

Abstract
In vertebrates, phagocytosis occurs mainly in specialized cells of the immune system and serves as a primary defense against invading pathogens, but it also plays a role in clearing apoptotic cells and in tissue remodeling during development. In contrast, unicellular eukaryotes, such as the ciliate Tetrahymena thermophila, employ phagocytosis to ingest and degrade other microorganisms to meet their nutritional needs. To learn more about the protein components of the multistep process of phagocytosis, we carried out an analysis of the Tetrahymena phagosome proteome. Tetrahymena cells were fed polystyrene beads, which allowed for the efficient purification of phagosomes. The protein composition of purified phagosomes was then analyzed by multidimensional separation coupled with tandem mass spectrometry. A total of 453 peptides were identified that resulted in the identification of 73 putative phagosome proteins. Twenty-eight of the proteins have been implicated in phagocytosis in other organisms, indicating that key aspects of phagocytosis were conserved during evolution. Other identified proteins have not previously been associated with phagocytosis, including some of unknown function. Live-cell confocal fluorescence imaging of Tetrahymena strains expressing green fluorescent protein-tagged versions of four of the identified phagosome proteins provided evidence that at least three of the proteins (including two with unknown functions) are associated with phagosomes, indicating that the bulk of the proteins identified in the analyses are indeed phagosome associated.
Phagocytosis is the process by which cells internalize particles that are too large to be taken up by pinocytosis or receptor-mediated endocytosis. Single-cell organisms, such as Dictyostelium discoideum and Entamoeba histolytica, use phagocytosis to provide nutrients for the cell (18, 59, 64). In mammals, phagocytosis is carried out primarily by cells of the immune system, including macrophages, neutrophils, and dendritic cells, whose extensive repertoire of cell surface receptors is responsible for their range of targets and their uptake efficiency (1, 82). Phagocytic ingestion of invading microbes by macrophages results in activation of the innate immune response, a necessary first step in the stimulation of the adaptive immune response to invading microorganisms. The ability of pathogens such as Mycobacterium tuberculosis and Staphylococcus aureus (25, 35, 65, 69, 81) to subvert phagocytosis for their survival and propagation underscores the importance of the phagocytic process for immune surveillance and integrity. Phagocytosis is also involved in additional functions in multicellular organisms, such as the removal of senescent or apoptotic cells and cell remodeling during development (57).
In mammalian cells, phagocytosis is initiated by ligand binding to cognate cell surface molecules, which include Fcγ and complement receptors (56). A nascent phagosome is then formed by lamellipodial extensions and invagination of the cell surface membrane in a process that involves a local restructuring of actin (17, 87). Once internalized, phagosomes proceed through a series of maturation steps that include transient and sequential interactions with early and late endosomal compartments (6, 7, 15, 37, 45, 53, 75) and which culminate in phagosome fusion with lysosomes to generate phagolysosomes. During the maturation process, phagosomes become acidified by proton-translocating vacuolar ATPases, and they acquire the hydrolytic enzymes that function in the phagolysosomal degradation of ingested phagosome cargo (14, 16). The complexity of phagocytosis is illustrated by recent analyses of the phagosome proteomes of mouse macrophages (30) and Entamoeba histolytica (58), where >140 and 85 proteins, respectively, were identified. While the roles of some of the proteins involved in mediating phagocytosis are clear, there remain many areas for which knowledge of the molecular mechanisms of the process needs to be enhanced or uncovered.
For this study, we analyzed the phagosome proteome of the ciliate Tetrahymena thermophila. In its natural habitat, this ciliated protozoan utilizes phagocytosis to ingest smaller food organisms, but phagocytosis appears to be rather nonspecific in that many types of particles can be ingested, including India ink and latex beads (5, 49, 84). A number of cytological analyses of phagocytosis in Tetrahymena have been carried out and indicate that there are similarities with the process in higher organisms (2, 44, 54, 55, 84). Particles are ingested by a specialized structure, the cytostome, at the base of the oral apparatus, and the phagosomes then travel to the posterior of the cell in a directed manner. Ultimately, the phagosomes fuse with the cytoproct at the posterior end of the cell, releasing their residual contents (see references 2 and 46).
Information is limited with regard to the proteins involved in phagocytosis in Tetrahymena. A gene encoding a cytoplasmic dynein (DYH1) has been implicated in phagocytosis (47), and Hosein et al. (39) reported that the directed motility of phagosomes from the cytostome to the cytoproct requires dynamic actin and Myo1p, a novel myosin (85). Phagosomes have been purified from Tetrahymena, and antibodies have been used to identify multiple small GTPases (52), which are known to be involved in phagosome maturation in other organisms (7). Calcium-binding proteins were also identified in early-stage phagosomes (49, 83), and Gonda et al. (31, 32) reported that Ca2+/calmodulin-binding proteins play a significant role in phagosome formation.
There are a number of features of T. thermophila that make it a strong model for the study of phagocytosis. First, there are numerous genetic and molecular genetic approaches that have been developed for this organism (reviewed in reference 78). Second, phagocytosis is nonessential in Tetrahymena, allowing for the isolation of mutations disrupting the process (63). Third, the utility of the system was recently augmented by the complete sequencing and preliminary annotation of the macronuclear genome (22, 73). The availability of this information has allowed proteomic analyses of isolated organelles, using tandem mass spectrometry (10, 71). In this study, we purified phagosomes from T. thermophila and characterized their protein composition by multidimensional separation coupled with tandem mass spectrometry (71, 86). A total of 73 proteins were identified that are viewed as strong candidates for constituents of the phagosome proteome. These include 28 proteins that have been implicated in phagocytosis in other organisms as well as 12 proteins of unknown function that are candidates as novel proteins involved in phagocytosis. Finally, genes encoding green fluorescent protein (GFP)-tagged versions of four of the identified proteins were introduced into Tetrahymena cells. Fluorescence confocal microscopy indicated a phagosomal association for at least three of the four tagged proteins, supporting the overall validity of the Tetrahymena phagosome proteome.
MATERIALS AND METHODS
Cells and cell culture.
Two T. thermophila strains that are impaired in exocytosis (cap negative), namely, MN173, which was kindly provided by Aaron P. Turkewitz (51), and Grl1 Ex4.1A (41), were employed, as well as the paclitaxel-sensitive strain CU522 (btu1-1/btu1-1) (27). Cells were maintained in SPPA medium (1% proteose peptone, 0.2% dextrose, 0.1% yeast extract, and 0.003% sequestrine [Novartis, Greensboro, NC]) containing 250 μg/ml penicillin G, 250 μg/ml streptomycin sulfate, and 0.25 μg/ml amphotericin B (all from Sigma-Aldrich, St. Louis, MO) (28). For strain Grl1 Ex4.1A, 180 μg/ml paromomycin sulfate (Sigma-Aldrich) was also included.
Phagosome isolation.
The polystyrene bead-mediated phagosome isolation procedures we employed were modifications of published protocols (5, 17, 34). For phagosome isolation, 0.5- or 1-liter cultures of cap-negative Tetrahymena cells were grown at 30°C with gentle rotation (∼100 rpm) to a density of 2 × 105 to 3 × 105 cells/ml. Red-fluorescing polystyrene microspheres (2.0-μm diameter; Duke Scientific, Palo Alto, Calif.) were added to the cultures at a final concentration of 0.002%, and incubation was continued without rotation for an additional 15, 30, or 60 min at 30°C. Cells were collected by centrifugation at 750 × g for 3 min at 8°C. The cell pellets were washed with 10 mM Tris-HCl, pH 7.5, and resuspended to a final volume of 10 ml in cold homogenization buffer, consisting of 250 mM sucrose, 3 mM imidazole, pH 7.4, and 1× Complete EDTA-free protease inhibitor cocktail plus 0.7 μg/ml pepstatin (both from Roche Applied Science, Indianapolis, IN). Cells were homogenized on ice with a 15-ml Dounce tissue grinder (Wheaton, Millville, NJ) until ≥90% of the cells were broken, as determined by fluorescence microscopy. ATP magnesium salt (ATP-Mg; Sigma-Aldrich) was added to a final concentration of 10 mM, and the homogenate was incubated for 15 min at 4°C. Phagosomes were isolated by sucrose step gradient ultracentrifugation essentially as described by Desjardins et al. (19), except that all solutions contained the protease inhibitors described above. The phagosomes were recovered from the 10 to 25% sucrose layer interface, ∼30 ml of phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, plus protease inhibitors) was added, and the phagosomes were pelleted by centrifugation at 100,000 × g for 20 min. The supernatant was removed, and phagosome pellets were stored at −70°C.
Western blot analysis.
Phagosomes prepared from cells that had been fed polystyrene beads for 15, 30, and 60 min were resuspended and pooled in a total volume of 100 μl of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) buffer (50 mM Tris-HCl, pH 6.8, 100 mM dithiothreitol, 2% SDS, 10% glycerol, 0.1% bromophenol blue; ∼5.5 × 106 cell equivalents/μl). To prepare a whole-cell protein extract, 2.5 ml of a cell culture without beads (∼2.5 × 105 cells/ml) was collected by centrifugation, washed in 10 mM Tris-HCl, pH 7.5, and resuspended in 208 μl SDS-PAGE buffer (∼3.0 × 103 cell equivalents/μl). The material was incubated in a boiling water bath for 10 min and centrifuged for 10 min at 12,000 × g before being loaded into the gel.
Proteins were separated by electrophoresis through either 12% or 18% SDS-PAGE gels, as described previously (67). For immunoblotting, proteins were transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore, Bedford, MA) according to the manufacturer's instructions. The membranes were blocked with 1% nonfat dry milk prepared in Tris-buffered saline (TBS; 20 mM Tris-HCl, pH 7.6, 137 mM NaCl) and incubated for 1 to 2 h at room temperature with polyclonal antibodies directed against Grl8p (9) (1:3,000 dilution) or histone H1 (12) (1:5,000) or a mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich) (1:1,000 dilution). Membranes were washed twice for 40 min each in TBS containing 0.1% Tween 20 (Sigma-Aldrich) and then three times for 5 min each with TBS. Antibody binding was detected using alkaline phosphatase-conjugated anti-rabbit immunoglobulin G (IgG) (Grl8p and histone H1; 1:10,000) or alkaline phosphatase-conjugated anti-mouse IgG (α-tubulin; 1:1000) and a 5-bromo-4-chloro-3-indolyl phosphate/Nitro Blue Tetrazolium liquid substrate system (Sigma-Aldrich).
rRNA isolation.
To assess ribosomal contamination of phagosome preparations, phagosomes were isolated by sucrose step gradient centrifugation as described above. Two-milliliter samples from each step of the gradient were extracted twice with an equal volume of phenol, and nucleic acids were precipitated with ethanol. Following centrifugation, the pellets were resuspended in 25 μl of RNA storage solution (Ambion, Austin, TX). Samples were separated by electrophoresis through a 1% agarose gel made and run in 89 mM Tris, 89 mM H3BO3, and 20 mM EDTA. Bands corresponding to the rRNAs were visualized by ethidium bromide staining.
Mass spectrometry.
Phagosome preparations were processed in four different ways prior to analysis by mass spectrometry. In each case, phagosomes prepared from equal numbers of cells incubated with polystyrene spheres for either 15, 30, or 60 min were pooled for analysis. In the first approach (Triton X-100 extraction), phagosomes derived from 3 liters of cells were resuspended and combined in HEPES-T buffer composed of 300 mM HEPES, pH 7.6, 200 mM KCl, 5 mM EDTA, 0.5% Triton X-100, 1× Complete EDTA-free protease inhibitor cocktail, and 0.7 μg/ml pepstatin (Roche). The resuspended pellets were incubated on ice for 30 min and centrifuged for 1 min at 13,000 × g at room temperature to pellet beads. The supernatant, containing 408 μg of protein, was transferred to a clean tube, fast frozen in ethanol-dry ice, and stored at −70°C. Most of the Triton X-100 was removed by three buffer exchanges into 10 mM Tris buffer, pH 7.5, using size-exclusion spin columns (Microcon-3) with a molecular size cutoff of 3,000 Da (Millipore). In the second approach (SDS extraction), phagosomes derived from 11.25 liters of cells were resuspended in HEPES buffer (HEPES-T without Triton X-100). SDS was added to a final concentration of 2%, and the samples were vortexed, incubated for 5 min at room temperature, and centrifuged as described above to pellet beads. Supernatants of each sample were combined and frozen at −70°C. SDS detergent removal columns were used following a procedure recommended by the manufacturer (The Nest Group, Southborough, MA). The third approach entailed a simple freeze-thaw of the phagosome preparation prior to direct digestion with trypsin.
Samples from these first three approaches were digested in solution with trypsin (Promega), using a modified version of a previously described procedure (74). Prior to digestion, the samples were denatured by heating at 60°C for 1 hour in the presence of 5 mM dithiothreitol. After being cooled to room temperature, the samples were alkylated by incubation with 10 mM iodoacetamide for 1 hour in the dark. Trypsin at a 1:20 concentration (wt/wt) was then added in an equal volume of 50 mM ammonium bicarbonate to the sample and incubated overnight at 37°C. Each of these samples was then analyzed by two-dimensional liquid chromatography-tandem mass spectrometry (LC-MS/MS). The first dimension of the LC separation, based on strong cation exchange, was performed offline, using an HP1050 high-performance LC system (Agilent, Palo Alto, CA) and an SF-2120 super fraction collector (Advantec MFS, Dublin, CA). The strong cation exchange column used was a 2.1-mm-internal-diameter (i.d.) by 100-mm-long poly(LC) polysulfoethyl A column packed with 5-μm beads with 300-Å pores with a 2.1-mm i.d. by 10-mm-long guard column of the same material plumbed upstream from the analytical column (The Nest Group). Separation was effected by a binary gradient at a flow rate of 0.2 ml/min. Eluent A consisted of a 10 mM KH2PO4 solution in 25% acetonitrile and 75% deionized water acidified to a pH of 3.0 with phosphoric acid. Eluent B consisted of a 10 mM KH2PO4 and 350 mM KCl solution in 25% acetonitrile and 75% deionized water acidified to a pH of 3.0 with phosphoric acid. A 1-hour run was set up with increasing eluent B concentrations, from 0% to 100%, from the 2-min to 58-min time points in a linear gradient. The run was terminated after 60 min. Fractions were collected every 2 minutes to give 30 fractions, with a 0.4-ml total volume in each fraction. The fractions were dried by a speed vacuum and stored at −20°C.
In the fourth approach, SDS-PAGE was employed as the first fractionation step (68). Phagosome samples were processed as described for Western blot analysis, 50 μl (845 μg) of the sample was boiled for 5 min in SDS-PAGE buffer, and proteins were separated in an 8.0-cm 12% SDS-PAGE gel. The gel was stained with Bio-Safe Coomassie stain (Bio-Rad, Hercules, CA), and a sterile razor blade was used to cut the gel lane into 24 3-mm slices, each of which was digested separately with trypsin, using an in-gel tryptic digestion procedure (70). The extracted tryptic peptides were dried by a speed vacuum as described above and stored at −20°C.
With the exception of the first five fractions from the second approach, all of the above fractions were then analyzed by nano-LC-MS/MS. A nanobore LC system from LC Packings (Amsterdam, The Netherlands) consisted of a Famos autosampler, a Switchos switching unit, and an Ultimate Nano LC system. It was interfaced to a QSTAR Pulsar hybrid quadrupole-time-of-flight mass spectrometer (Applied Biosystems/MDS SCIEX, Foster City, CA) using a Protana NanoES ion source (Protana Engineering A/S, Odense, Denmark). The spray capillary was a distally coated PicoTip SilicaTip emitter with a 10-μm-i.d. tip (New Objective, Woburn, MA). The nanobore LC column was a 75-μm-i.d. by 150-mm-long reverse-phase PepMap C18 nano-capillary column (LC Packings) packed with 3-μm beads with 100-Å pores. A 300-μm by 5-mm precolumn reverse-phase column of the same material was used for desalting the samples prior to separation. Each fraction was resuspended in 30 μl of 0.1% formic acid, 1 μl of which was loaded onto the precolumn and desalted by washing with eluent A for 3 to 4 min at a flow rate of 50 μl/min. Eluent A consisted of 94.9% deionized water, 5.0% acetonitrile, and 0.1% formic acid (pH ≈ 3). Eluent B consisted of 5.0% deionized water, 94.9% acetonitrile, and 0.1% formic acid. After being desalted, the precolumn was brought in line with the separation column, where separation was achieved using a binary mobile-phase gradient run for 2 h at a total flow rate of 200 nl/min. The gradient conditions used were as follows: 0 min, 5% eluent B; 8 min, 5% eluent B; 10 min, 15% eluent B; 20 min, 20% eluent B; 70 min, 40% eluent B; 80 min, 60% eluent B; 90 min, 80% eluent B; 102 min, 80% eluent B; 105 min, 5% eluent B; and 120 min, 5% eluent B.
MS data were acquired in information-dependent acquisition (IDA) mode with Analyst QS SP8, using Bioanalyst Extension 1.1 software (Applied Biosystems/MDS SCIEX). MS cycles comprised a time-of-flight MS survey scan with an m/z range of 400 to 1,500 Thomson (Th) for 1 s, followed by four product ion scans with an m/z range of 70 to 2,000 Th for 2 s each. Collision energy was automatically controlled by the IDA CE Parameters script. Switching criteria were set to ions greater than an m/z of 400 Th and smaller than an m/z of 1,500 Th with a charge state of 2 to 4 and an abundance of ≥10 counts. Former target ions were excluded for 60 s, and ions within a 4-Th window were ignored. Additionally, the IDA Extensions II script was set to one repetition before dynamic exclusion and to select a precursor ion nearest to a threshold of 10 counts on every fourth cycle.
During these analyses, protein concentrations were determined using bicinchoninic acid (BCA) protein assay reagents (Pierce Chemical, Rockford, IL) according to the manufacturer's instructions.
Bioinformatics.
Mass spectrometry data were analyzed by Mascot (Matrix Science Ltd., London, United Kingdom), using a conceptual translation of the entire Tetrahymena macronuclear genome sequence (71) or the set of proteins based on the preliminary gene predictions generated by The Institute of Genome Research (http://www.tigr.org/; http://seq.ciliate.org/cgi-bin/BLAST-tgd.pl) as a database. All peptides utilized were above the Mascot-recommended cutoff for significant sequence identity. In some cases, individual peptides were mapped to multiple Tetrahymena predicted proteins, which were typically members of a multigene family. However, for all such cases, there was always at least one additional identified peptide that uniquely matched only one member of the family. Only the family member that matched all peptides is reported.
Blastp (3; http://www.ncbi.nlm.nih.gov/BLAST/) was used to search the GenBank nonredundant protein database for predicted functional identification of the matched conceptually translated or predicted protein sequences, with E values of <1 × 10−7 considered significant. The GO (Gene Ontology [http://www.geneontology.org/]) database was used to search for matches to proteins localized to the lysosome or lytic vacuole compartment of the cell.
Molecular biological techniques.
Plasmid DNAs were isolated from bacterial cells, using a Magic Miniprep or Midiprep DNA purification system (Promega Corp., Madison, WI) according to the manufacturer's instructions. Whole-cell Tetrahymena DNA was isolated using a Wizard genomic purification kit (Promega) essentially according to the manufacturer's instructions for isolating genomic DNA from plant tissue, except that step 1 in the protocol was omitted. PCR products were isolated from low-melting-point agarose gels as described previously (60). DNA restriction digestions, dephosphorylations, and ligations were carried out under conditions recommended by the enzyme suppliers (New England Biolabs, Beverly, MA; GIBCO BRL Life Technologies, Inc., Rockville, MD) or by commonly used protocols (67). Sequencing was performed by the University of Connecticut Health Center Molecular Core Facility, using a BigDye Terminator sequencing kit (Applied Biosystems/MDS SCIEX).
PCR and GFP-tagged clone construction.
To generate C-terminally GFP-tagged versions of cathepsin B (Tetrahymena gene identifier 6.m00369) and unknown Tetrahymena phagosomal proteins Tpp2p, Tpp5p, and Tpp9p, their entire predicted coding regions were PCR amplified using the following primer pairs that incorporate BclI and MluI restriction sites (in bold), for CATHB, TPP2, and TPP9, or HindIII and MluI restriction sites (in bold), for TPP5: CathB/F (5′-GAATGATCATGAAACACTAAGCATTAATTATTACTGC-3′), CathB/R (5′-GAAACGCGTTAAGCAGGAAGAGCAGTAAC-3′), P2/F (5′-GAATGATCATGAGAAATTCAACCATTTTTACC-3′), P2/R (5′-GAAACGCGTTATTCTCTACCAACAACAAGG-3′), P9/F (5′-GAATGATCATGGTCAACGGCGGCTGTCCC-3′), P9/R (5′-GAAACGCGTTCAGTTTTTGCTTTACCAGG-3′), P5/F (5′-GAAAGCTTAATGCAACACCAAGATCTCAT-3′), and P5/R (5′-CTACGCGTGCATAATTGCTTAAGACGAAGC-3′). PCR was carried out with KlenTaq DNA polymerase (Sigma-Aldrich), essentially according to the manufacturer's instructions, using 50 to 100 ng of DNA obtained from a T. thermophila cDNA library (13) as the template for TPP2, TPP5, and TPP9. For CATHB, the coding region was amplified from genomic DNA by two successive PCR amplifications, the first of which used primers CathB-3/F (5′-GAAAACATGAAACACTAAGC-3′) and CathB/R, and the second of which used primers CathB/F and CathB/R.
PCR products were digested with BclI or HindIII and MluI and directionally inserted into the corresponding restriction sites of the MTT1-NRK2-GFP vector (8). Constructs were transformed into Escherichia coli TOP10 chemically competent cells (Invitrogen), and clones were verified by sequencing. For transformation into Tetrahymena, the plasmids were digested with XhoI and XbaI and introduced into strain CU522 cells by biolistic particle bombardment (11). Paclitaxel (20 μM; Sigma-Aldrich) was added to cultures to select for transformants in which the transgene was inserted into the BTU1 locus (29).
Live-cell microscopy.
Expression of GFP constructs was induced by growing cells in the presence of 0.5 to 2 μg/ml cadmium for 16 to 24 h at room temperature. One milliliter of cells at ∼2 × 105 cells/ml was centrifuged for 2 min at 13,000 × g, washed once in 10 mM Tris-HCl, pH 7.4 (buffer T), and resuspended in 100 μl buffer T. To examine actively phagocytosing cells, 30 μl of nonfluorescent 1.1-μm polystyrene latex beads (0.25% solids; Sigma-Aldrich) was added to a 1-ml culture, which was incubated for 15 to 120 min at room temperature prior to pelleting and resuspension in 100 μl buffer T as described above. Cells (3 μl) were then mixed with an equal volume of methyl cellulose (Connecticut Valley Biological Supply, Southampton, MA) that had been applied to a coverslip and viewed with a Zeiss Axiovert 100 M microscope coupled to a Zeiss 510 laser scanning system and software, using a Zeiss Plan-Apochromat 63× 1.4-numerical aperture differential interference contrast oil immersion lens. GFP fluorescence was excited with the 488-nm line of an argon laser. Images were processed with MetaMorph Offline, version 7.0 (Molecular Devices) and assembled with Adobe Photoshop, version 7.0. All images were obtained under the same microscopy conditions and uniformly processed.
RESULTS AND DISCUSSION
Phagosome isolation.
To isolate phagosomes for proteomic analysis, we adapted procedures that have proven effective for a number of organisms and cell types (17, 34, 58). The approach (Fig. 1) involved feeding 2-μm red fluorescent polystyrene beads to Tetrahymena organisms, which they readily ingest by phagocytosis (5). Following cell lysis by homogenization, the bead-containing phagosomes were purified from other cellular membranous compartments by sucrose density gradient centrifugation based on the lower density of the encased polystyrene beads. Phagosomes were purified from two Tetrahymena strains, namely, MN173 (51) and Grl1 Ex4.1A (41), which are defective in dense core granule or mucocyst discharge (77). These strains were employed because under our conditions, the dense core granules of wild-type cells undergo regulated secretion to form a sticky capsule around the cell that interferes with both cell lysis and phagosome recovery.
FIG. 1.

Summary of Tetrahymena phagosome proteome analysis. Tetrahymena micrographs are combined Nomarski and fluorescent images. Dark intracellular inclusions are bead-filled phagosomes (PGSMs), and the arrow indicates a single extracellular latex bead. See the text for additional details.
Previous work on Tetrahymena indicated that the entire phagocytosis process, from formation of the phagosome to postdigestive release of its contents, occurs over a period of 1 to 2 h at 30°C (5, 54). In our analyses, we sought to identify proteins from all stages of phagocytosis. Consequently, we prepared and pooled phagosome preparations from cells that had been fed polystyrene beads for 15, 30, and 60 min.
A number of analyses were performed to assess the purity of phagosome preparations. Antibodies directed against granule lattice protein 8 (Grl8p) (9, 77), macronuclear histone H1 (12), and α-tubulin were employed in Western blot analyses to assess contamination of the pooled phagosome preparations with dense core granules, nuclei, and cilia, respectively (Fig. 2A). Each of the proteins was readily detectable in total protein extracts prepared from Tetrahymena cells, but few, if any, of these proteins could be detected among the phagosomal proteins prepared from an equivalent number of cells. Even when 100-fold-greater amounts of the phagosome preparation were analyzed, α-tubulin and macronuclear histone H1 were not observed, indicating no detectable levels of contamination with nuclei and cilia (Fig. 2A). In the case of Grl8p, trace amounts of this protein, particularly the 45-kDa Grl8p proprotein (9), were observed when 100-fold-more cell equivalents of the phagosomal extract were analyzed, indicating that there was a low level of dense core granules within the preparation. Contamination by ribosomes was also assessed by directly examining fractions of the sucrose step gradient used to purify phagosomes for the presence of small- and large-subunit rRNAs (Fig. 2B). Nucleic acids were purified from a sample for each step in the gradient and analyzed by agarose gel electrophoresis to detect rRNA. The vast majority of the rRNA was present in the 49% and 62% sucrose layers at the bottom of the gradient. However, when increasing amounts of material were loaded in the gel, smaller amounts of rRNA were found to trail into at least the 25% sucrose layer, on which the phagosomes band. Overall, the results indicate that the isolation procedure results in a substantial enrichment of phagosomes from other cellular organelles, although at least small fractions of ribosomes and dense core granules copurify with the phagosomes.
FIG. 2.
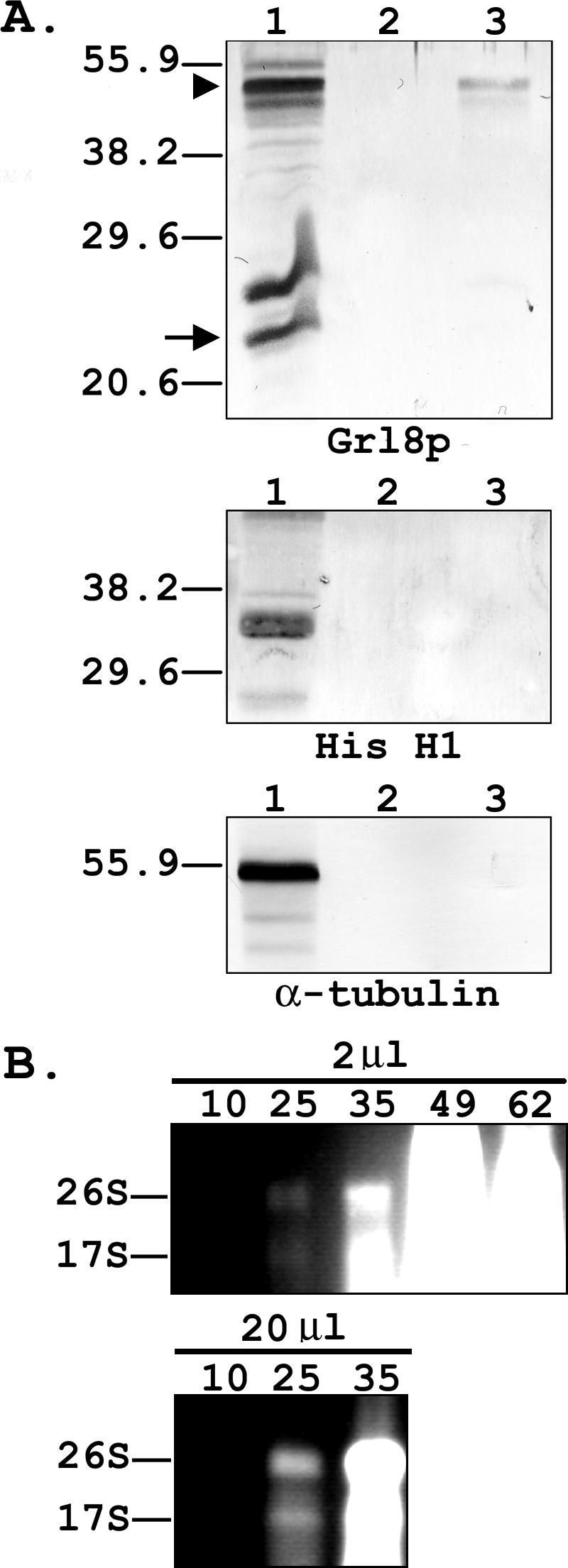
(A) Western blot analysis with antibodies to Grl8p, histone H1, and α-tubulin. Blots contain 104 cell equivalents of total Tetrahymena proteins (lanes 1) or 104 (lanes 2) or 106 (lanes 3) cell equivalents of phagosomes. In the Grl8p blot, the arrowhead denotes the position of the 45-kDa Grl8p proprotein, and the arrow indicates the 22-kDa mature form. (B) Ethidium bromide-stained agarose gels containing nucleic acids (2 μl or 20 μl) isolated from fractions of a sucrose gradient (10 to 62% sucrose) used to purify phagosomes. The positions of the 26S and 17S rRNAs are indicated.
Mass spectrometry of phagosome proteins.
Phagosome preparations were processed in four different ways prior to two-dimensional LC-MS/MS analyses in an attempt to maximize the number and types of proteins detected (Fig. 1). The first three approaches were (i) Triton X-100 solubilization of phagosomes, (ii) SDS extraction of proteins, and (iii) simple freeze-thawing of the phagosome preparation. A fourth approach employed an initial separation of SDS-solubilized proteins by SDS-PAGE in a 12% gel prior to LC-MS/MS analysis. A total of 453 nonredundant peptides were identified by these multiple approaches and were mapped by MASCOT to 183 proteins or predicted proteins in the Tetrahymena genome database. Blastp searches of the GenBank nonredundant protein database, using an expect (E) value of <10−7 as the cutoff for significant sequence similarity, were then carried out to identify homologs in other organisms and to provide preliminary annotation of proteins. Based on this standard, homologs were identified for 153 of the 183 identified Tetrahymena proteins.
Table 1 provides a listing of the 73 proteins that we consider strong candidates for components of the Tetrahymena phagosome proteome. The criteria for inclusion in the list included identification of the protein on the basis of two or more significantly scoring peptides in the MS analyses. The majority (52%) of these proteins were also identified by two or more of the approaches used in the MS analyses (Table 1). We chose to omit from the list a total of 25 ribosomal and dense core granule proteins identified on the basis of two or more matching peptides. These proteins were excluded because they are all expected to be abundant cellular proteins derived from the two cellular organelles (9, 21) for which we observed trace contamination in the phagosome preparations (see above). Additional information on the components of the phagosome proteome (e.g., refined gene predictions, expressed sequence tag support for gene predictions, and relevant Tetrahymena literature citations) are available at http://tetrahymenaphagocytosis.uchc.edu/.
TABLE 1.
Tetrahymena phagosomal proteins
| Tetrahymena gene IDa | No. of peptides (no. of analysesb) | BLASTp resultc
|
Other organisms with homologsd | ||
|---|---|---|---|---|---|
| Protein name | GenBank no. | E value | |||
| Strong candidates with homologs of known function in other organisms | |||||
| PreTt22225/6 | 6 (4) | 14-3-3 protein | 57017251 | 3.00E−121 | M |
| 160.m00088 | 2 (2) | Acetyl-coenzyme A acyltransferase (3-ketoacyl-coenzyme A thiolase) | 505533961 | 1.00E−79 | |
| 18.m00423 | 10 (3) | Acid alpha-glucosidase | 3023259 | 0 | GO |
| 45.m00189 | 5 (3) | Acid phosphatase | 50057591 | 1.00E−27 | E, GO |
| 25.m00418 | 6 (2) | Acid phosphatase | 12584854 | 4.00E−20 | E, GO |
| 120.m00106 | 5 (1) | ADP-ribosylation factor | 396808 | 2.00E−70 | |
| 18.m00296 | 10 (2) | ATP-binding cassette (ABC) transporter | 50057618 | 0 | |
| 32.m00165 | 10 (2) | ATP-binding cassette (ABC) transporter, subfamily C | 60099179 | 3.00E−119 | |
| 15.m00443 | 2 (1) | Bactericidal/permeability-increasing protein | 27155085 | 1.00E−08 | |
| 225.m00058 | 2 (1) | Calcium ATPase | 53801430 | 0 | E |
| 190.m00045 | 2 (2) | Carbonic anhydrase-like protein | 7268897 | 7.00E−11 | |
| 92.m00126 | 6 (4) | Cathepsin B | 27806671 | 9.00E−93 | M, GO |
| 6.m00369 | 2 (1) | Cathepsin B | 14582897 | 4.00E−85 | M, GO |
| 125.m00080 | 4 (3) | Cathepsin L/tetrain | 3273233 | 2.00E−133 | M, E, GO |
| 5.m00542 | 6 (4) | Cathepsin L-like protein | 24474971 | 2.00E−79 | M, GO |
| 13.m00464 | 2 (1) | Cathepsin L | 7239343 | 8.00E−65 | M, GO |
| 175.m00067 | 3 (2) | Chitinase-related protein | 62462538 | 5.00E−09 | |
| 46.m00201 | 4 (2) | Cytochrome b5-like, heme/steroid-binding domain | 34905998 | 2.00E−16 | |
| 9.m00561 | 3 (1) | Cytochrome P450 monooxygenase-related protein | 33113213 | 1.00E−21 | M |
| 96.m00145 | 6 (3) | Elongation factor 1-alpha | 416931 | 0 | M, E |
| 152.m00108 | 2 (1) | Elongation factor 1-beta | 56607110 | 2.00E−-68 | |
| 58.m00146 | 3 (1) | Endoglycoceramidase (cellulase domain) | 66826341 | 7.00E−-91 | |
| 3.m01761 | 4 (1) | α-Galactosidase/hydrolase | 22331822 | 2.00E−115 | GO |
| 10.m00541 | 3 (1) | GTP-binding protein (RHD3 family) | 66814646 | 1.00E−65 | |
| 51.m00272 | 4 (1) | GTP-binding protein (small; Sar1 family) | 74834470 | 1.00E−72 | |
| 7.m00462 | 4 (2) | Heat shock protein Hsp-70 | 74834195 | 0 | M, GO |
| 14.m00361 | 4 (1) | Heat shock protein Hsp-70 | 13359317 | 0 | M, GO |
| 25.m00321 | 2 (1) | Histone H2B2 | 578563 | 3.00E−42 | |
| 15.m00378 | 2 (1) | Histone H4 | 223273 | 5.00E−42 | |
| 40.m00220 | 3 (1) | Lysosomal acid lipase/gastric lipase | 27806551 | 3.00E−50 | M, E, GO |
| 94.m00154 | 3 (1) | Lysosomal acid phosphatase | 73982426 | 7.00E−23 | E, GO |
| 103.m00129 | 6 (2) | Lysosomal phospholipase A1/cathepsin L | 24474971 | 8.00E−174 | M, E, GO |
| 85.m00169 | 3 (1) | Methyltransferase-related protein | 78702333 | 3.00E−18 | |
| 3.m01844 | 10 (4) | Na+/K+-transporting ATPase, alpha subunit | 50057279 | 0 | |
| 101.m00128 | 3 (2) | Niemann-Pick C1 | 5714634 | 4.00E−88 | GO |
| 81.m00237 | 2 (1) | Palmitoyl-protein thioesterase | 40846454 | 3.00E−52 | M, GO |
| 65.m00149 | 3 (1) | Peroxisomal biogenesis factor 11A-related protein | 66804811 | 6.00E−13 | |
| 31.m00346 | 2 (2) | Peroxisomal multifunctional enzyme | 7658149 | 1.00E−107 | |
| 45.m00225 | 6 (3) | Phagosome protein 1, Tetrahymena (Php1p/P28p) | 24474973 | 3.00E−86 | |
| 45.m00228 | 2 (2) | Phagosome protein 1 (Php1p)-related, Tetrahymena | 24474973 | 2.00E−17 | |
| 108.m00178 | 2 (1) | Phospholipid scramblase | 2935163 | 2.00E−12 | |
| 57.m00252 | 15 (3) | Prolyl-4-hydroxylase (thioredoxin domains) | 50745403 | 1.00E−53 | |
| 76.m00142 | 3 (1) | Protein disulfide isomerase-related protein | 23394410 | 2.00E−18 | M |
| 350.m00011 | 2 (1) | Protein disulfide isomerase | 70990864 | 7.00E−16 | M |
| 31.m00241 | 4 (1) | Rab1 small GTP-binding protein | 74833747 | 2.00E−76 | E |
| PreTt22495 | 3 (1) | Rab7 GTPase | 6682935 | 5.00E−99 | M, E, GO |
| 19.m00274 | 2 (2) | Rab13 GTPase | 5738166 | 2.00E−84 | |
| 65.m00231 | 2 (2) | Reticulocyte binding-like protein 4 | 37725926 | 2.00E−09 | |
| 11.m00291 | 3 (2) | Secreted alpha beta hydrolase | 46229520 | 8.00E−16 | |
| 61.m00231 | 2 (1) | Sequestosome 1 | 31581536 | 2.00E−08 | |
| 84.m00113 | 5 (1) | SerH3 cell surface immobilization antigen | 6273279 | 0 | |
| 194.m00023 | 5 (2) | Squalene-hopene cyclase/terpenesynthase | 39982558 | 4.00E−46 | |
| 72.m00189 | 2 (1) | Surface protein type 51B-related protein, Paramecium | 1084998 | 1.00E−12 | |
| 3.m01929 | 2 (1) | Synaptobrevin/longin/VAMP-related protein | 31744980 | 5.00E−25 | |
| 73.m00202 | 4 (2) | Tubulin, alpha | 730899 | 0 | M |
| 36.m00227 | 5 (3) | Tubulin, beta | 730902 | 0 | M |
| 34.m00342 | 3 (2) | Vacuolar ATPase, subunit A | 66863387 | 0 | M, GO |
| 24.m00223 | 3 (3) | Vacuolar ATPase, subunit a | 74834076 | 0 | E |
| 48.m00253 | 2 (3) | Vacuolar ATPase, subunit d (C/AC39) | 67594935 | 1.00E−57 | |
| 120.m00116 | 9 (3) | Vacuolar H+-translocating inorganic pyrophosphatase | 2653446 | 0 | |
| 8.m00549 | 13 (3) | Vps13 (vacuolar protein sorting)/chorein | 66807841 | 4.00E−50 | |
| Strong candidates with unknown function | |||||
| 19.m00361 | 2 (1) | Tpp1p | |||
| 130.m00077 | 2 (2) | Tpp2p | |||
| 50.m00189 | 7 (3) | Tpp3p | |||
| 129.m00110 | 10 (2) | Tpp5p | 74834202 | 6.00E−43 | |
| 104.m00169 | 2 (1) | Tpp7p | |||
| 159.m00051 | 3 (1) | Tpp9p | |||
| 74.m00136 | 2 (1) | Tpp11p | |||
| 59.m00238 | 4 (1) | Tpp12p | |||
| 57.m00235 | 2 (2) | Tpp16p | |||
| 101.m00138 | 3 (3) | Tpp19p | |||
| 3.m01682 | 2 (1) | Tpp20p | |||
| 125.m00128 | 4 (1) | Tpp21p | 15236947 | 2.00E−59 | |
| Less well-supported candidates | |||||
| 48.m00238 | 1 (1) | Actin binding protein | 66807105 | 2.00E−37 | GO |
| 46.m00206 | 1 (1) | Actin binding protein | 66807105 | 6.00E−11 | GO |
| 42.m00199 | 1 (1) | Actin binding protein | 66807105 | 1.00E−09 | GO |
| 156.m00085 | 1 (1) | Cyclophilin | 47028327 | 2.00E−63 | E |
| 88.m00155 | 1 (1) | Cysteine proteinase | 15290508 | 1.00E−51 | GO |
| 54.m00236 | 1 (1) | Delta-9 fatty acid desaturase | 1620881 | 4.00E−178 | GO |
| 72.m00143 | 1 (1) | Glyceraldehyde-3-phosphate dehydrogenase 1 | 13377481 | 3.00E−158 | M, GO |
| 51.m00201 | 1 (1) | Heat shock protein Hsp-90 | 18855040 | 5.00E−180 | M |
| 36.m00298 | 1 (1) | Lipase | 71420307 | 2.00E−36 | M |
| 19.m00401 | 1 (1) | α-Mannosidase | 66801643 | 3.00E−156 | GO |
| 192.m00071 | 1 (1) | Protein disulfide isomerase-related protein | 56207705 | 1.00E−98 | M |
| 8.m00474 | 1 (1) | Sialidase (neuraminidase) | 135532 | 5.00E−40 | GO |
| 35.m00294 | 1 (1) | Ubiquitin | 1778712 | 9.00E−123 | E |
| 67.m00170 | 1 (1) | Vacuolar ATPase, subunit B | 14971015 | 0 | M, GO |
See Tetrahymena Genome Database (http://www.ciliate.org).
Number of different experimental approaches by which peptides from the protein were identified.
Only matches with expect (E) values of <10−7 were considered significant. The GenBank identification number for the top-scoring hit is given, while the protein name is based on all high-scoring hits.
M and E, homologs are found in the phagosome proteomes of the mouse (30) and Entamoeba histolytica (58), respectively; GO, homologs are localized to lysosomes or lytic vacuoles in the GO database (http://www.godatabase.org/).
The Tetrahymena phagosome proteome contains 61 proteins that produced significant hits in the Blastp analysis as well as 12 proteins that had no strong matches in the GenBank database. The latter group of novel proteins of unknown function are referred to as Tpp (Tetrahymena phagosomal proteome) proteins. The general validity of the list of Tetrahymena proteins identified as phagosome associated is supported by comparisons to the previously analyzed phagosome proteomes of the mouse (30) and Entamoeba histolytica (58). Proteins similar to 25 (34%) of the Tetrahymena phagosome proteins were identified in the phagosome proteomes of the other two organisms (Table 1). In addition, 18 of the Tetrahymena proteins had counterparts that are listed as localized to either the phagosome or the lytic vacuole (the Saccharomyces cerevisiae structure analogous to the phagolysosome) in the Gene Ontology (GO) database (http://www.geneontology.org/).
In addition to the 73 strong candidates, 14 Tetrahymena proteins were identified on the basis of a single peptide in the MS analysis that had counterparts in the mouse or E. histolytica phagosome proteome or the phagosome-related categories in the GO database (Table 1). While there is less experimental support for inclusion of these proteins in the Tetrahymena phagosome proteome per se, the identification of phagocytosis-related homologs in other organisms indicates that these proteins are candidates for phagosome proteins.
While these analyses provide an indication that many bona fide phagosome proteins have been identified in the analysis, there are also indications that the list contains at least a few nonphagosome proteins. For example, SerH3p is a well-characterized major cell surface antigen (20), and histones H2B2 and H4 are known nuclear proteins (50). The identification of these proteins is likely related to their abundance in the cell, as opposed to a novel function in phagocytosis, and their presence underscores the need for further analyses to either localize the identified proteins to phagosomes or obtain evidence for their function in phagocytosis. It should also be emphasized that the current and previously reported phagosome proteomes are almost certainly incomplete. While similar numbers of proteins were identified in the three analyses (mouse, ∼140 proteins [30]; and Entamoeba, 85 proteins [58]) and there is significant overlap between the proteins identified, there are also numerous proteins that were detected in only one of the analyses.
Components of the Tetrahymena phagosome proteome.
The Tetrahymena phagosome proteome includes a number of different classes of proteins involved in phagocytosis. One of the categories is hydrolytic enzymes, such as the cathepsin proteases and acid phosphatases that are components of phagolysosomes (23, 76). Several of the identified Tetrahymena proteins fall in this category, including proteins with strong matches to cathepsins L and B, acid alpha-glucosidase, and acid phosphatases. Also included in this category is lysosomal phospholipase A1, which has previously been shown to be secreted by Tetrahymena (38). Since it is unlikely that the phagosome preparation procedures we employed would have resulted in the recovery of proteins secreted into the culture medium, the detection of this hydrolytic enzyme in our analysis strongly suggests that it is also localized to phagosomes. In addition to these well-characterized components of phagosomes, a number of additional degradative enzymes were also identified, including a chitinase-related protein and an endoglycoceramidase/cellulase-related protein.
Proteins that are likely to be associated with the phagosome membrane and vesicular transport were also identified. These include three subunits of the vacuolar ATPase (V-type H+-ATPase subunits A, a, and d), which is a multisubunit complex that functions to generate and maintain the acidic environment of the phagolysosome (43). In addition, there are a number of proteins that are likely involved in the phagosome maturation process via their roles in vesicular trafficking and membrane fusion. Members of the Rab family of small GTPases (e.g., Rab5 and Rab7) and their effectors function in the sequential fusion and fission of nascent phagosomes with early and late endosomes during phagolysosome biogenesis (24, 33, 69, 72). The Tetrahymena phagosome proteome contains three Rab-like family members with greatest similarity to Rabs 1, 7, and 13, as well as two other putative GTP-binding proteins (Sar1 family and RHD3 family). Also identified was a protein with similarity to a synaptobrevin/longin/VAMP protein, or SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor), which is also involved in intracellular membrane trafficking in eukaryotic cells (66), as well as a member of the vacuolar protein sorting family, VPS13/chorein, that is thought to be involved in vesicle trafficking between the trans-Golgi network and the lysosome (48). There are also two ABC transporter proteins, and members of this protein family have been implicated in phagocytosis in multicellular organisms (see reference 4 and references therein).
VPS13/chorein is also an example of an identified Tetrahymena protein that has a human homologue(s) implicated in a genetic disease (61, 79, 80). Mutations in a member of the human VPS family, VPS13A, result in a range of clinical abnormalities, including peripheral blood acanthocytosis and adult-onset choreic involuntary movement. A second example is the Niemann-Pick C1-like protein, a transmembrane protein thought to be involved in endosome recycling. Niemann-Pick disease is characterized by an accumulation of cholesterol in the endo/lysosomal compartment (40, 42). Thus, further studies of the Tetrahymena proteins may provide new insights into their functions and the molecular bases of some genetic diseases.
Our analyses also identified a number of specific proteins previously implicated in phagocytosis in Tetrahymena. These include the previously mentioned lysosomal acid phosphatase as well as eukaryotic translation elongation factor 1α (EF-1α). In addition to EF-1α's role in translation, there is evidence that it is a calmodulin-binding protein involved in regulating the actin cytoskeleton (36, 62). Gonda et al. (32) demonstrated that in Tetrahymena, both EF-1α and calmodulin localize to the oral apparatus and the deep fiber, both of which are situated at the anterior end of the cell in the region where phagosomes form, and that a reagent that blocks Ca2+/calmodulin binding to its cognate partners inhibits phagosome formation. Finally, a previous sequencing analysis of peptides derived from a small number of Tetrahymena phagosome membrane proteins (49) identified four peptides that are derived from one of the proteins of unknown function (Tpp3p) (Table 1).
Overall, these results provide a clear indication that there are substantial similarities between phagocytosis processes in unicellular eukaryotes and mammals, implying that phagocytosis is an ancient innovation in the eukaryotic lineage. However, there are still likely to be some differences between ciliates and multicellular organisms. For example, lysosome-associated membrane proteins (LAMPs) are components of the phagolysosome in mammalian cells and were found in the mouse phagosome proteome analysis (23, 30) but were not observed in this study. In this instance, the explanation for the absence of these proteins appears to be that Tetrahymena lacks them, as no significant matches to LAMP proteins were found in searches of the Tetrahymena genome. More generally, unicellular eukaryotes appear to lack LAMPs, as searches of the E. histolytica, D. discoideum, and S. cerevisiae genomes failed to detect genes encoding similar proteins. It is unclear whether unicellular eukaryotes utilize alternative proteins to serve the function of LAMPs, which are thought to be involved in targeting of proteins to the lysosome, or whether they are able to dispense with the function of this class of proteins.
Analysis of GFP-tagged putative phagosome proteins.
To provide an initial assessment of protein localization and to determine if contamination is a significant issue, we constructed Tetrahymena strains expressing green fluorescent protein (GFP)-tagged versions of four putative phagosomal proteins, namely, cathepsin B and three of the proteins of unknown function (Tpp2p, Tpp5p, and Tpp9p). The proteins of unknown function were chosen in an essentially random manner, with the caveats that Tpp proteins of <200 amino acids (aa) were excluded and the existence of expressed sequence tags supporting expression of the locus was required. The entire predicted coding region of each of the four genes was cloned into the MTT1-NRK2-GFP vector (8), which contains the cadmium-inducible metallothionein (MTT1) gene promoter and provides a C-terminal GFP tag. In addition, this vector allows for the integration of the constructs into the nonessential β-tubulin 1 (BTU1) locus of Tetrahymena strain CU522. Transformants carrying each of the four GFP-tagged constructs were isolated, grown in the presence of cadmium to induce expression of the fusion constructs, and then fed polystyrene beads to induce phagocytosis. Under the conditions employed, the cells contained a mixture of both bead-containing phagosomes and phagosomes lacking beads (Fig. 3). The transformants showed no major differences in growth rate, morphology, or ability to ingest polystyrene beads in comparison to the parental CU522 cell line. It should be noted that while these results indicate that the GFP-tagged proteins are not toxic to the cells, they do not necessarily indicate that the tagged versions of the proteins function normally, as the endogenous, untagged loci are also present in the transformants.
FIG. 3.

Localization of GFP-tagged constructs in live Tetrahymena cells. Pairs of differential interference contrast (DIC) and fluorescence (FLUOR) confocal microscopy images are shown. (A) Control parental CU522 cell line. (B) GFP-tagged cathepsin B transformant cell line. (C and D) GFP-tagged Tpp2p transformant cell line (different confocal image sections of the same cell are shown in panels C and D). (E and F) GFP-tagged Tpp9p transformant cell line fed beads for 15 min and 2 h, respectively. (G) GFP-tagged Tpp5p transformant cell line. Images are oriented with the cell anterior directed toward the upper left corner of the image. Long arrows, bead-containing phagosomes; short arrows, phagosomes without beads; large arrowheads, contractile vacuoles (CV). MA, macronucleus. All images were obtained under the same microscopy conditions and were uniformly processed.
The cathepsin B protein (Tetrahymena gene 6.m00369) was chosen for analysis because this cysteine protease is a well-characterized component of phagosomes in other organisms. As one would expect, transformants expressing the cathepsin B::GFP fusion protein showed a fluorescent signal in the lumens of phagosomes (Fig. 3B). The phagosomes in the untransformed parental CU522 strain displayed a low level of autofluorescence (Fig. 3A), but the signals from the phagosomes of the CATHB::GFP strain were well above this low background level. Thus, as in other systems, this protein localizes to the lumens of phagosomes.
The TPP2 gene (130.m00077) is predicted to encode a 247-aa polypeptide with no strong matches in the protein database, but it does contain a likely signal sequence and a possible membrane-spanning domain near its C terminus. In the TPP2::GFP transformant, a fluorescent signal was observed in the spongiome (Fig. 3C), which is a membranous component of the contractile vacuole that is involved in osmoregulation (26). This localization was observed in both cells undergoing phagocytosis and cells grown under conditions where little phagocytosis occurred (data not shown). However, in cells with phagosomes, additional patches of GFP fluorescence were observed in association with the edges of phagosomes (Fig. 3D). Such a dual localization is not unprecedented, as the VatM subunit of the vacuolar H+-ATPase involved in phagosome acidification also localizes to the contractile vacuole in Dictyostelium discoideum (14). Our results with Tpp2p suggest that there are additional proteins shared between these two organelles. It is possible that Tpp2p is independently targeted to both the contractile vacuole and phagosomes, as appears to be the case for Dictyostelium VatM (14). An intriguing alternative is that there is some form of vesicular transport that links the two organelles, and this possibility merits further investigation.
The TPP9 gene (159.m00051) encodes a predicted protein of 202 aa with no discernible features. In the transformant expressing the TPP9::GFP fusion construct and fed beads for 15 min, fluorescence was observed primarily in patches that were often associated with the peripheries of phagosomes (Fig. 3E). Weaker fluorescence was also seen around the nucleus and the lumen of the contractile vacuole. When cells were examined 2 h after incubation with beads, many phagosomes displayed fluorescence more evenly around their borders, and puncta of more intense fluorescence were seen near the anterior end of the cell (Fig. 3F). While a definitive interpretation of the results for Tpp9p is not possible at this point, the results suggest that this protein might interact transiently with phagosomes and then be recycled to the oral apparatus of the cell so that it may again interact with nascent phagosomes.
The fourth gene analyzed, TPP5 (129.m00110), is predicted to encode a 311-aa protein. Tpp5p has no discernible functional features, but its sequence contains six repeats of a 42-aa sequence. There are two additional loci in the Tetrahymena genome that encode similar proteins, as well as two similar genes in the ciliate Paramecium tetraurelia, but homologs were not found in other organisms. The TPP5::GFP transformant displayed fluorescence throughout the cytoplasm and nucleus, but no signal was observed within phagosomes (Fig. 3G). The results, at face value, provide little evidence for any specific phagosome association, suggesting that Tpp5p represents a contaminating species isolated during the analysis. However, we cannot completely rule out the possibility that Tpp5p is phagosome associated, as the GFP fusion protein is likely overexpressed in our experimental system, and the presence of the GFP tag might result in mislocalization.
Overall, the GFP tagging results support or suggest a phagosome association for at least three of the four proteins analyzed. The results are consistent with our database searches and inspection of the list of proteins in the proteome; that is, many true phagosome-associated proteins are present, but a subset was likely derived from contaminating cellular structures in our phagosome preparation. This is a typical problem in proteomic analysis, but the genetic tools available for Tetrahymena, which include not only GFP tagging but also the ability to generate targeted gene knockouts (78), provide a means of further assessing the localization and function of candidate phagosome proteins. Indeed, the generation of the cathepsin B::GFP fusion construct provides a tool for analyzing mutations in other genes encoding candidate phagosome proteins, as it provides a marker for one step in the phagocytosis process, i.e., the delivery of degradative enzymes via lysosome fusion. The development of similar constructs to identify other steps in the pathway (e.g., use of one of the identified vacuolar H+-ATPase subunits to mark phagosome acidification) will allow a more detailed dissection of the effects of mutations in novel genes on phagocytosis. Coupled with the ability to knock out and modify genes, the current analysis of the Tetrahymena phagosome proteome offers the possibility of not only identifying new constituents of the phagocytic machinery but also investigating details of protein-protein interactions and the molecular mechanism underlying the process.
Acknowledgments
We thank Aaron Turkewitz and C. David Allis for providing cell lines and antibodies, Susan Krueger and Ann Cowan for assistance with microscopy, and Jacek Gaertig for providing GFP vectors.
This work was supported by National Science Foundation grant MCB-0343813 to L.A.K., by a Canadian Institutes for Health Research (CIHR) grant to R.E.P., and by a Natural Sciences and Engineering Research Council of Canada Collaborative Research and Development grant, with Eli Lilly Canada and MDS SCIEX as the industrial partners, to K.W.M.S. and R.E.P. Hardware support from the Ontario Research and Development Challenge Funds, Genome Canada, and Applied Biosystems/MDS SCIEX to K.W.M.S. is gratefully acknowledged.
Footnotes
Published ahead of print on 29 September 2006.
REFERENCES
- 1.Aderem, A., and D. M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593-623. [DOI] [PubMed] [Google Scholar]
- 2.Allen, R. D., and R. W. Wolf. 1979. Membrane recycling at the cytoproct of Tetrahymena. J. Cell Sci. 35:217-227. [DOI] [PubMed] [Google Scholar]
- 3.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bared, S. M., C. Buechler, A. Boettcher, R. Dayoub, A. Sigruener, M. Grandl, C. Rudolph, A. Dada, and G. Schmitz. 2004. Association of ABCA1 with syntaxin 13 and flotillin-1 and enhanced phagocytosis in tangier cells. Mol. Biol. Cell 15:5399-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batz, W., and F. Wunderlich. 1976. Structural transformation of the phagosomal membrane in Tetrahymena cells endocytosing latex beads. Arch. Microbiol. 109:215-250. [DOI] [PubMed] [Google Scholar]
- 6.Blocker, A., F. F. Severin, J. K. Burkhardt, J. B. Bingham, H. Yu, J.-C. Olivo, T. A. Schroer, A. A. Hyman, and G. Griffiths. 1997. Molecular requirements for bi-directional movement of phagosomes along microtubules. J. Cell Biol. 137:113-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bokoch, G. M. 2005. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 15:163-171. [DOI] [PubMed] [Google Scholar]
- 8.Boldrin, F., G. Santovito, J. Gaertig, D. Wloga, D. Cassidy-Hanley, T. Clark, and E. Piccinni. 2006. Metallothionein gene from Tetrahymena thermophila with a copper-inducible-repressible promoter. Eukaryot. Cell 5:422-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowman, G., N. C. Elde, G. Morgan, M. Winey, and A. P. Turkewitz. 2005. Core formation and the acquisition of fusion competence are linked during secretory granule maturation in Tetrahymena. Traffic 6:303-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowman, G. R., D. G. Smith, K. W. M. Siu, R. E. Pearlman, and A. P. Turkewitz. 2005. Genomic and proteomic evidence for a second family of dense core granule cargo proteins in Tetrahymena thermophila. J. Eukaryot. Microbiol. 52:291-297. [DOI] [PubMed] [Google Scholar]
- 11.Bruns, P. J., and D. Cassidy-Hanley. 2000. Biolistic transformation of macro- and micronuclei. Methods Cell Biol. 62:501-512. [DOI] [PubMed] [Google Scholar]
- 12.Chicoine, L. G., D. Wenkert, R. Richman, C. James, J. C. Wiggins, and C. D. Allis. 1985. Modulation of linker histones during development in Tetrahymena: selective elimination of linker histone during the differentiation of new macronuclei. Dev. Biol. 109:1-8. [DOI] [PubMed] [Google Scholar]
- 13.Chilcoat, N. D., N. C. Elde, and A. P. Turkewitz. 2001. An antisense approach to phenotype-based gene cloning in Tetrahymena. Proc. Natl. Acad. Sci. USA 98:8709-8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke, M., J. Koehler, Q. Arana, T. Liu, J. Heuser, and G. Gerisch. 2002. Dynamics of the vacuolar H(+)-ATPase in the contractile vacuole complex and the endosomal pathway of Dictyostelium cells. J. Cell Sci. 115:2893-2905. [DOI] [PubMed] [Google Scholar]
- 15.Collins, R. F., A. D. Schreiber, S. Grinstein, and W. S. Trimble. 2002. Syntaxins 13 and 7 function at distinct steps during phagocytosis. J. Immunol. 169:3250-3256. [DOI] [PubMed] [Google Scholar]
- 16.Desjardins, M. 1995. Biogenesis of phagolysosomes: the ‘kiss and run’ hypothesis. Trends Cell Biol. 5:183-186. [DOI] [PubMed] [Google Scholar]
- 17.Desjardins, M., and G. Griffiths. 2003. Phagocytosis: latex leads the way. Curr. Opin. Cell Biol. 15:498-503. [DOI] [PubMed] [Google Scholar]
- 18.Desjardins, M., M. Houde, and E. Gagnon. 2005. Phagocytosis: the convoluted way from nutrition to adaptive immunity. Immunol. Rev. 207:158-165. [DOI] [PubMed] [Google Scholar]
- 19.Desjardins, M., L. A. Huber, R. G. Parton, and G. Griffiths. 1994. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124:677-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doerder, F. P., and R. L. Hallberg. 1989. Identification of a cDNA coding for the SerH3 surface protein of Tetrahymena thermophila. J. Protozool. 36:304-307. [DOI] [PubMed] [Google Scholar]
- 21.Doudna, J. A., and V. L. Rath. 2002. Structure and function of the eukaryotic ribosome: the next frontier. Cell 109:153-156. [DOI] [PubMed] [Google Scholar]
- 22.Eisen, J. A., M. Wu, D. Wu, M. Thiagarajan, J. R. Wortman, J. H. Badger, Q. Ren, P. Amedeo, K. M. Jones, L. J. Tallon, A. L. Delcher, S. L. Salzberg, C. delToro, H. F. Rider, S. C. Williamson, R. A. Barbeau, J. C. Silva, B. J. Haas, W. H. Majoros, M. Farzad, J. M. Carlton, J. Garg, R. E. Pearlman, K. M. Karrer, L. Sun, R. K. Smith, Jr., G. Manning, N. C. Elde, A. P. Turkewitz, D. J. Asai, D. E. Wilkes, Y. Wang, H. Cai, K. Collins, K. Wilamowska, W. L. Ruzzo, Z. Weinberg, B. A. Stewart, S. R. Lee, D. Wloga, K. Rogowski, J. Frankel, J. Gaertig, C.-C. Tsao, M. A. Gorovsky, P. J. Keeling, R. F. Waller, N. J. Patron, M. Cherry, N. A. Stover, C. A. Krieger, E. P. Hamilton, E. Orias, and R. S. Coyne. 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 4:e286. doi: 10.1371/journal.pbio.0040286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eskelinen, E.-L., Y. Tanaka, and P. Saftig. 2003. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13:137-145. [DOI] [PubMed] [Google Scholar]
- 24.Feng, Y., B. Press, and A. Wandinger-Ness. 1995. Rab 7: an important regulator of late endocytic membrane traffic. J. Cell Biol. 13:1435-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foster, T. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948-958. [DOI] [PubMed] [Google Scholar]
- 26.Frankel, J. 2000. Cell biology of Tetrahymena thermophila. Methods Cell Biol. 62:27-125. [DOI] [PubMed] [Google Scholar]
- 27.Gaertig, J., Y. Gao, T. Tishgarten, T. G. Clark, and H. W. Dickerson. 1999. Surface display of a parasite antigen in the ciliate Tetrahymena thermophila. Nat. Biotechnol. 17:462-465. [DOI] [PubMed] [Google Scholar]
- 28.Gaertig, J., and G. Kapler. 2000. Transient and stable DNA transformation of Tetrahymena thermophila by electroporation. Methods Cell Biol. 62:485-500. [DOI] [PubMed] [Google Scholar]
- 29.Gaertig, J., T. H. Thatcher, L. Gu, and M. A. Gorovsky. 1994. Electroporation-mediated replacement of a positively and negatively selectable beta-tubulin gene in Tetrahymena thermophila. Proc. Natl. Acad. Sci. USA 91:4549-4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garin, J., R. Diez, S. Kieffer, J.-F. Dermine, S. Duclos, E. Gagnon, R. Sadoul, C. Rondeau, and M. Desjardins. 2001. The phagosome proteome: insight into phagosome functions. J. Cell Biol. 152:165-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonda, K., M. Katoh, K. Hanyu, Y. Watanabe, and O. Numata. 1999. Ca2+/calmodulin and p85 cooperatively regulate an initiation of cytokinesis in Tetrahymena. J. Cell Sci. 112:3619-3626. [DOI] [PubMed] [Google Scholar]
- 32.Gonda, K., M. Komatsu, and O. Numata. 2000. Calmodulin and Ca2+/calmodulin-binding proteins are involved in Tetrahymena thermophila phagocytosis. Cell Struct. Funct. 25:243-251. [DOI] [PubMed] [Google Scholar]
- 33.Gorvel, J.-P., P. Chavrier, M. Zerial, and J. Gruenberg. 1991. Rab5 controls early endosome fusion in vitro. Cell 64:915-925. [DOI] [PubMed] [Google Scholar]
- 34.Gotthardt, D., H. J. Warnatz, O. Henschel, F. Bruckert, M. Schleicher, and T. Soldati. 2002. High-resolution dissection of phagosome maturation reveals distinct membrane trafficking phases. Mol. Biol. Cell 13:3508-3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregory, D. J., and M. Olivier. 2005. Subversion of host cell signalling by the protozoan parasite Leishmania. Parasitology 130:S27-S35. [DOI] [PubMed] [Google Scholar]
- 36.Gross, S. R., and T. G. Kinzy. 2005. Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat. Struct. Mol. Biol. 12:772-778. [DOI] [PubMed] [Google Scholar]
- 37.Harrison, R. E., C. Bucci, O. V. Vieira, T. A. Schroer, and S. Grinstein. 2003. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol. Cell. Biol. 23:6494-6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartmann, M., A. Guberman, M. Florin-Christensen, and A. Tiedtke. 2000. Screening for and characterization of phospholipase A1 hypersecretory mutants of Tetrahymena thermophila. Appl. Microbiol. Biotechnol. 54:390-396. [DOI] [PubMed] [Google Scholar]
- 39.Hosein, R. E., S. A. Williams, and R. H. Gavin. 2005. Directed motility of phagosomes in Tetrahymena thermophila requires actin and Myo1p, a novel unconventional myosin. Cell Motil. Cytoskelet. 61:49-60. [DOI] [PubMed] [Google Scholar]
- 40.Ikonen, E., and M. Hölttä-Vuori. 2004. Cellular pathology of Niemann-Pick type C disease. Semin. Cell Dev. Biol. 15:445-454. [DOI] [PubMed] [Google Scholar]
- 41.Jacobs, M. E., D. E. Cortezzo, and L. A. Klobutcher. 2004. Assessing the effectiveness of coding and non-coding regions in antisense ribosome inhibition of gene expression in Tetrahymena. J. Eukaryot. Microbiol. 51:536-541. [DOI] [PubMed] [Google Scholar]
- 42.Karten, B., R. B. Campenot, D. E. Vance, and J. E. Vance. 2006. The Niemann-Pick C1 protein in recycling endosomes of pre-synaptic nerve terminals. J. Lipid Res. 47:504-514. [DOI] [PubMed] [Google Scholar]
- 43.Kawasaki-Nishi, S., T. Nishi, and M. Forgac. 2003. Proton translocation driven by ATP hydrolysis in V-ATPases. FEBS Lett. 545:76-85. [DOI] [PubMed] [Google Scholar]
- 44.Kitajima, Y., and J. G. A. Thompson. 1977. Differentiation of food vacuolar membranes during endocytosis in Tetrahymena. J. Cell Biol. 75:436-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kjeken, R., M. Egeberg, A. Haberman, M. Kuehnel, P. Peyron, M. Floetenmeyer, P. Walther, A. Jahraus, H. Defacque, S. A. Kuznetsov, and G. Griffiths. 2004. Fusion between phagosomes, early and late endosomes: a role for actin in fusion between late, but not early endocytic organelles. Mol. Biol. Cell 15:345-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klobutcher, L. A., K. Ragkousi, and P. Setlow. 2006. The Bacillus subtilis spore coat provides “eat resistance” during phagocytic predation by the protozoan Tetrahymena thermophila. Proc. Natl. Acad. Sci. USA 103:165-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee, S., J. C. Wisniewski, W. L. Dentler, and D. J. Asai. 1999. Gene knockouts reveal separate functions for two cytoplasmic dyneins in Tetrahymena thermophila. Mol. Biol. Cell 10:771-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lemmon, S. K., and L. M. Traub. 2000. Sorting in the endosomal system in yeast and animal cells. Curr. Opin. Cell Biol. 12:457-466. [DOI] [PubMed] [Google Scholar]
- 49.Maicher, M. T., and A. Tiedtke. 1999. Biochemical analysis of membrane proteins from an early maturation stage of phagosomes. Electrophoresis 20:1011-1016. [DOI] [PubMed] [Google Scholar]
- 50.Medzihradszky, K. F., X. Zhang, R. J. Chalkley, S. Guan, M. A. McFarland, M. J. Chalmers, A. G. Marshall, R. L. Diaz, C. D. Allis, and A. L. Burlingame. 2004. Characterization of Tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS). Mol. Cell. Proteomics 3:872-886. [DOI] [PubMed] [Google Scholar]
- 51.Melia, S. M., E. S. Cole, and A. P. Turkewitz. 1998. Mutational analysis of regulated exocytosis in Tetrahymena. J. Cell Sci. 111:131-140. [DOI] [PubMed] [Google Scholar]
- 52.Meyer, M., T. Mayer, and A. Tiedtke. 1998. Maturation of phagosomes is accompanied by specific patterns of small GTPases. Electrophoresis 19:2528-2535. [DOI] [PubMed] [Google Scholar]
- 53.Müller-Taubenberger, A., A. N. Lupas, H. Li, M. Ecke, E. Simmeth, and G. Gerisch. 2001. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 20:6772-6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nilsson, J. R. 1977. On food vacuoles in Tetrahymena pyriformis GL. J. Protozool. 24:502-507. [Google Scholar]
- 55.Nilsson, J. R. 1987. Structural aspects of digestion of Escherichia coli in Tetrahymena. J. Protozool. 34:1-6. [Google Scholar]
- 56.Nimmerjahn, F., and J. V. Ravetch. 2006. Fcγ receptors: old friends and new family members. Immunity 24:19-28. [DOI] [PubMed] [Google Scholar]
- 57.Ogden, C., and K. Elkon. 2006. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr. Dir. Autoimmun. 9:120-142. [DOI] [PubMed] [Google Scholar]
- 58.Okada, M., C. D. Huston, B. J. Mann, W. A. J. Petri, K. Kita, and T. Nozaki. 2005. Proteomic analysis of phagocytosis in the enteric protozoan parasite Entamoeba histolytica. Eukaryot. Cell 4:827-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okada, M., and T. Nozaki. 2006. New insights into molecular mechanisms of phagocytosis in Entamoeba histolytica by proteomic analysis. Arch. Med. Res. 37:244-252. [DOI] [PubMed] [Google Scholar]
- 60.Quan, L., and M. Wilkinson. 1991. DNA fragment purification from LMP agarose. BioTechniques 10:737-738. [PubMed] [Google Scholar]
- 61.Rampoldi, L., C. Dobson-Stone, J. P. Rubio, A. Danek, R. M. Chalmers, N. W. Wood, C. Verellen, X. Ferrer, A. Malandrini, G. M. Fabrizi, R. Brown, J. Vance, M. Pericak-Vance, G. Rudolf, S. Carrè, E. Alonso, M. Manfredi, A. H. Németh, and A. P. Monaco. 2001. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 28:119-120. [DOI] [PubMed] [Google Scholar]
- 62.Rasmussen, C., and C. Wiebe. 1999. Cloning of a Schizosaccharomyces pombe homologue of elongation factor 1 alpha by two-hybrid selection of calmodulin-binding proteins. Biochem. Cell Biol. 77:421-430. [PubMed] [Google Scholar]
- 63.Rasmussen, L., and E. Orias. 1975. Tetrahymena: growth without phagocytosis. Science 190:464-465. [DOI] [PubMed] [Google Scholar]
- 64.Rezabek, B. L., J. M. Rodriguez-Paris, J. A. Cardelli, and C. P. Chia. 1997. Phagosomal proteins of Dictyostelium discoideum. J. Eukaryot. Microbiol. 44:284-292. [DOI] [PubMed] [Google Scholar]
- 65.Rosenberger, C. M., and B. B. Finlay. 2003. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 4:385-396. [DOI] [PubMed] [Google Scholar]
- 66.Rossi, V., D. K. Banfield, M. Vacca, L. E. P. Dietrich, C. Ungermann, M. D'Esposito, T. Galli, and F. Filippini. 2004. Longins and their longin domains: regulated SNAREs and multifunctional SNARE regulators. Trends Biochem. Sci. 29:682-688. [DOI] [PubMed] [Google Scholar]
- 67.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 68.Schirle, M., M. A. Heurtier, and B. Kuster. 2003. Profiling core proteomes of human cell lines by one-dimensional PAGE and liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 12:1297-1305. [DOI] [PubMed] [Google Scholar]
- 69.Scott, C. C., R. J. Botelho, and S. Grinstein. 2003. Phagosome maturation: a few bugs in the system. J. Membr. Biol. 193:137-152. [DOI] [PubMed] [Google Scholar]
- 70.Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68:850-858. [DOI] [PubMed] [Google Scholar]
- 71.Smith, J. C., J. G. Northey, J. Garg, R. E. Pearlman, and K. W. M. Siu. 2005. Robust method for proteome analysis by MS/MS using an entire translated genome: demonstration on the ciliome of Tetrahymena thermophila. J. Proteome Res. 4:909-919. [DOI] [PubMed] [Google Scholar]
- 72.Stenmark, H., and V. M. Olkkonen. 2001. The Rab GTPase family. Genome Biol. 2:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stover, N. A., C. J. Krieger, G. Binkley, Q. Dong, D. G. Fisk, R. Nash, A. Sethuraman, S. Weng, and J. M. Cherry. 2006. Tetrahymena Genome Database (TGD): a new genomic resource for Tetrahymena thermophila research. Nucleic Acids Res. 34:D500-D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tirumalai, R. S., K. C. Chan, D. A. Prieto, H. J. Issaq, T. P. Conrads, and T. D. Veenstra. 2003. Characterization of the low molecular weight human serum proteome. Mol. Cell. Proteomics 2:1096-1103. [DOI] [PubMed] [Google Scholar]
- 75.Tse, S. M., W. Furuya, E. Gold, A. D. Schreiber, K. Sandvig, R. D. Inman, and S. Grinstein. 2003. Differential role of actin, clathrin, and dynamin in Fcγ receptor-mediated endocytosis and phagocytosis. J. Biol. Chem. 278:3331-3338. [DOI] [PubMed] [Google Scholar]
- 76.Turk, V., B. Turk, and D. Turk. 2001. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 20:4629-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turkewitz, A. P. 2004. Out with a bang! Tetrahymena as a model system to study secretory granule biogenesis. Traffic 5:63-68. [DOI] [PubMed] [Google Scholar]
- 78.Turkewitz, A. P., E. Orias, and G. Kapler. 2002. Functional genomics: the coming of age for Tetrahymena thermophila. Trends Genet. 18:35-40. [DOI] [PubMed] [Google Scholar]
- 79.Ueno, S.-I., Y. Maruki, M. Nakamura, Y. Tomemori, K. Kamae, H. Tanabe, Y. Yamashita, S. Matsuda, S. Kaneko, and A. Sano. 2001. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 28:121-122. [DOI] [PubMed] [Google Scholar]
- 80.Velayos-Baeza, A., A. Vettori, R. R. Copley, C. Dobson-Stone, and A. P. Monaco. 2004. Analysis of the human VPS13 gene family. Genomics 84:536-549. [DOI] [PubMed] [Google Scholar]
- 81.Vergne, I., J. Chua, S. B. Singh, and V. Deretic. 2004. Cell biology of Mycobacterium tuberculosis phagosome. Annu. Rev. Cell Dev. Biol. 20:367-394. [DOI] [PubMed] [Google Scholar]
- 82.Vieira, O. V., R. J. Botelho, and S. Grinstein. 2002. Phagosome maturation: aging gracefully. Biochem. J. 366:689-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vosskuehler, C., and A. Tiedtke. 1993. Magnetic separation of phagosomes of defined age from Tetrahymena thermophila. J. Eukaryot. Microbiol. 40:556-562. [Google Scholar]
- 84.Weidenbach, A. L., and G. A. Thompson, Jr. 1974. Studies of membrane formation in Tetrahymena pyriformis. VIII. On the origin of membranes surrounding food vacuoles. J. Protozool. 21:745-751. [DOI] [PubMed] [Google Scholar]
- 85.Williams, S. A., R. E. Hosein, J. A. Garces, and R. H. Gavin. 2000. MYO1, a novel, unconventional myosin gene affects endocytosis and macronuclear elongation in Tetrahymena thermophila. J. Eukaryot. Microbiol. 47:561-568. [DOI] [PubMed] [Google Scholar]
- 86.Wolters, D. A., M. P. Washburn, and J. R. Yates III. 2001. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 73:5683-5690. [DOI] [PubMed] [Google Scholar]
- 87.Yan, M., R. F. Collins, S. Grinstein, and W. S. Trimble. 2005. Coronin-1 function is required for phagosome formation. Mol. Biol. Cell 16:3077-3087. [DOI] [PMC free article] [PubMed] [Google Scholar]