

Abstract
Fungal pathogens usually have multiple genes that encode extracellular hydrolytic enzymes that may degrade the physical barriers in their hosts during the invasion process. Nectria hematococca, a plant pathogen, has two inducible pectate lyase (PL) genes (pel) encoding PL that can help degrade the carbohydrate barrier in the host. pelA is induced by pectin, whereas pelD is induced only in planta. We show that the disruption of either the pelA or pelD genes alone causes no detectable decrease in virulence. Disruption of both pelA and pelD drastically reduces virulence. Complementation of the double disruptant with pelD gene, or supplementation of the infection droplets of the double disruptant with either purified enzyme, PLA, or PLD, caused a recovery in virulence. These results show that PL is a virulence factor. Thus, we demonstrate that disruption of all functionally redundant genes is required to demonstrate the role of host barrier-degrading enzymes in pathogenesis and that dismissal of the role of such enzymes based on the effects of single-gene disruption may be premature.
The role of fungal extracellular enzymes that can degrade physical barriers during the invasion of the host has been controversial. A variety of lines of evidence for their role in pathogenesis, including the correlation between the level of such enzymes and virulence, immunocytochemical evidence for the secretion of the enzyme during infection, protection of the host by inhibition of the enzymes with selective inhibitors or specific antibodies, and enhancement of virulence by gene transfer, have been challenged on the basis of the results of disruption experiments on genes that encode cutinase and cell wall-degrading enzymes (1–14). In the case of animal pathogens, single and double proteinase gene disruption failed to show a clear decrease in virulence (15–17). The inability to demonstrate a drastic decrease in virulence by such gene disruption has been interpreted as casting doubt on the role of extracellular enzymes that degrade physical barriers of the host in pathogenesis (18). However, highly pathogenic fungi usually have multiple genes encoding enzymes that degrade the polymer barriers of the host (5–7, 9, 14, 19), and some may be expressed only in the host (20). Therefore, the results from gene disruption experiments may not be able to reveal the true role of the extracellular enzymes in pathogenesis unless the genes that can encode all of the functionally redundant enzymes are disabled. Here we report the first example of loss of virulence by disrupting genes encoding both of the two functionally redundant enzymes, either of which can function alone as virulence factor.
A plant pathogen, Nectria hematococca, which infects pea plants, produces several pectin-degrading enzymes, presumably to help penetrate cell-wall carbohydrate barriers in the host during infection. Such enzymes include endopectate lyases that are encoded by at least four pectate lyase (PL) genes (pel) that belong to a new class of lyases, based on the lack of the consensus signature sequences of other PL and the inability to fit the structure-based alignments of other PL (20). Two are inducible: pelA by pectin (21) and pelD only by the host (20), whereas pelB and pelC are only constitutively expressed and only at very low levels. Thus, the inducible pelA and pelD genes were selected for study as candidates as virulence factors. Here we report that disruption of either pelA or pelD alone does not reduce virulence, but disruption of both pelA and pelD reduces virulence drastically. Complementation of the double gene disruptant with a pelD gene or supplementation of the infection droplets containing the double disruptant with either of the two purified enzymes, PLA or PLD, causes a recovery of virulence, demonstrating that PL is a virulence factor.
Materials and Methods
Fungal Strain and Growth Conditions.
N. hematococca (Fusarium solani f. sp. pisi) was a field isolate, T8, obtained from H. D. VanEtten (University of Arizona, Tucson, AZ) (22).
Vectors, Enzymes, and Chemicals.
The pSV40/Zeo and zeocin were from Invitrogen. Plasmids were propagated in Escherichia coli DH5α. Restriction and modifying enzymes, Taq polymerase, DH5α cells, 123-bp DNA ladder, and TRIzol reagent were from Life Technologies (Gaithersburg, MD). Expand High Fidelity Taq polymerase was from Boehringer Mannheim, Novozyme 234 from InterSpex Products (Foster City, CA), Driselase and α-glucuronidase were from Sigma and hygromycin, from Calbiochem. Primers were from Integrated DNA Technologies (Coralville, IA). Rediprime random-primed labeling kit and enhanced chemiluminescence Western blotting detection system were from Amersham International.
Construction of Gene Replacement Vector pPELA∷hph.
pG3S2 (21) carrying a 4.7-kb SstI fragment of the pelA gene was digested with SacII to generate a 895-bp fragment that included 312 bp of the 3′ end of the pelA open reading frame (ORF) and a 6.69-kb fragment containing the pBluescript vector flanked by 3.1 kb of the 5′ end of the pelA gene (including 417 bp of the ORF) and 625 bp of the 3′ noncoding region. The 6.69-kb fragment was filled in with Klenow and dNTPs. The 2.5-kb hph construct carrying hyg gene with a Cochliobolus heterostrophus promoter (23) was restricted from a pBluescript vector with BamHI, filled in with Klenow, and ligated to the blunt-ended pelA fragment to yield the pelA-disrupted construct pS2H2 with hph in the opposite orientation to pelA. To extend the 3′ flanking region of the pelA gene in the construct pS2H2, a 2.75-kb SstI and XbaI fragment flanking the 3′ noncoding region of pelA was ligated to the BamHI/SstI-digested pS2H2, yielding the 8.5-kb pelA disruption construct, pPELA∷hph (Fig. 1A).
Figure 1.
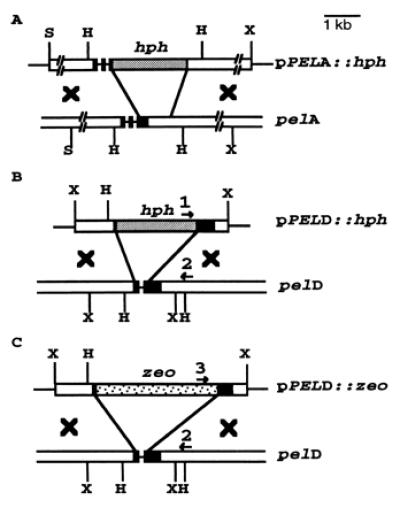
The replacement vectors for the pel genes in N. hematococca. Open boxes, the 5′ and 3′ flanking regions of the pel genes; black boxes, the pel gene exons; restriction sites, H, HindIII; X, XbaI; and S, SstI. Primers 1 and 2 were used for detecting homologous recombination of hph-disrupted pelD and primers 2 and 3 for zeo-disrupted pelD.
Construction of Gene Replacement Vector pPELD∷hph.
The EcoRI site in the pelD gene clone (20), pX55, was eliminated, and a fragment of pX55 with a deletion of 250 bp from the coding region of pelD was amplified by an inverse PCR with a sense primer, 5′-GCG CGC GAA TTC GTC TTC ATC CTC GAG GAG-3′, and an antisense primer, 5′-GGC CGG ACT AGT CGA GGA CAG TGA CGA TGC-3′, with Expand High Fidelity Taq DNA polymerase. The 5-kb PCR product was treated with T4 DNA polymerase and dNTP to remove any overhang A-nucleotide, and a SalI fragment carrying the hyg gene with Aspergillus nidulans trpC promoter from pCSN43 (24) was ligated to generate pPELD∷hph (Fig. 1B).
Construction of Gene Replacement Vector pPELD∷zeo to Produce pelA, pelD Double Disruptant.
A sense primer 5′-GGA GGG CCA CCA TGG CCA-3′ and an antisense primer 5′-TGC CAT GGG GTC GGC GTC GGT CAG TC-3′ were used to amplify the zeocin resistance gene with Pfu polymerase and pSV40/Zeo vector as a template. The product was cut with NcoI and cloned into the NcoI site of pNOM102 (25) to generate pNOM102∷zeo, placing the expression of the zeo gene under the control of the Aspergillus nidulans gpd promoter. pNOM102∷zeo was cut with HindIII, partially filled in with Klenow in the presence of dATP and dGTP, and then cut with EcoRI to release the zeocin cassette. The 5-kb pelD PCR fragment obtained above by inverse PCR was cut with SpeI and partially filled in by Klenow in the presence of dCTP and dTTP, then cut with EcoRI and the zeocin cassette was ligated to generate pPELD∷zeo (Fig. 1C).
Complementation of pelA, pelD Double Disruptant with pelD Gene.
The SalI fragment (2.0 kb) released from pX55 (20), which contains 1.0 kb of 5′-flanking region, coding region, and 300 bp of 3′-flanking region of pelD, was ligated to XhoI-digested pCSN43, and the resulting vector was used to transform pelA, pelD double disruptant. Because the double disruptant used does not show a high degree of hygromycin resistance, complemented transformants could be selected on hygromycin (200 μg/ml)-containing PDA. Transformants were tested for intact pelD by PCR with pelD gene primer: 5′-GCT CCG CAG CAA TCT CCA G-3′ and a primer from hph gene: 5′-CTA GCT CCA GCC AAG CCC-3′. pelD gene expression was tested by reverse transcription–PCR (RT-PCR) with pelD-coding region primers.
Transformation of Protoplasts.
Protoplast preparation and transformation were performed as before (22). For the double disruptant, the pelA disruptant was transformed with the pelD disruption construct pPELD∷zeo. DNA and PEG4000-treated protoplasts (105) in 200 μl of STC (1.2 M sorbitol/10 mM Tris, pH 7.5/10 mM CaCl2) were mixed with 10 ml of regeneration medium (mineral medium containing 1.2 M sorbitol/2% glucose/1% agar) at 45°C and overlaid on a previously poured layer (12 ml) of regeneration medium containing 300 μg/ml zeocin. Transformants were regrown on selection medium, and conidia were produced on nonselective medium.
PCR Screening Directly on Spores as the Template Source.
Spores (104-105) were mixed with PCR buffer/1.5 mM MgCl2/200 μM of each dNTP/4% DMSO/25 ng of each primer in a total volume of 48 μl and heated at 94°C for 15 min; 2.5 units of Taq DNA polymerase was then added to each reaction mixture (36 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 45 s, and polymerization at 72°C for 45 s, final extension reaction at 72°C for 10 min). The primers used for the identification of pelA disruptants were sense, 5′-GTT CAC TGC TGC TTT CGT-3′ from the 5′ end of the ORF of pelA, and antisense, 5′-GGA GCC AGT AAC AGT GCA G-3′, from the 3′ end of the pelA ORF that was deleted in making the construct. For pelD disruption with hph, a sense primer from hph gene, 5′-GAG CTC TCC AAC ATT GAT G-3′, and an antisense primer from the pelD gene, 5′-GAG CTC TCC AAC ATT GAT G-3′ (outside the region used for gene replacement construct), were used to amplify an expected junction PCR product of 995 bp; for pelD disruption with zeo, a sense primer from zeo gene, 5′-CTT CAA GTC AGC CAA CTG C-3′, and an antisense primer from pelD gene, 5′-GAG CTC TCC AAC ATT GAT G-3′ (outside the segment used for gene replacement construct), were used to amplify an expected junction PCR product of 980 bp.
Genomic Southern Analysis.
Genomic DNA of wild-type and single or double disruptants of pelA and pelD was prepared (26), digested with HindIII, and subjected to Southern blot analysis (27).
Gene-Specific RT-PCR with RNA from Pea Stems Infected with N. hematococca.
The preparation of pea stem and infection with conidia of N. hematococca were performed as described (28). Total RNA was isolated from the infected plant tissue as described (20). RNA (1 μg) was treated with amplification-grade RNase-free DNase and reverse transcribed into cDNA with SuperScript II H− reverse transcriptase with oligo (dT) in a 40-μl reaction mixture. A 5-μl aliquot of the first-strand cDNA was used for PCR in a 50-μl reaction mixture. The sense and antisense primers used were 5′-CAA GAC TCT CCC CAA GAG-3′ and 5′-GGA GCC AGT AAC AGT GCA-3′ for pelA (expected PCR product, 654 bp); 5′-TAC CGT TCT TCC CGC TTC-3′ and 5′-ACC GGA AGA GCG GTC GAT-3′ for pelB (expected PCR product, 208 bp); 5′-GCA GCA AGT CTC TGA GCG-3′ and 5′-GCA GTG CCG CTG CCA AGG-3′ for pelD (expected PCR product, 332 bp). Aliquots of the PCR products were analyzed on 2% agarose gels. In all cases, the identities of the PCR products were verified by DNA blot hybridization.
Immunoblot Analysis.
Multiple droplets of conidial suspension of the wild-type and pel gene-disrupted N. hematococca (5 × 104 per 5 μl) were placed on sections of etiolated pea stem as described (28). After 4 days, the infected pea stems were ground in liquid N2, resuspended in an equal volume of H2O, and centrifuged at 12,000 × g for 10 min. The supernatants were dialyzed against H2O and concentrated 10-fold with a Centricon 10 concentrator. Aliquots were subjected to SDS/PAGE with 12% resolving gel for Western blot by using antibody raised against pectate lyase C; the four PL are immunologically crossreactive (20, 29).
PL Induction and Assay.
Spores (5 × 107) in water were incubated with sterile pea stems for 4 days at 24°C. Mycelia and infected stems were frozen and ground in liquid N2, and the extract supernatant prepared as above was dialyzed, concentrated, and assayed for PL activity (30).
Virulence Assays.
Bioassay on etiolated pea stem segments was performed as described (28). Where indicated, the conidial suspension also contained purified enzyme, either PLA or PLD. Degree of penetration was assessed by microscopic examination of stained cross section of the tissue from the middle of the lesion. A hand-sectioned slice fixed in 50% ethanol containing 5% glacial acetic acid and 13% formalin for 20 min was stained with cotton blue-lactophenol for 10 min followed by removal of excess stain by washing with lactophenol. The number of cell layers penetrated in at least 30 sections was scored under the microscope, and the average is shown for each assay.
The second bioassay involved a modification of the whole-plant assay (31). In the modified-growth assay, sterile soil was used in place of vermiculite, and the pea seeds were soaked in aerated water for 15 h, germinated for 24 h at 24°C, and then planted in the spore-impregnated soil.
Results
Disruption of Only pelA.
N. hematococca contains a pectin-inducible pelA gene (25) and a host-inducible pelD gene (20). pelA was disrupted by using a construct containing about 3 kb each of 5′- and 3′-flanking regions of pelA gene with a 895-bp internal segment replaced with a hygromycin resistance gene (hyg) (27). The transformants were screened by PCR with the spores directly as the source of the template. One of the primers used was from the 3′ end of the ORF of pelA that was replaced with hph gene in the disruption construct and therefore the disruptant did not give the 848-bp PCR product expected from the native pelA gene (data not shown). PCR with hph gene-specific primers gave a product at 880 bp, as expected (data not shown). Gene disruption was confirmed by Southern hybridization that showed the 2.2-kb band from HindIII digestion of the wild type was shifted to ≈4.0 kb in the disruptant as expected (Fig. 2A Left). RT-PCR of RNA isolated from infected pea stems showed the absence of pelA transcripts and presence of pelD transcripts in the fungus growing on the host (Fig. 2B). No PL activity or immunologically cross-reacting protein could be detected when the pelA-disrupted mutant was cultured in the presence of pectin. Furthermore, on growth on the pea epicotyl tissue that induces both pelA and pelD in the wild type, the expected 23-kDa PLD and 26-kDa PLA were detected by immunoblot whereas the pelA disruptant showed only 23 kDa (PLD) (Fig. 3A). PL B and C were not seen in these immunoblots, probably because of very low levels of expression. To test whether pelA disruption affected virulence, lesion formation caused by the conidia of the pelA disruptant on pea epicotyl segments was assessed (28). The pelA disruptant and the wild type showed identical time course of formation of equally severe lesions (Fig. 3B). Inclusion of antibodies prepared against PLA in the inoculation droplet protected the host from infection by both the wild type and pelA disruptant (Fig. 3B). This result suggested that, even in the absence of PLA, another immunologically similar PL probably was produced in sufficient amounts to achieve penetration through the pectinaceous barrier of the host. In fact, an extract of pea stem infected by a pelA disruptant showed 60% of the lyase activity observed with the wild type.
Figure 2.
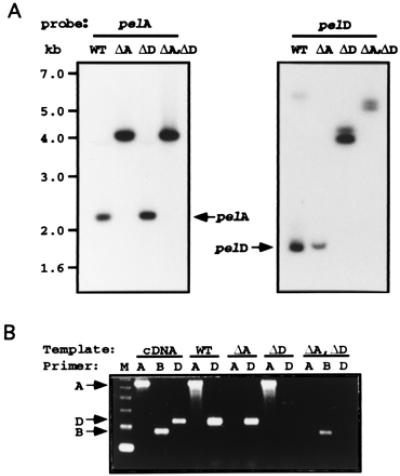
(A) Genomic Southern blots showing the absence of native genes and appearance of new fragments from the disrupted genes in the mutants. Genomic DNA (10 μg each) was digested with HindIII and probed with cDNAs of pelA (Left) and pelD (Right). Size markers shown on the left apply for both panels. (B) RT-PCR showing the absence of transcripts from the disrupted genes and the presence of transcripts from the native genes. Gene-specific RT-PCR amplification of pelA, pelB, or pelD was done by using gene-specific primers (20) and RNA isolated from N. hematococca-infected pea epicotyl. WT, wild type; ΔA, pelA disruptant; ΔD, pelD disruptant; and ΔA, ΔD: pelA and pelD double-gene disruptant. Position of the expected products from the three genes are shown by the arrows. Molecular size marker (M), 123-bp DNA ladder.
Figure 3.
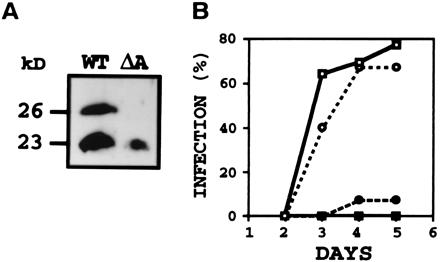
(A) Immunoblot analysis of proteins from N. hematococca wild type (WT) or pelA disruptant (ΔA) grown on pea epicotyl sections showing products of pelA (26 kDa) and pelD (23 kDa) in WT and only pelD product in the pelA disruptant. After 5 days of growth at 24°C (28), the soluble proteins were subjected to immunoblot by using anti-PLC (29) and enhanced chemiluminescence detection system. (B) Time course of infection of pea epicotyl sections by WT or pelA disruptant in the presence of antibody to PLA or preimmune IgG. Assay was done with 104 conidia and 10 μg of IgG (28, 31). Infection in the presence of water only was identical to that in the presence of preimmune IgG. (○) WT, preimmune; (□) ΔA, preimmune; (●) WT, anti-PLA; and (■) ΔA, anti-PLA. % infection, % of epicotyl sections that showed lesion.
Disruption of Only pelD.
The pelD gene was disrupted by protoplast transformation with a construct made from a 2.7-kb pelD gene fragment with the replacement of a 250-bp internal coding segment of pelD with the hyg gene (24). The hygromycin-resistant transformants were screened by PCR with spores directly as the source of the template. PCR, by using a sense primer from the hph gene and an antisense primer from the 3′-side of the pelD gene from a region outside that which was used for making the gene replacement construct, yielded a 995-bp product expected from homologous recombination (data not shown). When the HindIII digestion product of the genomic DNA from the putative disruptant was subjected to Southern hybridization, the pelD-containing fragment was found at 4 kb, whereas the native pelD gene was found at 1.7 kb, showing pelD disruption (Fig. 2A Right). RT-PCR of RNA isolated from infected pea stems did not show pelD transcripts with the pelD-disrupted mutant, whereas the pelA transcript was readily detected; with wild type, both pelA and pelD transcripts were found (Fig. 2B). These results showed that the disruptant did not express pelD in the host.
Double Disruption of Both pelA and pelD.
To disrupt both pelA and pelD, pelA disruptant was transformed with a pelD disruption construct containing a zeocin resistance gene under the control of gpd promoter from A. nidulans (25) that replaced a 250-bp internal coding segment of a 2.7-kb pelD gene segment. The transformants resistant to zeocin were screened for double disruptants by PCR on the conidia of the transformants. With primers representing the zeo gene and a segment of pelD gene outside that used for making the gene replacement construct, the 980-bp product, expected from homologous recombination, was found in the disruptant (data not shown). Southern hybridization of the genomic DNA of the pelA, pelD double disruptant showed the expected absence of hybridization bands representing the genomic fragments containing the native pelA and pelD (Fig. 2A). To test for the expression of the pel genes, spores of the disruptants were used to inoculate pea epicotyl tissue and the extracted RNA was subjected to RT-PCR with gene-specific primers (20). The double disruptant showed the lack of a 654-bp product expected from native pelA gene and lack of a 332-bp product expected from native pelD gene (Fig. 2B), but showed the expected low level of product (at 208 bp) from a constitutively expressed pelB (32) (Fig. 2B). Immunoblots of proteins from host tissue incubated with the double disruptant showed the absence of both pelA and pelD products (data not shown).
Tests for Virulence of the pel Gene Disruptants.
Virulence of the wild-type N. hematococca and its gene-disrupted mutants was tested in two different ways. When conidial spore suspension was placed on pea epicotyl segments (24), the wild-type fungus and the pelA or pelD single-gene disrupted mutants produced lesions on most epicotyl sections (Fig. 4 a, b, and c), suggesting that absence of one of the two inducible lyases, PLA or PLD, had no impact on the virulence. The pelA, pelD double gene disruptant showed very few mild lesions that did not extend deep into the tissue (Fig. 4d). Examination of the cross section of the inoculated areas after staining the fungus with lactophenol cotton blue revealed that the wild-type and pelA or pelD single gene-disrupted mutants penetrated extensively and deeply into the host, whereas the pelA, pelD double gene-disrupted mutant did not penetrate deep into the tissue (Fig. 4h). As infection advanced, a great amount of tissue degradation and maceration could be seen extending deep into the tissue, reaching the central vascular bundles with the wild-type and single gene disruptants. At the same time, the double-gene disruptant seldom reached more than one or two cells in depth into the tissue with little tissue damage.
Figure 4.
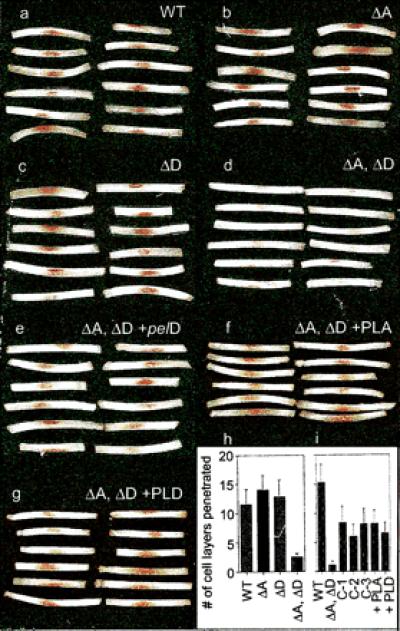
Infection of pea epicotyl sections by (a) wild type; (b) pelA disruptant; (c) pelD disruptant; (d) pelA, pelD disruptant; (e) pelD-complemented pelA, pelD disruptant of N. hematococca; (f) infection droplets of pelA, pelD double-disruptant supplemented with PLA; or (g) double disruptant supplemented with PLD – lesion formation is seen in all cases except with pelA, pelD double-gene disruptant. (h) Mycelial penetration of pea epicotyl sections inoculated with the wild type or the indicated single and double disruptants. (i) Penetration by the wild-type, double-disruptant, three pelD-complemented double disruptants (C-1, C-2, and C-3), and double-disruptant spore suspension supplemented with purified PLA (100 ng) or PLD (40 ng). Assay with 106 conidia (28).
In a second test for virulence, conidia were mixed with the soil before planting pea seeds, and the seedlings were examined for infection. With the wild type or pelA or pelD single-gene disruptants, severe lesions were visible, with stunted growth for those seedlings that emerged from the soil. The seedlings from the soil inoculated with pelA, pelD double disruptant showed the same degree of growth as the seedlings in the noninoculated soil and had only a few very mild lesions (Fig. 5). Under conditions of more severe infection, no seedlings emerged from the soil when the wild-type or single-gene disrupted mutants were used, but pelA, pelD double disruptant allowed the growth of seedlings with some lesions (data not shown). Clearly, pelA, pelD double disruptant showed drastically decreased virulence with both tests for virulence.
Figure 5.
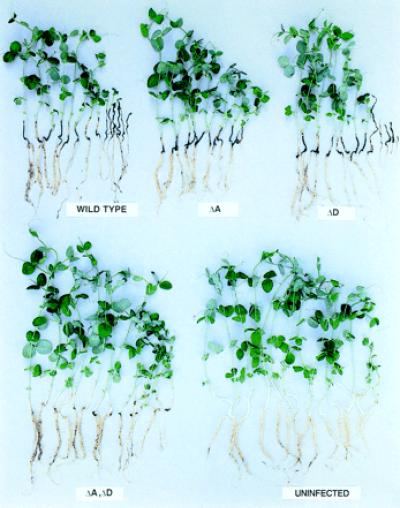
Virulence of wild-type and pel gene disruptants as shown by the growth of pea seedlings raised from seed in soil mixed with the conidia; 3.7 × 105 conidia/g soil, 12 days of growth as in ref 30. Designations of disruptants as in Fig. 2.
Complementation of the Double-Gene Disruptant with pelD.
If the loss of virulence resulting from the double-gene disruption is in fact caused by the inability to produce PL needed for penetration, introduction of pelD gene into the double-gene disruptant should enable it to infect its host. In fact, expression of lyase in such complemented transformant clearly showed recovery of pathogenicity (Fig. 4e) and host penetration (Fig. 4i). The PL activity found in the extracts of pea stem infected with the wild-type and gene-disrupted and -complemented mutants strongly supports the conclusion that the observed loss and recovery of virulence resulted from the loss and recovery of the ability to produce lyase, respectively. For example, extracts of pea stem infected with either pelA or pelD disruptants showed 60 and 80%, respectively, of the activity observed with the wild type, whereas the double-gene disruptant did not show detectable lyase activity. On the other hand, pelD-complemented transformants showed 10–20% of the lyase activity observed with the wild type and a recovery of virulence because most stem segments inoculated with the complemented mutants showed lesions (Fig. 4e). Microscopic examination of tissue slices showed that the complemented double mutants penetrated to about half of the depth penetrated by the wild type, whereas the double disruptant hardly penetrated into the tissue (Fig. 4i). Because the complemented mutants did not produce PL levels anywhere near that of the wild type, it is not surprising that the observed degree of recovery of virulence was not complete. However, the observed recovery is a clear indication that the loss of virulence resulting from the double disruption was caused by the absence of the lyase.
Supplementation of the Infection Droplets of the Double-Gene Disruptant with Purified PLA or PLD.
Inclusion of purified PLA or PLD in the infection droplet resulted in a recovery of virulence, as seen by lesion formation on virtually all host segments (Fig. 4 f and g). Enzyme droplets without conidia showed no symptoms. With exogenous lyase, penetration of the double disruptant into the host tissue reached about 50% of that observed with the wild type (Fig. 4i), whereas the double disruptant failed to penetrate into the tissue or produce visible lesions in the absence of enzyme supplementation. Exogenous enzyme added into the droplet in which the conidia are placed is not targeted specifically to the point of contact between the germ tube and the host wall and therefore cannot be expected to be as fully effective as the enzyme naturally secreted at the point of penetration by the germ tube.
Discussion
The occurrence of multiple genes that encode functionally redundant enzymes that degrade the physical barriers in the host is a common feature of fungal pathogens that infect plants and animals. For example, Candida is known to have at least seven genes that encode secreted aspartic proteinases (9, 33). Aspergilli that cause invasive aspergillosis are known to express genes that encode serine proteinases (34–37), metalloproteinases (38–40), and aspartic proteinases (41). Disruption of one or two of these does not show a significant decrease in virulence (15–17). When one aspartic proteinase gene was disrupted, immunologically crossreactive proteinase(s) was found to be associated with the fungal wall (42), suggesting the presence of multiple aspartic proteinases. Multiple genes for cutinases and pectin-degrading enzymes are found in plant pathogenic fungi (5, 19, 43, 44). In such cases, single-gene disruption is not likely to reveal decreases in virulence. In addition, some genes that encode degradative enzymes such as the pelD of N. hematococca may be expressed only in the host (20), or different genes may be expressed on different organs or during different stages of growth or infection (45, 46). In such cases, even finding such enzymes may be difficult, as the commonly used culture conditions would not allow expression of such genes. The type of homology that allowed us to clone pelD does not have to hold true for functionally homologous enzymes. Another limitation of the gene disruption approach, in cases where multiple genes are involved, is the possibility of compensatory enhancement of expression of other genes accompanying disruption of a gene that encodes an enzyme with the same function. Disruption of serine proteinase caused an increase in metalloproteinase in Aspergillus flavus (47).
The presence of multiple genes that encode enzymes that can degrade the physical barriers of the host provides the pathogen an evolutionary advantage in that the differential regulation of expression of such genes would allow the pathogen to express the appropriate gene when the ecological need requires its expression. Usually, there is a low level of constitutive expression of the degradative enzymes and therefore the conidium would arrive at the host surface with a small amount of the enzyme. The cutinase and PL carried by N. hematococca are examples. From this organism, we have cloned pelB (32) and cut2 (T. Sirakova and P. E. Kolattukudy, unpublished work) that are constitutively expressed. On contact with the polymeric barrier of the host, the constitutively expressed enzyme would generate small amounts of hydrolytic products. These products would then activate the transcription of the inducible gene, generating a high level of the enzyme that can assist the pathogen to penetrate through the physical barriers of the host. This concept has been demonstrated for cutinase, and the transcription factors involved in the expression of the induced gene (48, 49) and the constitutively expressed gene (T. Sirakova, D. Li, and P.E.K., unpublished work) have been cloned from N. hematococca. In this fungus, the constitutively expressed gene, pelB, was suggested to provide the oligomers that induce pelA, because antisense expression of pelB prevented induction of pelA (W. Guo and P.E.K., unpublished work). To penetrate through the polymeric barrier of the host to infect it or grow saprophytically on the polymer, the microbe can use the same strategy of using a low level of constitutively expressed enzymes to sense the nature of the polymeric nutrient present in the immediate environment. The monomers could then transcriptionally activate the inducible gene to produce the enzyme either for infection or saprophytic growth. Separate polymer-degrading enzymes specializing in saprophytic growth or pathogenesis (45, 46) may also exist.
Even though the presence of multiple genes can make gene disruption ineffective in decreasing virulence, antibodies prepared against a barrier-degrading enzyme may show protection of the host against infection, if the products of the multiple genes are crossreactive as seen with both PL (20, 31) and cutinases (5, 6, 28) of N. hematococca. Enzyme-active site-directed inhibitors can show protection, if the multiple gene products use the same catalytic mechanism, as seen with cutinases that might assist penetration into plant hosts (5, 6) and with Candida aspartic proteinases that assist infection of animals (33). In such cases, inhibitors of the enzymes have been found to protect the plant or animal hosts from infection (5, 6, 33). However, disruption of one or more genes cannot reveal the role of the extracellular enzymes if other genes are expressed to produce functionally redundant enzymes. The results obtained with the single-gene disruptants illustrate this point (1–4, 10, 11, 16, 17, 38). There are cases where single-gene disruption caused some decrease in virulence (43, 50). In other cases, multiple-gene disruption failed to significantly decrease virulence (8, 12, 13). Therefore, conclusive evidence for or against a role for any particular enzyme activity in any aspect of pathogenesis has been difficult to obtain (51). A major decrease in virulence would require disruption of genes that encode all of the functionally redundant barrier-degrading enzymes that can play an essential role in pathogenesis. The present results clearly show how multiple gene products that catalyze degradation of host barriers, including those that might be uniquely induced by the host, can play an important role in pathogenesis and how single-gene disruption experiments can fail to reveal the true role of the extracellular polymer-degrading enzymes in fungal pathogenesis (18). The extracellular enzymes that degrade the physical barriers of the host may be suitable targets for antifungal therapy.
Acknowledgments
This work was supported in part by National Science Foundation Grant IBN-9816868.
Abbreviations
- pel
pectate lyase gene
- PL
pectate lyase
- RT-PCR
reverse transcription–PCR
- ORF
open reading frame
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160271497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160271497
References
- 1.Scott-Craig J S, Panaccione D G, Cervone F, Walton J D. Plant Cell. 1990;2:1191–1200. doi: 10.1105/tpc.2.12.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stahl D J, Schafer W. Plant Cell. 1992;4:621–629. doi: 10.1105/tpc.4.6.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweigard J A, Chumley F G, Valent B. Mol Gen Genet. 1992;232:183–190. [PubMed] [Google Scholar]
- 4.Bowen J K, Templeton M D, Dharrock K R, Crowhurst R N, Rikkerink E H. Mol Gen Genet. 1995;246:196–205. doi: 10.1007/BF00294682. [DOI] [PubMed] [Google Scholar]
- 5.Kolattukudy P E, Gamble D L. In: Pathogenesis and Host Specificity in Plant Diseases. Kohmoto K, Singh U S, Singh R P, editors. Vol. 2. New York: Pergamon; 1995. pp. 83–101. [Google Scholar]
- 6.Kolattukudy P E, Li D, Hwang C-S, Flaishman M A. Can J Bot. 1995;73,Suppl. 1:S1160–S1168. [Google Scholar]
- 7.Bouchara J P, Tronchin G, Larcher G, Chabasse D. Trends Microbiol. 1995;3:327–330. doi: 10.1016/s0966-842x(00)88965-9. [DOI] [PubMed] [Google Scholar]
- 8.Apel-Birkhold P C, Walton J D. Appl Environ Microbiol. 1996;62:4129–4135. doi: 10.1128/aem.62.11.4129-4135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoegl L, Ollert M, Korting H C. J Mol Med. 1996;74:135–142. doi: 10.1007/BF01575445. [DOI] [PubMed] [Google Scholar]
- 10.Crowhurst R N, Binnie S J, Bowen J K, Hawthorne B T, Plummer K M, Rees-George J, Rikkerink E H, Templeton M D. Mol Plant–Microbe Interact. 1997;10:355–368. doi: 10.1094/MPMI.1997.10.3.355. [DOI] [PubMed] [Google Scholar]
- 11.van Kan J A, van't Klooster J W, Wagemakers C A, Dees D C, van der Vlugt-Bergmans C J. Mol Plant–Microbe Interact. 1997;10:30–38. doi: 10.1094/MPMI.1997.10.1.30. [DOI] [PubMed] [Google Scholar]
- 12.Wu S-C, Ham K-S, Darvill A G, Albersheim P. Mol Plant–Microbe Interact. 1997;10:700–708. [Google Scholar]
- 13.Scott-Craig J S, Cheng Y-Q, Cervone F, de Lorenzo G, Pitkin J W, Walton J D. Appl Environ Microbiol. 1998;64:1497–1503. doi: 10.1128/aem.64.4.1497-1503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abad-Zapatero C, Goldman R, Muchmore S W, Hutchins C, Oie T, Stewart K, Cutfield S M, Cutfield J F, Foundling S I, Ray T L. Adv Exp Med Biol. 1998;436:297–313. doi: 10.1007/978-1-4615-5373-1_41. [DOI] [PubMed] [Google Scholar]
- 15.Monod M, Paris S, Sarfati J, Jaton-Ogay K, Ave P, Latge J P. FEMS Microbiol Lett. 1993;106:39–46. doi: 10.1111/j.1574-6968.1993.tb05932.x. [DOI] [PubMed] [Google Scholar]
- 16.Tang C M, Cohen J, Krausz T, van Noorden S, Holden D W. Infect Immun. 1993;61:1650–1656. doi: 10.1128/iai.61.5.1650-1656.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaton-Ogay K, Paris S, Huerre M, Quadroni M, Falchetto R, Togni G, Latge J P, Monod M. Mol Microbiol. 1994;14:917–928. doi: 10.1111/j.1365-2958.1994.tb01327.x. [DOI] [PubMed] [Google Scholar]
- 18.Keen N T. Phytoparasitica. 1995;23:281–284. [Google Scholar]
- 19.Koller W, Yao C, Trial F, Parker D M. Can J Bot. 1995;73:S1109–S1118. [Google Scholar]
- 20.Guo W, González-Candelas L, Kolattukudy P E. Arch Biochem Biophys. 1996;332:305–312. doi: 10.1006/abbi.1996.0346. [DOI] [PubMed] [Google Scholar]
- 21.González-Candelas L, Kolattukudy P E. J Bacteriol. 1992;174:6343–6349. doi: 10.1128/jb.174.20.6343-6349.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soliday C L, Dickman M B, Kolattukudy P E. J Bacteriol. 1989;171:1942–1951. doi: 10.1128/jb.171.4.1942-1951.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turgeon B G, Garber R C, Yoder O C. Mol Cell Biol. 1987;7:3297–3305. doi: 10.1128/mcb.7.9.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staben C, Jensen B, Singer M, Pollock J, Schechtman M, Kinsey J, Selker E. Fungal Genet Newsl. 1989;36:79–81. [Google Scholar]
- 25.Roberts I N, Oliver R P, Punt P J, van den Hondel C A M J J. Curr Genet. 1989;15:177–180. doi: 10.1007/BF00435503. [DOI] [PubMed] [Google Scholar]
- 26.Kämper J, Kämper U, Rogers L M, Kolattukudy P E. J Biol Chem. 1994;269:9195–9204. [PubMed] [Google Scholar]
- 27.Bajar A, Podila G K, Kolattukudy P E. Proc Natl Acad Sci USA. 1991;88:8208–8212. doi: 10.1073/pnas.88.18.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiti I B, Kolattukudy P E. Science. 1979;205:507–508. doi: 10.1126/science.205.4405.507. [DOI] [PubMed] [Google Scholar]
- 29.Guo W, González-Candelas L, Kolattukudy P E. Arch Biochem Biophys. 1995;323:352–360. doi: 10.1006/abbi.1995.9954. [DOI] [PubMed] [Google Scholar]
- 30.Crawford M S, Kolattukudy P E. Arch Biochem Biophys. 1987;258:196–205. doi: 10.1016/0003-9861(87)90336-5. [DOI] [PubMed] [Google Scholar]
- 31.Rogers L M, Flaishman M A, Kolattukudy P E. Plant Cell. 1994;6:935–945. doi: 10.1105/tpc.6.7.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo W, González-Candelas L, Kolattukudy P E. J Bacteriol. 1995;177:7070–7077. doi: 10.1128/jb.177.24.7070-7077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fallon K, Bausch K, Noonan J, Huguenel E, Tamburini P. Infect Immun. 1997;65:551–556. doi: 10.1128/iai.65.2.551-556.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichard U, Buttner H, Eiffert H, Staib F, Rüchel R. J Med Microbiol. 1990;33:243–251. doi: 10.1099/00222615-33-4-243. [DOI] [PubMed] [Google Scholar]
- 35.Monod M, Togni G, Rahalison L, Frenk E. J Med Microbiol. 1991;35:23–28. doi: 10.1099/00222615-35-1-23. [DOI] [PubMed] [Google Scholar]
- 36.Kolattukudy P E, Lee J D, Rogers L M, Zimmerman P, Ceselski S, Fox B, Stein B, Copelan E A. Infect Immun. 1993;61:2357–2368. doi: 10.1128/iai.61.6.2357-2368.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramesh M V, Sirakova T, Kolattukudy P E. Infect Immun. 1994;62:79–85. doi: 10.1128/iai.62.1.79-85.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monod M, Paris S, Sanglard D, Jaton-Ogay K, Bille J, Latge J P. Infect Immun. 1993;61:4099–4104. doi: 10.1128/iai.61.10.4099-4104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markaryan A, Morozova I, Yu H, Kolattukudy P E. Infect Immun. 1994;62:2149–2157. doi: 10.1128/iai.62.6.2149-2157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sirakova T A, Markaryan A, Kolattukudy P E. Infect Immun. 1994;62:4208–4218. doi: 10.1128/iai.62.10.4208-4218.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J D, Kolattukudy P E. Infect Immun. 1995;63:3796–3803. doi: 10.1128/iai.63.10.3796-3803.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reichard U, Monod M, Odds F, Ruchel R. J Med Vet Mycol. 1997;35:189–196. doi: 10.1080/02681219780001131. [DOI] [PubMed] [Google Scholar]
- 43.ten Have A, Mulder W, Visser J, van Kan J A. Mol Plant–Microbe Interact. 1998;11:1009–1016. doi: 10.1094/MPMI.1998.11.10.1009. [DOI] [PubMed] [Google Scholar]
- 44.Templeton M D, Sharrock K R, Bowen J K, Crowhurst R N, Rikkerink E H. Gene. 1994;142:141–146. doi: 10.1016/0378-1119(94)90369-7. [DOI] [PubMed] [Google Scholar]
- 45.Yao C, Koller W. Mol Plant–Microbe Interact. 1995;8:122–130. [Google Scholar]
- 46.Fan C Y, Koller W. FEMS Microbiol Lett. 1998;158:33–38. [Google Scholar]
- 47.Ramesh M V, Kolattukudy P E. J Bacteriol. 1996;178:3899–3907. doi: 10.1128/jb.178.13.3899-3907.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li D, Kolattukudy P E. J Biol Chem. 1995;270:11753–11756. doi: 10.1074/jbc.270.20.11753. [DOI] [PubMed] [Google Scholar]
- 49.Li D, Kolattukudy P E. J Biol Chem. 1997;272:12462–12467. doi: 10.1074/jbc.272.19.12462. [DOI] [PubMed] [Google Scholar]
- 50.Sheih M-T, Brown R L, Whitehead M P, Cary J W, Cotty P J, Cleveland T E, Dean R A. Appl Environ Microbiol. 1997;63:3548–3552. doi: 10.1128/aem.63.9.3548-3552.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tonukari N J, Scott-Craig J S, Walton J D. Plant Cell. 2000;12:237–447. doi: 10.1105/tpc.12.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]