation was surprising because xrcc1 mutant cells are much more sensitive to DNA alkylating agents than IR (30,31). Although IR directly causes DNA strand breaks, oxygen free radicals generated by the IR account for a significant fraction of DNA lesions. To determine whether oxidative stress is involved in the dephosphorylation of DNA ligase IIIα, cells were incubated with hydrogen peroxide, menadione and glucose oxidase. As shown in Figure 5D, each of these agents efficiently induced DNA ligase IIIα dephosphorylation indicating that active oxygen species are involved in this event.
ATM mediates oxidative stress-induced dephosphorylation of DNA ligase IIIα
Three protein kinases, DNA-PKcs, ATR and ATM, which resemble PI3 kinases, have been implicated in the cellular response to DNA damage (32). To determine whether one or more of these enzymes is involved in the oxidative stress-induced dephosphorylation of DNA ligase IIIα, we utilized wortmannin, an inhibitor of PI3 kinases (33). As shown in Figure 6A, wortmannin inhibited hydrogen peroxide-induced dephosphorylation of DNA ligase IIIα (Figure 6A), indicating the involvement of a PI3 kinase in this event. Next we used genetically defined pairs of cell lines to determine the role of ATM (25), ATR (34) and DNA-PK (35) in the oxidative stress-induced dephosphorylation of DNA ligase IIIα. Neither the overexpression of a dominant negative, kinase-dead version of ATR (34) nor the absence of DNA-PKcs (35) had any effect on the hydrogen peroxide-induced dephosphorylation of DNA ligase IIIα (data not shown). In contrast, hydrogen peroxide-induced dephosphorylation of DNA ligase IIIα was greatly attenuated in AT fibroblasts that harbor mutations in the ATM gene (Figure 6B). As expected, this defect was corrected in the AT fibroblast cell line complemented by expression of wild type ATM cDNA (Figure 6B), confirming the participation of the ATM kinase in the oxidative stress-induced dephosphorylation of DNA ligase IIIα.
Figure 6 .
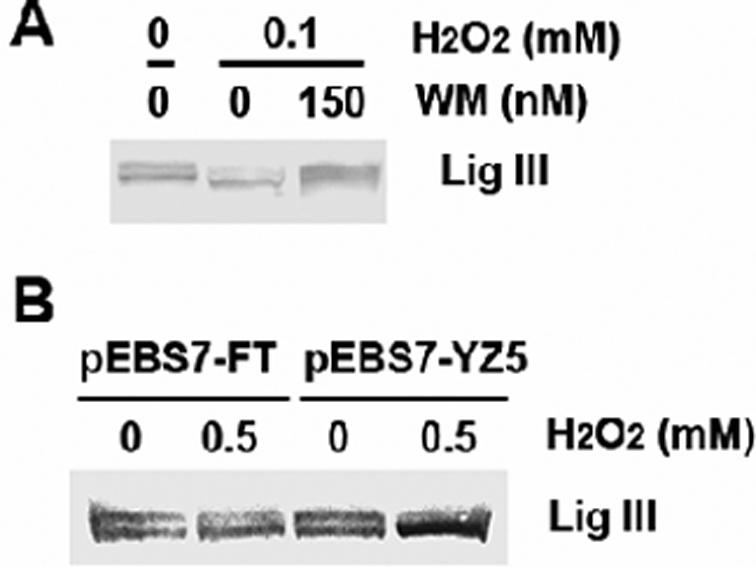
ATM mediates DNA ligase IIIα dephosphorylation induced by Oxidative damage. (A) S phase-enriched HeLa cells were treated with hydrogen peroxide (H2O2) either in the presence or absence of wortmannin as indicated. (B) ATM-deficient (pEBS7-FT) and ATM-complemented (pEBS7-YZ5) cell lines were either mock treated or treated with hydrogen peroxide (H2O2) as described in Materials and Methods section. DNA ligase III was detected in cell extracts by immunoblotting.
The steady state level of protein phosphorylation is determined by the opposing actions of kinases and phosphatases. Treatment of an S phase-enriched population of HeLa cells with hydrogen peroxide resulted in a marked reduction of Cdk2 activity (Figure 7, upper panel). By contrast, the DNA alkylating agent EMS did not significantly affect Cdk2 activity (Figure 7, upper panel). This prompted us to examine whether the inhibition of Ckd2 activity by oxidative stress was dependent upon ATM., Hydrogen peroxide treatment did not result in inhibition Cdk2 activity in ATM-deficient cells (Figure 7, middle panel) but this effect was restored by expression of wild type ATM in the mutant cells (Figure 7, lower panel). Together these results show that Cdk2 kinase activity is inhibited by an ATM-dependent pathway that is activated by hydrogen peroxide.
Figure 7 .

Oxidative stress inhibits Cdk2 activity in an ATM-dependent manner. Cdk2 was immunoprecipitated from extracts of S phase-enriched HeLa cells, and ATM-deficient (pEBS7-FT) and ATM-complemented (pEBS7-YZ5) cells that had been treated with either hydrogen peroxide (H2O2) or EMS (EMS) as described in Materials and Methods. Immunoprecipitated Cdk2 was incubated with purified histone H1 (Sigma) and [γ-32P]ATP. After separation by SDS–PAGE, total histione H1 was visualized by Coomassie blue staining (H1, upper panel) and labeled histone H1 by phosphorimager analysis (32P, middle panel). Cdk2 protein in the immunoprecipitates was detected by imuunoblotting (lower panel, Cdk2).
DISCUSSION
Mammalian DNA ligase IIIα participates in nuclear base excision and single-strand break repair pathways that play a critical role in the repair of DNA damage in non-dividing cells (20,36). In this study, we show that DNA ligase IIIα is phosphorylated by the cell cycle kinase, Cdk2 both in vitro and in vivo. The major site in DNA ligase IIIα phosphorylated by Cdk2 is Ser123. We have purified recombinant DNA ligase IIIβ from baculovirus-infected insect cells that often reiterate the post translational modifications of mammalian proteins. This protein had similar biochemical properties to unmodified DNA ligase IIIβ purified from E.coli (28,29), suggesting that phosphorylation does not significantly impact catalytic activity. In support of this idea, Ser123 is not located within the conserved C-terminal catalytic domain of DNA ligase III. Previously we have shown that an N-terminal fragment of DNA ligase IIIα encompassing Ser123 interacts with both poly(ADP-ribose) polymerase 1 and poly (ADP-ribose) (30), suggesting that Ser123 phosphorylation may regulate the binding of DNA ligase IIIα with poly(ADP-ribose) polymerase 1 and possibly other protein partners. Although this modification does not appear to affect complex formation with XRCC1, it is possible that it reflects the switching off of housekeeping repair pathways involving DNA ligase IIIα/XCCC1, such as short patch BER, that operate in non-dividing cells when replication-associated repair pathways, such as long patch BER, are activated. This would not impact the participation of free XRCC1 in S phase repair pathways that may be mediated via its interaction with PCNA (20,37).
If phosphorylation of DNA ligase IIIα in an unperturbed S phase turns off its repair function, a likely response to DNA damage would be the reactivation of this enzyme. Consistent with this idea, DNA damage induces the dephosphorylation of phosphorylated DNA ligase IIIα. Since DNA ligase III-deficient CHO cells exhibit hypersensitivity to simple DNA alkylating agents (30), it was surprising that oxidative stress rather than DNA alkyalting agents was more effective at inducing DNA ligase IIIα dephosphorylation. However, the notion that DNA ligase IIIα plays a critical role in the repair of oxidative DNA damage is supported by protein–protein interactions between the subunits of the DNA ligase IIIα /XRCC1 complex and the DNA glycosylases, OGG1 and NEIL1 that repair oxidized base lesions via different BER subpathways (12,14).
Previous studies have shown that there is increased oxidative stress in atm cells suggesting that the lack of the ATM kinase disrupts cellular redox homeostasis (38–40). Here we show that oxidative stress-induced dephosphorylation of DNA ligase IIIα is dependent upon the ATM kinase. These results suggest that ATM is activated by oxidative stress and are consistent with emerging evidence that ATM may sense other cellular changes in addition to DNA breaks (41). The ATM-dependent dephosphorylation of DNA ligase IIIα is likely to involve two distinct mechanisms. As has been observed previously in Xenopus egg extracts (42), activation of ATM results in the inhibition of Cdk2. In addition, it has been shown recently that protein phosphatase PP1is activated by IR in an ATM-dependent manner (43). Thus we suggest that oxidative stress-induced dephosphorylation of DNA ligase IIIα is a consequence of Cdk2 inhibition and the activation of a cellular phosphatase such as PP1.
It has been hypothesized that DNA damage caused by oxygen free radicals may contribute to cancer formation, neurodegeneration and aging. Here we have shown that DNA ligase IIIα, a key enzyme in the BER and single strand break repair, is modified in response to oxidative stress in an ATM-dependent manner. Since cerebellar ataxia is a characteristic symptom of ataxia telangiectasia (44) and, recently, defects in single-strand break repair have been linked with hereditary forms of spinocerebellar ataxia (45), our results contribute to the emerging evidence that the repair of oxidative DNA damages, such as base lesions and single strand breaks, protects against neurodegeneration.
Acknowledgments
We thank Drs Phang-Lang Chen and Ed Harlow for reagents and Dr Fey Rassool for critical reading of this manuscript. This work was supported by the National Institutes of Health Research Grants ES 012512 (AET) and CA92584 (AET). Funding to pay the Open Access publication charges for this article was provided by the NIEHS (ES012512).
Conflict of interest statement. None declared.
REFERENCES
- 1.Tomkinson A.E., Mackey Z.B. Structure and function of mammalian DNA ligases. Mutat. Res. 1998;407:1–9. doi: 10.1016/s0921-8777(97)00050-5. [DOI] [PubMed] [Google Scholar]
- 2.Lakshmipathy U., Campbell C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 1999;19:3869–3876. doi: 10.1128/mcb.19.5.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackey Z.B., Ramos W., Levin D.S., Walter C.A., McCarrey J.R., Tomkinson A.E. An alternative splicing event which occurs in mouse pachytene spermatocytes generates a form of DNA ligase III with distinct biochemical properties that may function in meiotic recombination. Mol. Cell. Biol. 1997;17:989–998. doi: 10.1128/mcb.17.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caldecott K.W., McKeown C.K., Tucker J.D., Ljunquist S., Thompson L.H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nash R.A., Caldecott K., Barnes D.E., Lindahl T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry. 1997;36:5207–5211. doi: 10.1021/bi962281m. [DOI] [PubMed] [Google Scholar]
- 6.Taylor R.M., Wickstead B., Cronin S., Caldecott K.W. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr. Biol. 1998;8:877–880. doi: 10.1016/s0960-9822(07)00350-8. [DOI] [PubMed] [Google Scholar]
- 7.Caldecott K.W., McKeown C.K., Tucker J.D., Stanker L., Thompson L.H. Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res. 1995;23:4836–4843. doi: 10.1093/nar/23.23.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldecott K.W., Aoufouchi S., Johnson P., Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubota Y., Nah R.A., Klungland A., Schar P., Barnes D.E., Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 10.Masson M., Niedergang C., Schreiber V., Muller S., Menissier de Murcia J., de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marsin S., Vidal A.E., Sossou M., Menissier-de Murcia J., Le Page F., Boiteux S., de Murcia G., Radicella J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003;278:44068–44074. doi: 10.1074/jbc.M306160200. [DOI] [PubMed] [Google Scholar]
- 12.Vidal A.E., Boiteux S., Hickson I.D., Radicella J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein–protein interactions. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitehouse C.J., Taylor R.M., Thistlethwaite A., Zhang H., Karimi-Busheri F., Lasko D.D., Weinfeld M., Caldecott K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 14.Wiederhold L., Leppard J.B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A.E., Izumi T., Prasad R., Wilson S.H., et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Clements P.M., Breslin C., Deeks E.D., Byrd P.J., Ju L., Bieganowski P., Brenner C., Moreira M.C., Taylor A.M., Caldecott K.W. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair. 2004;3:1493–1503. doi: 10.1016/j.dnarep.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 16.Date H., Igarashi S., Sano Y., Takahashi T., Takahashi T., Takano H., Tsuji S., Nishizawa M., Onodera O. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem. Biophys. Res. Commun. 2004;325:1279–1285. doi: 10.1016/j.bbrc.2004.10.162. [DOI] [PubMed] [Google Scholar]
- 17.Gueven N., Becherel O.J., Kijas A.W., Chen P., Howe O., Rudolph J.H., Gatti R., Date H., Onodera O., Taucher-Sholz G., et al. Aprataxin, a novel protein that protects against genotoxic stress. Hum. Mol. Genet. 2004;13:1081–1093. doi: 10.1093/hmg/ddh122. [DOI] [PubMed] [Google Scholar]
- 18.Moreira M.C., Barbot C., Tachi N., Kozuka N., Uchida E., Gibson T., Mendonca P., Costa M., Barros J., Yanagisawa Y., et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nature Genet. 2001;29:189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 19.Luo H., Chan D.W., Yang T., Rodriguez M., Chen B.P., Leng M., Mu J.J., Chen D., Songyang Z., Wang Y., et al. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol. Cell. Biol. 2004;24:8356–8365. doi: 10.1128/MCB.24.19.8356-8365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor R.M., Moore D.J., Whitehouse J., Johnson P., Caldecott K.W. A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Mol. Cell. Biol. 2000;20:735–740. doi: 10.1128/mcb.20.2.735-740.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakshmipathy U., Campbell C. Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic Acids Res. 2000;28:3880–3886. doi: 10.1093/nar/28.20.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loizou J.I., El-Khamisy S.F., Zlatanou A., Moore D.J., Chan D.W., Qin J., Samo S., Meggio F., Pinna L.A., Caldecott K.W. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y., Farmer A.A., Chen C.-F., Jones D.C., Chen P.-L., Lee W.-H. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer Res. 1996;56:3168–3172. [PubMed] [Google Scholar]
- 24.Ferrari G., Rossi R., Arosio D., Vindigni A., Biamonti G., Montecucco A. Cell cycle-dependent phosphorylation of human DNA ligase I at the cyclin-dependent kinase sites. J. Biol. Chem. 2003;278:37761–37777. doi: 10.1074/jbc.M304462200. [DOI] [PubMed] [Google Scholar]
- 25.Ziv Y., Bar-Shira A., Pecker I., Russell P., Jorgensen T.J., Tsarfati I., Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–167. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 26.Guo C.Y., Wang Y., Brautigan D.L., Larner J.M. Histone H1 dephosphorylation is mediated through a radiation-induced signal transduction pathway dependent on ATM. J. Biol. Chem. 1999;274:18715–18720. doi: 10.1074/jbc.274.26.18715. [DOI] [PubMed] [Google Scholar]
- 27.van den Heuvel S., Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 28.Leppard J., Dong Z., Mackey Z.B., Tomkinson A.E. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol. Cell. Biol. 2003:5919–5927. doi: 10.1128/MCB.23.16.5919-5927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackey Z.B., Niedergang C., Murcia J.M., Leppard J., Au K., Chen J., de Murcia G., Tomkinson A.E. DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly(ADP-ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J. Biol. Chem. 1999;274:21679–21687. doi: 10.1074/jbc.274.31.21679. [DOI] [PubMed] [Google Scholar]
- 30.Thompson L.H., Brookman K.W., Dillehay L.E., Carrano A.V., Mazrimas J.A., Mooney C.L., Minkler J.L. A CHO-cell strain having hypersensitivity to mutagens, a defect in DNA strand-break repair, and an extraordinary baseline frequency of sister-chromatid exchange. Mutat. Res. 1982;95:427–440. doi: 10.1016/0027-5107(82)90276-7. [DOI] [PubMed] [Google Scholar]
- 31.Thompson L.H., Brookman K.W., Jones N.J., Allen S.A., Carrano A.V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell. Biol. 1990;10:6160–6171. doi: 10.1128/mcb.10.12.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J., Yu Y., Hamrick H.E., Duerksen-Hughes P.J. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis. 2003;24:1571–1580. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- 33.Sarkaria J.N., Tibbets R.S., Busby E.C., Kennedy A.P., Hill D.E., Abraham R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- 34.Cliby W.A., Roberts C.J., Cimprich K.A., Stringer C.M., Lamb J.R., Schreiber S.L., Friend S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lees-Miller S.P., Godbout R., Chan D.W., Weinfeld M., Day R.S., Barron G.M., Allalunisturner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–1185. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 36.Frosina G., Fortini P., Rossi O., Carrozzino F., Raspaglio G., Cox L.S., Dane D.P., Abbondandolo A., Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 37.Fan J., Otterlei M., Wong H.-K., Tomkinson A.E., Wilson D.M., III XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004;32:2193–2201. doi: 10.1093/nar/gkh556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reichenbach J., Schubert R., Schindler D., Muller K., Bohles H., Zielen S. Elevated oxidative stress in patients with ataxia telangiectasia. Antioxid. Redox Signal. 2002;4:465–469. doi: 10.1089/15230860260196254. [DOI] [PubMed] [Google Scholar]
- 39.Rotman G., Shiloh Y. Ataxia-telangiectasia: is ATM a sensor of oxidative damage and stress? Bioessays. 1997;19:911–917. doi: 10.1002/bies.950191011. [DOI] [PubMed] [Google Scholar]
- 40.Watters D.J. Oxidative stress in ataxia telangiectasia. Redox Rep. 2003;8:23–29. doi: 10.1179/135100003125001206. [DOI] [PubMed] [Google Scholar]
- 41.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 42.Costanzo V., Robertson K., Ying C.Y., Kim E., Avvedimento E., Gottesman M., Grieco D., Gautier J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol. Cell. 2000;6:649–659. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 43.Guo C.Y., Brautigan D.L., Larner J.M. Ionizing radiation activates nuclear protein phosphatase-1 by ATM-dependent dephosphorylation. J. Biol. Chem. 2002;277:41756–41761. doi: 10.1074/jbc.M207519200. [DOI] [PubMed] [Google Scholar]
- 44.Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Ann. Rev. Genet. 1997;31:635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- 45.Caldecott K.W. DNA single-strand break repair and spinocerebellar ataxia. Cell. 2003;112:7–10. doi: 10.1016/s0092-8674(02)01247-3. [DOI] [PubMed] [Google Scholar]