

Abstract
Cholesterol-rich microdomains (or “lipid rafts”) within the plasma membrane have been hypothesized to exist in a liquid-ordered phase and play functionally important roles in cell signaling; however, these microdomains defy detection using conventional imaging. To visualize domains and relate their nanostructure and dynamics to mast cell signaling, we use two-photon (760 nm and 960 nm) fluorescence lifetime imaging microscopy and fluorescence polarization anisotropy imaging, with comparative one-photon anisotropy imaging and single-point lifetime and anisotropy decay measurements. The inherent sensitivity of ultrafast excited-state dynamics and rotational diffusion to the immediate surroundings of a fluorophore allows for real-time monitoring of membrane structure and organization. When the high affinity receptor for IgE (FcɛRI) is extensively cross-linked with anti-IgE, molecules associated with cholesterol-rich microdomains (e.g., saturated lipids (the lipid analog diI-C18 or glycosphingolipids)) and lipid-anchored proteins coredistribute with cross-linked IgE-FcɛRI. We find an enhancement in fluorescence lifetime and anisotropy of diI-C18 and Alexa 488-labeled IgE-FcɛRI in the domains where these molecules colocalize. Our results suggest that fluorescence lifetime and, particularly, anisotropy permit us to correlate the recruitment of lipid molecules into more ordered domains that serve as platforms for IgE-mediated signaling.
INTRODUCTION
Cholesterol-rich microdomains, or “lipid rafts”, within the plasma membrane have been implicated in essential cell functions such as signaling and membrane trafficking (1–3) and are proposed to exist in a liquid-ordered phase that is enriched in saturated phospholipids and sphingolipids (3). However, noncaveolar cholesterol domains have not been directly observed in vivo without substantial perturbation (e.g., long-term incubation in the cold or extensive cross-linking of cholesterol-rich microdomain markers) (4–7); thus, much controversy about their size, shape, lifetime, and molecular composition remains. In model membrane systems such as giant unilamellar vesicles (8,9) and supported lipid bilayers and monolayers (10–13), the liquid-ordered phase has been widely visualized and characterized, but these models lack the full complexity and reactivity associated with the plasma membrane of living cells.
A variety of spectroscopic and microscopic techniques have been used to investigate the structure and dynamics of lipid microdomains in both model and cellular systems. Spectrofluorimetry (14), single-point dynamics with a temporal resolution of approximately tens of nanoseconds (15), electron spin resonance (16,17), and NMR (18) spectroscopies have provided valuable insights into the structural order of small and giant unilamellar vesicles and isolated detergent-resistant membranes (DRMs) that have been extracted from various cell types. Nevertheless, these ensemble measurements offer either no information on site-specific dynamics or no spatio-temporal information at the single-cell level. For single cell experiments, fluorescence recovery after photobleaching (FRAP) (5), fluorescence correlation spectroscopy (FCS) (19), and single-particle tracking (SPT) (20,21) have been used to analyze the biophysical properties of lipid microdomains. These translational diffusion-based techniques are limited by optical diffraction and ensemble averaging (FRAP and FCS) or by the valency of the probe (SPT), as well as by the timescale (microseconds to seconds) associated with translational diffusion. In contrast, fluorescence lifetime and rotational diffusion approaches are exquisitely sensitive to molecular structure (i.e., organization and conformation) and rigidity of the surroundings (22,23) and may provide insight into the short-range dynamics (picoseconds to nanoseconds timescale) and molecular order of the membrane nanostructure. This molecular-level environmental sensitivity provides an opportunity to bypass the diffraction limit of the intensity-dependent measurements typically used to characterize cholesterol-rich lipid domains in vivo.
Here, we investigate specialized cholesterol-rich domains in individual RBL-2H3 mast cells and probe the influence of cross-linking induced changes on membrane nanostructure, and therefore biological function. Our integrated biophotonics techniques include confocal and two-photon fluorescence microscopies, two-photon fluorescence lifetime imaging (2P-FLIM), and fluorescence polarization anisotropy, with complementary one-photon measurements. When the high-affinity immunoreceptor for IgE (FcɛRI) on mast cells and basophils is cross-linked with multivalent antigen, it is phosphorylated by the Src family tyrosine kinase, Lyn, in a cholesterol-dependent manner (7,24). We are able to visualize large-scale domains of IgE-FcɛRI when these complexes are cross-linked with secondary antibody (anti-IgE, α-IgE) in the cold (5–7). Although this extensive cross-linking represents a gross, nonphysiological perturbation, it permits a clear comparison of cells in the presence or absence of cross-linking and ease of identification of regions of interest for single-point measurements. Thus, these conditions establish a fundamental basis for more physiological studies, while allowing for a correlation between dynamic membrane nanostructure and the location of specific signaling molecules (i.e., IgE-FcɛRI).
MATERIALS AND METHODS
Cell preparation
RBL-2H3 mast cells were maintained and harvested as previously described (7,25). Cells (3 × 106 cells/mL) suspended in bovine serum albumin (BSA)-containing buffered saline solution (BSA/BSS: 20 mM HEPES pH 7.4, 135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, 1 mg/mL BSA) were sensitized with a 10-fold molar excess of mouse anti-2,4-dinitrophenyl IgE (provided by Dr. David Holowka, Cornell University). IgE was used either unlabeled or conjugated to AlexaFluor 488 (A488-IgE) via a commercially available protein labeling kit (Invitrogen, Eugene, OR), which labels the monomeric IgE at exposed primary amines (the dye/IgE ratio was 5.5 as determined by absorption spectroscopy). Cells were labeled with a 50-fold dilution of 0.2 mg/mL 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (diI-C18, Invitrogen) stock solution prepared in dimethylsulfoxide (DMSO). For this protocol, the dye/lipid ratio in cells was previously estimated to be ∼1:220 (or 0.46%) (5). RBL cells were incubated (2 h, 4°C) either in the presence or absence of 10 μg/mL polyclonal rabbit anti-IgE (α-IgE) (provided by Dr. Holowka), as described previously (5), to form extensive patches of IgE-FcɛRI (“cross-linked” cells), or not (“uncross-linked” or “monomeric” cells). Before imaging, 25 μL of cell suspension was pipetted onto a 22 mm × 22 mm glass cover slip, covered with a 3” × 1” glass slide, sealed with VALAP (Vaseline/lanolin/paraffin (2:1:1, wt/wt)), and incubated for 10–15 min. at room temperature (∼20°C) for immobilization. Cells dually labeled with diI-C18 and A488-IgE were used for steady-state confocal imaging. For all other experiments, cells were fluorescently labeled with either diI-C18 (in the presence of unlabeled IgE for possible cross-linking) or A488-IgE. For solution experiments, A488-IgE was diluted in phosphate-buffered saline (PBS, 10 mM Na2HPO4, pH 7.4, 150 mM NaCl) and diI-C18 in DMSO.
Fluorescence microscopy and ultrafast spectroscopy
The experimental setup and data analysis will be described in more detail elsewhere (26). Briefly, we describe the different modalities of fluorescence microscopy and ultrafast spectroscopy used in these studies.
Confocal microscopy
Three-channel detection confocal microscopy, consisting of a fiber-coupled laser system, a scanner (FluoView300, Olympus, Melville, NY), differential interference contrast (DIC) optics, an inverted microscope (IX81, Olympus), and an Olympus 60×, 1.2 numerical aperture (NA) water-immersion objective, was used in these studies. The scanner was also modified to allow for 2P laser scanning required for 2P-FLIM applications. A488-IgE was imaged using 488 nm argon ion laser excitation and a 525/30 bandpass filter, while diI-C18 was imaged with 543 nm HeNe laser excitation and a 605/70 bandpass filter. DIC imaging was used to monitor cell viability before and after all experiments to ensure that no photodamage had occurred. Unlabeled RBL cells showed negligible autofluorescence (data not shown). Imaging and spectroscopy experiments were carried out at room temperature (∼20°C).
2P-fluorescence lifetime imaging and one-photon time-resolved lifetime
For 2P-fluorescence and lifetime imaging, a femtosecond laser system (∼120 fs, 76 MHz) was used, which consists of a solid-state Ti:sapphire laser (Mira 900F, Coherent, Santa Clara, CA) that is pumped with a 10 W diode laser (Verdi, Coherent) and provides 700–1000 nm excitation. The laser beam was conditioned and steered toward the laser scanner, before being focused on the sample with a 60×, 1.2 NA water-immersion objective. The pulse power at the sample ranged from ∼0.5–5 mW (i.e., energy per pulse ∼10–100 pJ). The two-photon, epifluorescence signal was detected via an exit port of the microscope using a dichroic mirror. For two-channel imaging, the fluorescence signal was split (50/50) into two detection channels, filtered (BGG22, Chroma, Rockingham, VT) to isolate further scattered laser light, polarization-analyzed using Glan-Thompson polarizers, and then detected using microchannel plates (R3809U-50, Hamamatsu, Hamamatsu City, Japan). For lifetime imaging, only one of the channels was used and the polarizer was set at the magic angle (54.7°) with respect to the laser polarization. The signal was amplified and fluorescence lifetime images were constructed using a time-correlated single-photon counting module (SPC830, Becker & Hickl, Berlin, Germany). The module was synchronized using a fast photodiode, which was triggered with ∼5% of the laser pulses. For 2P-FLIM of RBL cells, both A488-IgE and diI-C18 were excited at 760 nm, based on the 2P excitation spectra of these dyes (data not shown and (27)). At this wavelength, autofluorescence from unlabeled RBL cells was negligible. To further eliminate the possibility of autofluorescence, we also conducted additional 2P-FLIM experiments at 960 nm excitation to avoid intrinsic autofluorescence from NADH and FAD, and comparable results for both A488-IgE and diI-C18 were obtained (Supplemental Fig. 1). 2P-FLIM images were recorded using 256 × 256 pixels with 65 time bins per pixel (i.e., 259 ps/bin) at both the equatorial plane and the top surface of cells. Under these conditions, a typical FLIM image was recorded within 120 s, without photodamage or cellular movement. Experiments at 760 nm were performed over several days on different cell preparations that had (n = 12 cells for A488-IgE, 6 cells for diI-C18) or had not (n = 12 cells for A488-IgE, 12 cells for diI-C18) been incubated with α-IgE.
The fluorescence lifetime of a given fluorophore provides valuable information about its molecular structure and the surrounding local environment (23,28). Generally, the time-resolved fluorescence decay of a given fluorophore at pixel (x,y) measured at the magic angle (i.e., without rotational effects) can be best described as a sum of exponentials with time constants (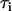 ) and amplitudes (
) and amplitudes (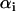 ) as follows (26):
) as follows (26):
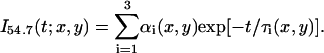 |
(1) |
These fluorescence decays were deconvoluted with the system response function (full-width at half-maximum, FWHM ∼60 ps) and the average fluorescence lifetime was calculated as 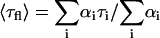 . The fluorescence quantum yield (
. The fluorescence quantum yield (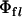 ) and the excited-state fluorescence decay rate (
) and the excited-state fluorescence decay rate (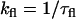 ) of a given fluorophore depend on the radiative (
) of a given fluorophore depend on the radiative (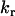 ) and nonradiative (
) and nonradiative (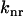 ) rates following
) rates following 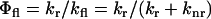 . Considering the complexity of cellular environments, various nonradiative pathways are likely to compete with fluorescence emission of a given fluorophore. For example, when a fluorophore is incorporated into a lipid membrane, its excited-state lifetime is affected by the overall organization or order of the membrane (29), the compositional diversity of the lipids (heterogeneity), and the solvent-fluorophore interactions. Isomerization, which explains the enhanced fluorescence properties of diI-C18 upon restriction in an organic phase (i.e., the membrane), is a primary example of such a pathway. As a result, the nature of the fluorescence decays enables us to quantify the excited state dynamics and to assign the competing nonradiative pathways present under given environmental conditions. Here, we used excited-state dynamics to reveal the effect of IgE cross-linking on local membrane nanostructure and protein-protein interactions in RBL cells, as probed by the lipid analog diI-C18 and A488-IgE, respectively.
. Considering the complexity of cellular environments, various nonradiative pathways are likely to compete with fluorescence emission of a given fluorophore. For example, when a fluorophore is incorporated into a lipid membrane, its excited-state lifetime is affected by the overall organization or order of the membrane (29), the compositional diversity of the lipids (heterogeneity), and the solvent-fluorophore interactions. Isomerization, which explains the enhanced fluorescence properties of diI-C18 upon restriction in an organic phase (i.e., the membrane), is a primary example of such a pathway. As a result, the nature of the fluorescence decays enables us to quantify the excited state dynamics and to assign the competing nonradiative pathways present under given environmental conditions. Here, we used excited-state dynamics to reveal the effect of IgE cross-linking on local membrane nanostructure and protein-protein interactions in RBL cells, as probed by the lipid analog diI-C18 and A488-IgE, respectively.
To enhance the signal/noise ratio and temporal resolution, we also conducted complementary single-point measurements (i.e., the laser was parked without scanning) using 1024 time channels with 16 ps/channel. In this case, one-photon (1P) laser pulses were generated (480 nm, 4.2 MHz) using a pulse picker (Mira 9200, Coherent) and second harmonic generation (using SHG-4500, Coherent). In this case, the laser was steered toward the sample via the rear exit port of the microscope and strategically positioned at a selected area on the cell membrane. For all single-point experiments, a uniformly labeled location on the cell equator of uncross-linked cells was chosen, whereas for cross-linked cells the beam was positioned on an equatorial membrane patch. The single-point fluorescence decays reported here were measured (using magic-angle detection to eliminate any rotational effect on the observed excited-state dynamics) and/or calculated from simultaneously measured parallel and perpendicular polarizations (see below). Since the fluorescent labels lack perfect centers of symmetry in (diI-C18) or on (A488-IgE) the membrane, the first excited state can be excited with either one or two photons, thus our comparative 1P single-point time-resolved lifetime and 2P-FLIM measurements are likely equivalent.
Steady-state fluorescence polarization anisotropy imaging and 1P time-resolved anisotropy
To correlate the observed dependence of excited-state dynamics with membrane nanostructure, we performed steady-state and time-resolved fluorescence anisotropy measurements. Two-channel fluorescence intensity images were recorded simultaneously at two different polarizations, one parallel (0°) and the other perpendicular (90°) with respect to the excitation polarization. During the excited-state lifetime, a fluorophore undergoes rotational diffusion that is sensitive to its hydrodynamic volume and the nature of the surrounding environment. After photoselective excitation of a fluorophore at pixel coordinates (x,y) in a cell, the time-resolved fluorescence polarization anisotropy, r(t;x,y), can be described generally as (26):
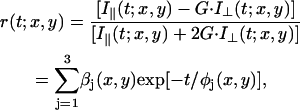 |
(2) |
where 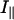 and
and 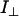 are the parallel and perpendicular fluorescence emission intensities, with respect to the excitation polarization vector, as selected using Glan-Thompson polarizers. The time-resolved anisotropy depends on the rotational correlation time (φj) and the preexponential factor (
are the parallel and perpendicular fluorescence emission intensities, with respect to the excitation polarization vector, as selected using Glan-Thompson polarizers. The time-resolved anisotropy depends on the rotational correlation time (φj) and the preexponential factor (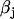 ) whose sum is equal to the initial anisotropy, r0 (23). In some cases, φ3 approaches infinity so that β3 is referred to as the residual anisotropy, r∞, which is due to the hindrance of fluorophore rotation and is present at times that are long compared to the fluorescence lifetime of the probe (23). The degree of orientational constraint of a fluorophore is defined by the ratio, r∞/r0, which describes its range of angular rotation and is related to the order parameter, S, with S = (r∞/r0)1/2 (30,31). The G-factor accounts for possible polarization bias in the detection efficiency (23). The denominator in Eq. 2 is equivalent to the magic-angle fluorescence decay, but with the advantages of direct correlation between the excited-state and rotational dynamics of a fluorophore at the same cellular location and minimal cellular photodamage.
) whose sum is equal to the initial anisotropy, r0 (23). In some cases, φ3 approaches infinity so that β3 is referred to as the residual anisotropy, r∞, which is due to the hindrance of fluorophore rotation and is present at times that are long compared to the fluorescence lifetime of the probe (23). The degree of orientational constraint of a fluorophore is defined by the ratio, r∞/r0, which describes its range of angular rotation and is related to the order parameter, S, with S = (r∞/r0)1/2 (30,31). The G-factor accounts for possible polarization bias in the detection efficiency (23). The denominator in Eq. 2 is equivalent to the magic-angle fluorescence decay, but with the advantages of direct correlation between the excited-state and rotational dynamics of a fluorophore at the same cellular location and minimal cellular photodamage.
For 1P steady-state confocal anisotropy imaging, the G-factor (0.96) was estimated, using reference fluorophores, by comparative measurements of steady-state anisotropy carried out in a spectrofluorimeter with polarization capabilities and on our confocal microscope. In these control experiments, free fluorescent molecules (∼1 μM) in solution (e.g., green fluorescent protein, DsRed, rhodamine green, or rhodamine 6G in water) and in viscous media (e.g., rhodamine green or rhodamine 6G in 100% glycerol) were excited by either 488 nm or 543 nm excitation. As an example, we obtained steady-state anisotropies of 0.31 ± 0.01 and 0.30 ± 0.01 for 1 μM green fluorescent protein in water using fluorimetry and confocal microscopy, respectively, which is in agreement with Rocheleau et al. (32). As a result of these experiments, depolarization due to the high NA objective was determined to be negligible using an underfilled objective. For complementary 2P-steady-state anisotropy imaging (Supplemental Fig. 2) and single-point time-resolved anisotropy measurements, the G-factor (0.7) was determined using the tail-matching approach (23,28) for solutions of free fluorescein (in water), Alexa 488 (in water), or diI-C18 (in DMSO). The measured initial anisotropies were always lower than the projected theoretical values (0.4 and 0.57 for 1P and 2P, respectively), which implies a nonzero interdipole angle between the absorbing and emitting dipoles (23,33,34). In fact, the 1P initial anisotropies of diI-C18 (r0 = 0.381, as calculated from the reported polarization of 0.480 (35)) and Alexa 488 (r0 = 0.376 (36)) in highly ordered environments (encapsulated in Sylgard 182 silicone elastomer and 100% glycerol, respectively) suggest that, although the absorption and emission dipoles are not completely parallel, these fluorophores have the structural range to report on the order of their surroundings.
Data analysis
The measured magic angle fluorescence decays per pixel in 2P-FLIM images were analyzed using a nonlinear, least-squares fitting routine with deconvolution of the system response function (FWHM ∼60 ps) in the SPCImage software package (Becker & Hickl). The response function was measured using the second harmonic signal of the femtosecond pulses or generated by SPCImage based on the rise time of the measured decay. The residual and a reduced chi square (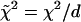 , where
, where 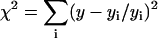 , y is the original data value for the point, yi is the fitted value for a given point, and d = degrees of freedom) were used to assess the goodness of the fit. In general, fluorescence decays fit with
, y is the original data value for the point, yi is the fitted value for a given point, and d = degrees of freedom) were used to assess the goodness of the fit. In general, fluorescence decays fit with 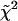 = 1.0–1.3 were considered satisfactory. The same nonlinear, least-squares fitting routine was also applied to the single-point fluorescence decays acquired at the magic angle. In some cases, the magic angle fluorescence decays were calculated from simultaneously measured parallel and perpendicularly polarized time-resolved fluorescence (see above). Nonlinear least-squares fitting of these decays were carried out using either OriginPro 7 (OriginLab, Northhampton, MA) or IgorPro (WaveMetrics, Lake Oswego, OR) without deconvolution of the system response function, especially when the decay time constants were larger than the FWHM of the response function (i.e., for A488-IgE). The goodness of the fit was evaluated using χ2 calculated by OriginPro 7 or IgorPro (
= 1.0–1.3 were considered satisfactory. The same nonlinear, least-squares fitting routine was also applied to the single-point fluorescence decays acquired at the magic angle. In some cases, the magic angle fluorescence decays were calculated from simultaneously measured parallel and perpendicularly polarized time-resolved fluorescence (see above). Nonlinear least-squares fitting of these decays were carried out using either OriginPro 7 (OriginLab, Northhampton, MA) or IgorPro (WaveMetrics, Lake Oswego, OR) without deconvolution of the system response function, especially when the decay time constants were larger than the FWHM of the response function (i.e., for A488-IgE). The goodness of the fit was evaluated using χ2 calculated by OriginPro 7 or IgorPro (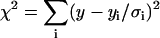 , where y and yi are as previously described and σi is the standard error for the point). In this case, decays fit with
, where y and yi are as previously described and σi is the standard error for the point). In this case, decays fit with 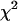 = 0–0.01 were considered satisfactory. Comparative lifetime measurements of diI-C18 in cells (the sample with the shortest decay time constant of all) and various free fluorescent molecules in solution (data not shown) indicated negligible differences in the time constants using both techniques (with or without deconvolution). We also performed unpaired, two-tailed Student's t-tests using Excel (Microsoft, Redmond, WA) to assess whether the variation of fluorescence decay parameters observed for diI-C18 and A488-IgE upon cross-linking was statistically significant (p ≤ 0.05). Single-point time-resolved lifetime experiments were performed, on three separate days, on cells that had (n = 21 cells for diI-C18, 19 cells for A488-IgE) or had not (n = 23 cells for diI-C18, 18 cells for A488-IgE) been incubated with α-IgE.
= 0–0.01 were considered satisfactory. Comparative lifetime measurements of diI-C18 in cells (the sample with the shortest decay time constant of all) and various free fluorescent molecules in solution (data not shown) indicated negligible differences in the time constants using both techniques (with or without deconvolution). We also performed unpaired, two-tailed Student's t-tests using Excel (Microsoft, Redmond, WA) to assess whether the variation of fluorescence decay parameters observed for diI-C18 and A488-IgE upon cross-linking was statistically significant (p ≤ 0.05). Single-point time-resolved lifetime experiments were performed, on three separate days, on cells that had (n = 21 cells for diI-C18, 19 cells for A488-IgE) or had not (n = 23 cells for diI-C18, 18 cells for A488-IgE) been incubated with α-IgE.
For steady-state fluorescence anisotropy image analysis, we developed a custom MATLAB (The MathWorks, Natick, MA) program to calculate steady-state initial anisotropy images (r0(t;x,y)) of diI-C18 and A488-IgE labeled cells. The measured parallel and perpendicularly polarized fluorescence images were recorded simultaneously and used in these calculations. 1P steady-state anisotropy images of diI-C18 were acquired at the equatorial plane and at the top surface of the cells, over four individual days, on cells that had (n = 27 cells) or had not (n = 20) been incubated with α-IgE. For single-point time-resolved fluorescence polarization anisotropy, the decays were fit (Eq. 2) using IgorPro, without deconvolution of the system response function. Because the anisotropy decays were calculated from time-resolved parallel and perpendicular fluorescence decays, such deconvolution is rather difficult. As an alternative approach, the first few time channels from the zero-time of these calculated decays were omitted and the fit was started beyond the FWHM (∼60 ps) of the system response function. The χ2 generated by the IgorPro fitting software was again used to determine the goodness of the fit. Our a priori expectation was that membrane anisotropy (as probed by diI-C18) and the anisotropy of A488-IgE would increase upon IgE receptor cross-linking, so unpaired, one-tailed Student's t-tests were performed in Excel on all of the anisotropy decay fit parameters to determine whether the mean values were significantly different (p ≤ 0.05) for cross-linked versus uncross-linked cells. Single-point time-resolved anisotropy experiments were performed, on three separate days, on cells that had (n = 11 cells for diI-C18, 12 cells for A488-IgE) or had not (n = 11 cells for diI-C18, 13 cells for A488-IgE) been incubated with α-IgE.
RESULTS AND DISCUSSION
The so-called “raft hypothesis”, which attributes functional roles in signaling and trafficking to cholesterol-rich domains, has remained controversial, in part due to the difficulty of observing domains in vivo without significant nonphysiological perturbations (e.g., low temperatures or extensive cross-linking). The elusive nature of these domains is likely due to lipid-lipid and lipid-protein interactions that are short-lived and thermodynamically unstable in the unstimulated state and to the possible transient coalescence of domains to nucleate functional domains upon receptor engagement (e.g., antigen-induced cross-linking of IgE-FcɛRI) and allow signal initiation. Baird, Holowka, and co-workers have investigated FcɛRI phosphorylation by the tyrosine kinase, Lyn, which occurs upon IgE-FcɛRI cross-linking by antigen (7). Their studies have demonstrated that such phosphorylation proceeds in a cholesterol-dependent manner (7), which is due to the protective nature of cholesterol against tyrosine phosphatases (37). As a first step toward our goal of following the association of the IgE receptor and other fluorescently labeled signaling molecules with changes in membrane nanostructure as signaling proceeds, we used RBL mast cells that were extensively cross-linked with α-IgE to form micron-sized patches or domains. Although these conditions are nonphysiological, this system allows us to test whether changes in lifetime and anisotropy correlate with the formation and location of these receptor-containing domains.
Confocal microscopy reveals IgE cross-linking-induced coredistribution of a lipid analog with IgE-FcɛRI in RBL cells
Under resting conditions, A488-IgE-FcɛRI complexes are preferentially localized on the plasma membrane of live RBL mast cells, which is labeled with diI-C18 (Fig. 1). To prevent cross talk between the two imaging channels (Alexa 488 and diI-C18), we used different excitation wavelengths and emission filters. When IgE-sensitized RBL mast cells are extensively cross-linked with α-IgE for long periods in the cold, we observe the formation of large domains of IgE-FcɛRI (Fig. 1 D). The majority of the lipid analog diI-C18 (our lipid microdomain marker) coredistributes with IgE-FcɛRI into these well-defined patches (Fig. 1 E), as observed in previous studies (5,7). The patches are several microns in size and are markedly different from the more uniform distributions of IgE-FcɛRI (Fig. 1 A) and diI-C18 (Fig. 1 B) in the absence of cross-linking by the secondary antibody. Upon cross-linking induced domain formation, an apparent enhancement in fluorescence intensity is observed. These results are in agreement with prior work on coredistribution of a variety of “raft markers” such as gangliosides (7,25), glycosylphosphatidylinositol-anchored proteins (6), kinases (7), and lipid analogs (5) with IgE-FcɛRI.
FIGURE 1.

Confocal fluorescence imaging and steady-state fluorescence anisotropy imaging of RBL-2H3 cells. diI-C18 (λex = 543 nm) in the plasma membrane (B and E) colocalizes with A488-IgE (λex = 488 nm) on the cell surface (A and D) upon extensive cross-linking by α-IgE. Steady-state anisotropy images of diI-C18 (C and F) are shown in the absence or presence of α-IgE, respectively. Higher anisotropy, or membrane order, increases toward the red end of the color scale. Bar, 5 μm.
These steady-state images, nevertheless, lack information concerning the molecular dynamics associated with the changes in membrane nanostructure at the vicinity of specific signaling molecules (e.g., IgE-FcɛRI). Furthermore, the observed enhancement of fluorescence intensity at cross-linking induced patches can be attributed to either an increase in the local concentration of the fluorescent labels upon cross-linking and/or an enhanced fluorescence quantum yield of the fluorophore due to changes in its immediate surroundings. To understand the underlying structural bases of the observed fluorescence enhancement, we conducted 2P-FLIM where the excited state fluorescence lifetime is independent of the fluorophore concentration. In addition, we carried out polarization anisotropy imaging to visualize the orientation distribution of the dipole moment of diI-C18 in the plasma membrane. These noninvasive techniques that are best-suited for short-scale dynamics (both in space and time) increase our chances for real-time monitoring of these fleeting microdomains in vivo.
Ultrafast excited state dynamics report environmental changes with increased lifetime of diI-C18 and A488-IgE upon IgE-FcɛRI cross-linking
Fluorescence lifetime (i.e., quantum yield) is sensitive to conformational changes in both the molecular structure and surrounding environment of a given fluorophore. We exploited this property to probe potential differences in membrane nanostructure as a function of receptor cross-linking. 2P-FLIM (λex = 760 and 960 nm, 76 MHz) was performed with high spatial and temporal resolution on RBL cells labeled with diI-C18 or A488-IgE-FcɛRI, in the presence and absence of extensive cross-linking with α-IgE. To our knowledge, this work represents the first application of 2P-FLIM to the association of IgE-FcɛRI with cholesterol-rich membrane domains. To overcome the inherently low signal/noise (256 × 256 pixels) and temporal resolution present in 2P-FLIM decays (65 time channels, 195 ps/channel), we performed complementary single-point lifetime experiments (1024 time channels, 16 ps/channel), using 1P excitation (λex = 480 nm, 4.2 MHz), in which the laser was focused on an equatorial region of interest. The relative size of the membrane patches in cells with cross-linked IgE-FcɛRI and the 1P-excitation using an underfilled 1.2 NA objective makes it challenging to directly compare 1P and 2P-FLIM results; however, we observe the same trend in our results with both types of excitation.
2P-FLIM
Upon IgE cross-linking, 2P-FLIM reveals bright equatorial patches of either diI-C18 or A488-IgE-FcɛRI (Fig. 2), similar to our confocal images. Corresponding DIC images (Fig. 2, A, C, E, and G) were captured to examine cell morphology and viability after 2P-FLIM. The fluorescence decays per pixel in these FLIM images were fit to a triexponential for diI-C18 and a biexponential for A488-IgE labeled cells and revealed longer fluorescence lifetimes for both probes upon FcɛRI cross-linking (Fig. 2, F and H, respectively) as compared with uncross-linked cells (Fig. 2, B and D). Representative data from the selected areas (noted by the arrows in Fig. 2) were acquired per pixel and the average lifetime, 〈τfl〉, was calculated.
FIGURE 2.

2P-FLIM of RBL-2H3 cells. Representative 2P-FLIM images of diI-C18 (D and H) and A488-IgE (B and F) in the plasma membrane of cells that have been cross-linked (F and H) with α-IgE or not (B and D) (λex = 760 nm). The average lifetime (〈τfl〉, indicated by the color scale) was determined by fitting the lifetime decays at the pixels (binning factor = 1) highlighted by arrows, yielding A488-IgE (− α-IgE) = 1.14 ± 0.09 ns; A488-IgE (+ α-IgE) = 1.34 ± 0.03 ns; diI-C18 (− α-IgE) = 0.57 ± 0.04 ns; and diI-C18 (+ α-IgE) = 0.71 ± 0.03 ns. Corresponding DIC images (A, C, E, and G) show cell viability after 2P-FLIM. Histograms demonstrate an increase in the average lifetime distributions of diI-C18 (J) and A488-IgE (I) that occur for cells upon cross-linking (curves 1 and 3) versus uncross-linked cells (curves 2 and 4). The free fluorescent markers in solution decay as a single exponential with average lifetimes of 0.43 ± 0.04 ns (n = 3) for 10 μM diI-C18 in DMSO and 3.1 ± 0.2 ns (n = 3) for 10 μM A488 in water (images not shown). Bar, 5 μm.
The measured average lifetime of diI-C18 is 0.71 ± 0.02 ns (area highlighted by an arrow in Fig. 2 H) and 0.57 ± 0.03 ns (highlighted area in Fig. 2 D) in the presence and absence of α-IgE, respectively. Fig. 2 J shows histograms of the average lifetime distributions for diI-C18 in the cells depicted in Fig. 2, H and D. For cross-linked cells (curve 3, Fig. 2 J (histogram peak ∼0.75 ns)), the average lifetime increases as compared to uncross-linked cells (curve 4, Fig. 2 J (histogram peak ∼0.50 ns)). Recently, using 1P-FLIM (128 × 128 pixels and 180 s acquisition, with λex = 473 nm), Owen et al. (29) measured a lower lifetime distribution for the fluorescent probe, di-4-ANEPPDHQ, in liquid-disordered (1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC) large unilamellar vesicles, as compared with liquid-ordered (egg n-palmitoyl-sphingomyelin:cholesterol, 0.7:0.3, mol/mol) vesicles. With this probe, they also observed a decreased lifetime and reduction of ordered phase in HEK293 cells upon temperature increase and cholesterol depletion. Together, our results and the results of Owen et al. suggest that the higher fluorescence lifetime of diI-C18 that we observe upon IgE cross-linking is likely a result of the constrained probe moving into more ordered regions in the plasma membrane of mast cells. Our results further indicate that this redistribution may cause an increase in the fluorescence lifetime (or quantum yield) of diI-C18, and thus an increase in fluorescence intensity as we observed with confocal imaging (Fig. 1). As a result, we conclude that cross-linking induced changes in membrane nanostructure can be reported by 2P-FLIM.
FLIM images of A488-IgE-FcɛRI show cross-linking induced equatorial patching and overall increased fluorescence lifetime as well (Fig. 2). The average lifetime of A488-IgE-FcɛRI in cross-linked cells is 1.34 ± 0.07 ns (area highlighted by an arrow in Fig. 2 F), as compared with 1.14 ± 0.03 ns (highlighted area in Fig. 2 B) for the monomeric receptor. The bimodal distribution observed for the average lifetimes of A488-IgE on cross-linked cells (see Fig. 2 F and curve 1, Fig. 2 I (histogram peaks ∼0.90 ns and 1.35 ns)) indicates one subpopulation of A488-IgE with a considerably longer lifetime and a second population with a lifetime distribution similar to that of cells that have not been cross-linked with the secondary antibody (Fig. 2 B and curve 2, Fig. 2 I (histogram peak ∼0.85 ns)). These differences demonstrate that changes resulting from protein-protein interactions can also be identified using FLIM and suggest that α-IgE, which is a polyclonal antibody with various affinities, does not saturate the A488-IgE binding sites available on the cell surface. Because FLIM is a concentration-independent measurement, the enhanced lifetimes observed for both probes report a change in the local environment of the fluorophores, rather than a cross-linking induced increase in local concentration of the fluorophores. The broad FWHM of the lifetime distributions in the pixel histograms indicate a heterogeneous environment of both the lipid and IgE labels in living cells. Measurements using 2P-FLIM with 960 nm excitation are comparable for both diI-C18 and A488-IgE (Supplemental Fig. 1), suggesting a negligible cellular autofluorescence contribution under our experimental conditions and minimal excitation wavelength dependence.
1P single-point time-resolved fluorescence
The excited-state of the lipid analog diI-C18, which preferentially partitions into the cross-linked IgE-FcɛRI domains, experiences a longer average fluorescence lifetime in cross-linked cells (〈τfl〉 = 0.8 ± 0.1 ns, p = 2.36 × 10−5; Table 1) as compared with cells that do not undergo receptor cross-linking (〈τfl〉 = 0.60 ± 0.04 ns; Table 1). These average lifetime values are in close agreement with those obtained by 2P-FLIM for diI-C18 labeled cells, with and without IgE-FcɛRI cross-linking. In single-point experiments, diI-C18 fluorescence decays as a triexponential, whether in cells in the presence (τ1 = 0.13 ± 0.03 ns (p = 2.2 × 10−4), α1 = 0.45 ± 0.03 (p = 1.0 × 10−4), τ2 = 0.75 ± 0.08 ns (p = 2.2 × 10−4), α2 = 0.40 ± 0.05 (p = 0.016), τ3 = 3.1 ± 0.4 ns, and α3 = 0.15 ± 0.04 (p = 0.017)) or absence of FcɛRI cross-linking (τ1 = 0.08 ± 0.01 ns, α1 = 0.53 ± 0.05, τ2 = 0.62 ± 0.05 ns, α2 = 0.35 ± 0.04, τ3 = 2.9 ± 0.2 ns, and α3 = 0.12 ± 0.02; Table 1). Fluorescence decays were also calculated from parallel and perpendicularly polarized fluorescence decays (following the denominator of Eq. 2) and fit without deconvolution, starting a few time channels beyond time zero. As a result, the shortest lifetime components (τ1) had lower amplitudes and the average lifetimes were consequently higher, whereas all of the time constants remained within standard deviation of the values obtained with deconvolution. For example, in cells treated with α-IgE (n = 5), diI-C18 had an average lifetime of 1.3 ± 0.1 ns (p = 7.65 × 10−3), with τ1 = 0.09 ± 0.01 ns, α1 = 0.29 ± 0.06, τ2 = 0.92 ± 0.08 ns (p = 0.015), α2 = 0.50 ± 0.03, τ3 = 3.7 ± 0.4 ns (p = 0.013), and α3 = 0.21 ± 0.06 (p = 0.017). For those not cross-linked (n = 6), diI-C18 again had a lower average lifetime of 0.98 ± 0.09 ns, with τ1 = 0.07 ± 0.02 ns, α1 = 0.32 ± 0.07, τ2 = 0.7 ± 0.1 ns, α2 = 0.46 ± 0.08, τ3 = 2.8 ± 0.5 ns, and α3 = 0.22 ± 0.06. These complementary single-point lifetime measurements enable us to directly correlate the observed excited-state dynamics of the diI-C18 lipid and A488-IgE labels with their surrounding environment. As shown in Fig. 3 A, the magic angle fluorescence decays of diI-C18 for these two cellular conditions are different in shape, with diI-C18 in cross-linked cells demonstrating a slower lifetime decay (Fig. 3 A, curve 1) than in uncross-linked cells (Fig. 3 A, curve 2). This observation suggests that the diI-C18 lipid analog localizes to regions of differing membrane structure upon treatment with α-IgE and that environmental constraint increases in cross-linking induced domains. These single-point lifetime measurements follow the same trend we observed in the FLIM experiments—that is, lifetime increases for both diI-C18 and A488-IgE upon FcɛRI cross-linking (Table 1).
TABLE 1.
Single-point, time-resolved fluorescence lifetime of diI-C18 and A488-IgE in RBL cells with and without IgE-FcɛRI cross-linking using α-IgE
| τ1/ns | α1 | τ2/ns | α2 | τ3/ns | α3 | τfl/ns | n | |
|---|---|---|---|---|---|---|---|---|
| diI-C18 labeled cells | ||||||||
| − α-IgE | 0.08(1) | 0.53(5) | 0.62(5) | 0.35(4) | 2.9(2) | 0.12(2) | 0.60(4) | 10 |
| + α-IgE | 0.13(3)* | 0.45(3)* | 0.75(8)* | 0.40(5)* | 3.1(4) | 0.15(4)* | 0.8(1)* | 11 |
| A488-IgE labeled cells | ||||||||
| − α-IgE | – | – | 1.0(1) | 0.81(2) | 3.4(3) | 0.19(2) | 1.5(2) | 5 |
| + α-IgE | – | – | 1.10(6)* | 0.77(1)* | 3.6(1) | 0.23(1)* | 1.67(4)* | 7 |
| Free markers in solution | ||||||||
| Alexa 488 (water†) | – | – | – | – | 4.1(2) | 1.0 | 4.1(2) | 2 |
| A488-IgE (PBS†) | – | – | 1.0(1)* | 0.52(2) | 3.38(2)* | 0.48(2) | 2.13(3)* | 2 |
| diI-C18 (DMSO) | – | – | – | – | 0.61(3) | 1.0 | 0.61(3) | 2 |
| Fluorescein (water†) | – | – | – | – | 3.77(2) | 1.0 | 3.77(2) | 2 |
| Fluorescein (PBS†) | – | – | – | – | 3.94(1) | 1.0 | 3.94(1) | 2 |
λex = 480 nm, T ≈ 20°C. The fitting parameters represent an average over a number of cells (n) with the standard deviation in the last digit shown in parentheses to reflect the cell-to-cell variation of the individual fit parameters. All data shown, except for A488-IgE labeled cells, are from single-point decays acquired at the magic angle, using deconvolution with the system response function (FWHM ∼60 ps) as in 2P-FLIM experiments (see text). A comparison was made with the fitting parameters of fluorescence decays calculated from parallel and perpendicularly polarized fluorescence decays (see text).
p < 0.05, as determined from unpaired, two-tailed Student's t-tests, indicating a statistically significant difference in the means as compared to cells in the absence of receptor cross-linking.
The pH of water is ∼5–5.5, and the pH of PBS is 7.4.
FIGURE 3.

1P time-resolved fluorescence lifetime of diI-C18 and A488-IgE in living mast cells, under different conditions of IgE cross-linking. Representative single-point fluorescence lifetime decays for diI-C18 (A) in the plasma membrane of mast cells in the presence (curve 1) or absence (curve 2) of α-IgE; and A488-IgE (B) on the surface of mast cells with (curve 3) or without (curve 4) cross-linking (λex = 480 nm for both diI-C18 and A488-IgE). Triexponential diI-C18 decays were measured at magic angle polarization and biexponential A488-IgE decays were calculated from the measured parallel and perpendicularly polarized fluorescence decays (following the denominator of Eq. 2). In the calculated magic angle fluorescence decays, the fitting was started beyond the FWHM of the system response function. All decays were fit following Eq. 1, and the fit parameters of time-resolved fluorescence decays are summarized in Table 1. Comparison between both lifetime methods (with and without deconvolution) is included in the text where appropriate. With samples having long excited state lifetimes (i.e., Alexa 488), there are no significant differences in the fitting parameters with each method.
To validate the cross-linking induced enhancement in diI-C18 lifetime and the underlying mechanism of membrane ordering, we compared our cell results with time-resolved fluorescence of diI-C18 in giant unilamellar vesicles (GUVs) of various lipid phases (F. Ariola, J. Heetderks, P.S. Weiss, and A.A. Heikal, unpublished results). In fluid phase (DOPC) GUVs, diI-C18 fluorescence decays as a biexponential with τ1 = 0.87 ± 0.02 ns, α1 = 0.83 ± 0.01, τ2 = 1.5 ± 0.1 ns, and α2 = 0.17 ± 0.01, and an estimated average lifetime of 0.97 ± 0.01 ns (n = 5). For diI-C18 in GUVs of a liquid-ordered composition (DPPC/cholesterol, 0.7:0.3, mol/mol) that is near the 2:1 ratio of phospholipid/cholesterol found in RBL plasma membranes (14), we measured an average lifetime of 1.22 ± 0.07 ns (n = 5). These results agree with previous studies by Packard and Wolf (38), who reported an increase in the average lifetime of diI-C18 as lipid order increased, with 0.80 ns for diI-C18 in fluid phase (DOPC), 0.96 ns in egg phosphatidylcholine, 1.13 ns in gel phase (DPPC) and 1.54 ns in the liquid-ordered composition (DPPC/cholesterol, 0.7:0.3, mol/mol). The GUV results also support our argument that the increased fluorescence lifetime of diI-C18 in cross-linked mast cell membranes is due to greater lipid order (i.e., 〈τfl〉 = 0.60 ± 0.04 ns in uncross-linked cells and 0.8 ± 0.1 ns in cells cross-linked with α-IgE). Furthermore, the lifetime components of diI-C18 in either natural (Table 1) or model membranes are longer than that of the free dye in DMSO, which only decays as a single exponential (〈τfl〉 = 0.61 ± 0.03 ns and 0.54 ± 0.02 ns from fitting measured and calculated fluorescence decays, respectively). When diI-C18 is intercalated within the membrane, the conformational changes of the fluorophore become restricted due to steric hindrance of neighboring lipid molecules (26).
Similarly, fluorescence lifetime measurements were also conducted on A488-IgE labeled RBL mast cells for comparison. The excited-state fluorescence lifetime of A488-IgE increases when cells undergo IgE receptor cross-linking, as compared with uncross-linked conditions. The measured average lifetime for cross-linked A488-IgE-FcɛRI is significantly larger (〈τfl〉 = 1.67 ± 0.04 ns, p = 0.004; Table 1) when compared to monomeric IgE-FcɛRI (〈τfl〉 = 1.5 ± 0.2 ns; Table 1). The fluorescence of A488-IgE in cells in the presence of cross-linking decays as a biexponential (τ1 = 1.10 ± 0.06 ns (p = 0.025), α1 = 0.77 ± 0.01 (p = 6.49 × 10−4), τ2 = 3.6 ± 0.1 ns and α2 = 0.23 ± 0.01 (p = 6.10 × 10−3); Table 1), similar to A488-IgE fluorescence in the absence of cross-linking (τ1 = 1.0 ± 0.1 ns, α1 = 0.81 ± 0.02, τ2 = 3.4 ± 0.3 ns, and α2 = 0.19 ± 0.02; Table 1). The overall profile of the lifetime decays (Fig. 3 B) and the observed fitting parameters in single-point time-resolved fluorescence measurements support the trend in 2P-FLIM experiments regarding the local environments of diI-C18 and IgE in cells in the presence or absence of IgE-FcɛRI cross-linking. The longer fluorescence decays that occur when α-IgE is present are further substantiated by our time-resolved anisotropy results, as well as by polarized fluorescence depletion experiments (microseconds timescale) in which the lifetime of IgE on mast cells increased upon cross-linking (39).
As a comparison, time-resolved fluorescence measurements on the fluorophores of interest were carried out in solution. As previously noted, the excited-state fluorescence of free diI-C18 (in DMSO) decays as a single exponential (Table 1) with an average lifetime of 0.61 ± 0.03 ns. This ultrafast excited state lifetime is consistent with the low fluorescence quantum yield of diI-C18 in solution, and the nonradiative pathway for the excited-state depopulation of diI-C18 is likely to include isomerization (40). The fluorescence of free Alexa 488 (in water) decays as a single exponential with an average lifetime of 4.1 ± 0.2 ns (Table 1), which is longer than when the dye is covalently attached to IgE and similar in magnitude to the 3.75 ± 0.05 ns obtained by Agronskaia et al. (41), who analyzed a 50 μM aqueous solution of Alexa 488 with 2P time-correlated single-photon counting (λex = 800 nm). The longer excited-state lifetime of Alexa 488 in water (Table 1), as compared to when it is covalently linked to receptor-bound IgE, indicates either an active nonradiative pathway is available in the cellular environment of Alexa 488 or a greater stabilization of the excited state of A488 by the polar solvent molecules. The average lifetime of A488-IgE in PBS (2.13 ± 0.03 ns) is appropriately shorter than free Alexa 488 in solution, yet significantly longer than A488-IgE-FcɛRI in uncross-linked cells (p = 0.016). A clear assignment of the active nonradiative mechanism for Alexa 488 upon covalently modifying IgE is rather difficult to define due to the random labeling of A488 on IgE. Although previously published results of lifetime measurements on fluorescently-labeled IgE bound to FcɛRI (using comparable techniques) are not available, we hypothesize that the observed lifetime of A488-IgE represents an average over a variety of dye environments that vary in both solvent accessibility and possible quenching by protein amino acids, whereas the environment of free A488 in aqueous solution is much more homogeneous. To confirm the structural bases of the observed increases of both diI-C18 and A488-IgE fluorescence lifetimes upon IgE cross-linking, we carried out time-resolved fluorescence anisotropy measurements.
Dipole-moment distribution and rotational diffusion report increased membrane order upon IgE-FcɛRI cross-linking
In previous fluorimetry experiments, Gidwani et al. (14) measured the steady-state fluorescence anisotropy of plasma membrane blebs and DRMs isolated from RBL cells. In that study, both biomembrane systems were labeled with a diphenylhexatrienyl phospholipid analog and revealed sensitivity to cholesterol-dependent membrane ordering. The plasma membrane blebs and lipids extracted from plasma membrane vesicles exhibited an enhanced steady-state anisotropy (40% greater) compared to liquid-disordered (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) vesicles and a decreased steady-state anisotropy (60% less) as compared to liquid-ordered (DPPC/cholesterol, 0.66:0.33, mol/mol) vesicles (14). However, these cuvette-based experiments lack the dynamics information, as well as the spatial resolution, for signaling molecules in intact, living cells. To overcome these limitations, we used steady-state fluorescence anisotropy imaging (Fig. 1) to evaluate the spatial distribution of lipid order. In addition, complementary single-point time-resolved fluorescence anisotropy measurements (Fig. 4) were carried out using pulsed 1P excitation (λex = 480 nm, 4.2 MHz), and simultaneous detection of parallel and perpendicular polarized emissions, of singly labeled cells to follow the dynamics of diI-C18 within the membrane of living RBL cells and the dynamics of the protein-protein interactions between A488-IgE-FcɛRI and α-IgE.
FIGURE 4.

1P time-resolved fluorescence anisotropy of diI-C18 and A488-IgE in living mast cells, under different conditions of IgE cross-linking. Representative single-point time-resolved anisotropy decays of diI-C18 (A) in the plasma membrane of mast cells with (curve 1) or without (curve 2) IgE cross-linking and A488-IgE (B) on the surface of cells in the presence (curve 3) or absence (curve 4) of α-IgE (λex = 480 nm for both diI-C18 and A488-IgE). Curves were fit, following Eq. 2, as a single (diI-C18 labeled cells) or biexponential (A488-IgE labeled cells), both with an additional residual anisotropy component. Points of the decays at times well beyond the excited state lifetime of the probe where noise levels were high (3 ns for diI-C18) were not included in the fit. The fit parameters of time-resolved fluorescence anisotropy decays are summarized in Table 2. The observed associated-anisotropy feature (47) in diI-C18 without cross-linking was not reproducible in all our measurements and may be attributed to the low signal/noise at times much longer than the excited-state lifetime (see Fig. 3).
Steady-state fluorescence anisotropy imaging
We used steady-state fluorescence anisotropy imaging to visualize local ordering of diI-C18 (Fig. 1 and Supplemental Fig. 2) and A488-IgE (Supplemental Fig. 2) within the cell membrane of live mast cells, treated and untreated with α-IgE. Such measurements enable us to further evaluate how the local nanostructure of the plasma membrane is altered upon IgE-FcɛRI cross-linking with secondary antibody and to provide a structural basis for the observed increase in fluorescence lifetime. The confocal fluorescence images reveal colocalization of diI-C18 (Fig. 1 E) with A488-IgE (Fig. 1 D) in the presence of FcɛRI cross-linking, without a striking visual difference in the average anisotropy of diI-C18 in cells in the presence (Fig. 1 F) or absence (Fig. 1 C) of cross-linking. Cells with α-IgE (e.g., Fig. 1 F) exhibit the expected patching with a more homogeneous anisotropy distribution of diI-C18 within the IgE receptor domains. In contrast, the diI-C18 anisotropy images of cells in the absence of α-IgE (e.g., Fig. 1 C) generally display more heterogeneity in the fluorescent label dipole-moment orientation throughout the plasma membrane. Higher anisotropy was consistently observed at the domain perimeters, which may be a reflection of increased interfacial tension or barriers between the more ordered domains and the less ordered bulk plasma membrane. As mentioned in Materials and Methods, to avoid artifacts due to polarization-biased detection sensitivity, we estimated the G-factor (0.96) from comparative measurements of the steady-state anisotropy of several free fluorescent molecules in solution and in viscous media performed in a spectrofluorimeter with polarization capabilities and on our confocal microscope. Based on the selected set of filters and dichroic mirrors, the contribution of scattered light to our images is negligible, and because the parallel and perpendicularly polarized images were recorded simultaneously, the effects of potential cell movement can also be ruled out.
Similar trends were also observed using 2P-fluorescence polarization anisotropy imaging (Supplemental Fig. 2), which offers the advantages of a larger dynamic range (i.e., maximum theoretical r0-value of 0.57, compared with 0.4 for 1P excitation), an accurately determined G-factor, and negligible cellular autofluorescence and scattered light. These single-cell anisotropy images indicate heterogeneous local lipid and protein environments under the different states of IgE-FcɛRI cross-linking in live RBL cells. The spatial resolution inherent in steady-state anisotropy offers new information that may be averaged out in previous bulk studies on cell blebs or plasma membrane vesicles studied in suspension using spectrofluorimetry (14). Although the diI-C18 steady-state anisotropy images provide insight into the spatial organization of the plasma membrane, they unfortunately lack the dynamics information related to changes in membrane nanostructure and to the effect of IgE-FcɛRI cross-linking on the membrane. To obtain such information, we conducted time-resolved anisotropy studies of diI-C18 and A488-IgE-FcɛRI in mast cells, as a function of IgE receptor cross-linking with a secondary antibody.
1P single-point time-resolved fluorescence anisotropy
Single-point fluorescence anisotropy measurements, with picosecond temporal resolution, reveal notably higher restrictions on the rotational mobility of diI-C18 upon cross-linking (Fig. 4 and Table 2). The fluorescence anisotropy of diI-C18 decays as a single exponential with a residual anisotropy (r∞) component, indicating a wobbling (or tumbling) motion of the fluorophore around a flexible tether to/within the membrane. Although there is a significant variation from cell to cell, the characteristic rotational correlation time of diI-C18 differs with the cross-linking state of IgE-FcɛRI (φ1 = 0.13 ± 0.01 ns (p = 0.048) and 0.10 ± 0.01 ns in the presence and absence of α-IgE, respectively; Table 2), indicating an increase in membrane nanostructure with IgE cross-linking. Under our experimental conditions, such a fast rotational time constant may not be assigned to free diI-C18 fluorophores due to the significant observed residual anisotropy (0.19 ± 0.07 and 0.21 ± 0.04 for cells with and without IgE cross-linking, respectively). The overall initial anisotropy of diI-C18 is comparable in the membrane of cells incubated with α-IgE (r0 = 0.30 ± 0.06) and in cells without α-IgE (r0 = 0.33 ± 0.02; Fig. 4 A, Table 2). Our initial anisotropy values for diI-C18 in RBL cells are somewhat larger than the ∼0.25 obtained for the diphenylhexatrienyl phospholipid analog in RBL plasma membrane blebs and DRM vesicles at room temperature using steady-state anisotropy measurements in a spectrofluorimeter (14), which may be due to differences in the membrane curvature of the blebs and vesicles versus RBL cells, the ensemble averaging in the cuvette experiments as compared with the single-cell approach reported here, or to the use of a different anisotropy probe. The degree of orientational constraint, as reflected by the r∞/r0 ratio (30, 31), is also similar for diI-C18 in the plasma membrane of cross-linked cells as compared to uncross-linked cells (0.64 ± 0.02 and 0.64 ± 0.09, respectively; Table 2). In contrast to diI-C18 in mast cells (Table 2), free diI-C18 in DMSO has an initial anisotropy of 0.30 ± 0.01 and a rotational correlation time of 0.73 ± 0.06 ns, consistent with its molecular mass (934 Da) and the viscosity of DMSO (∼2 cP). The longer rotational diffusion of diI-C18 upon receptor cross-linking correlates well with the increased lifetime of the probe in the same cells. These results are further substantiated by previous FRAP studies that showed a restriction of diI-C16 lateral diffusion (∼30–40% decrease in mobile fraction) within domains of RBL cells that had been extensively cross-linked in the cold, as compared to cells that had not been cross-linked (5,42). These time-resolved anisotropy studies provide support for the hypothesis that the diI-C18 lipid analog redistributes into IgE-FcɛRI-containing membrane domains of greater structural order upon cross-linking.
TABLE 2.
Single-point time-resolved fluorescence polarization anisotropy of diI-C18 and A488-IgE in RBL cells, with and without IgE-FcɛRI cross-linking by α-IgE
| φ1/ns | β1 | φ2/ns | β2 | r∞ | r0 | r∞/r0 | n | |
|---|---|---|---|---|---|---|---|---|
| diI-C18 labeled cells | ||||||||
| − α-IgE | 0.10(1) | 0.12(3) | – | – | 0.21(4) | 0.33(2) | 0.64(9) | 7 |
| + α-IgE | 0.13(1)* | 0.10(3) | – | – | 0.19(7) | 0.30(6) | 0.64(2) | 7 |
| A488-IgE labeled cells | ||||||||
| − α-IgE | 0.13(2) | 0.10(3) | 1.56(2) | 0.07(2) | 0.08(3) | 0.18(5) | 0.45(9) | 13 |
| + α-IgE | 0.16(3)* | 0.07(1) | 1.83(5)* | 0.07(1) | 0.11(4) | 0.19(4) | 0.59(1)* | 11 |
| Free markers in solution | ||||||||
| Alexa 488 (water‡) | 0.11(6) | 0.33(7) | – | – | – | 0.33(7) | – | 2 |
| A488-IgE (PBS‡) | 0.17(2) | 0.12(3) | 2.01(2)* | 0.06(1) | 0.03(1)† | 0.15(1) | 0.22(3)* | 4 |
| diI-C18 (DMSO) | 0.73(6) | 0.30(1) | – | – | – | 0.30(1) | – | 4 |
| Fluorescein (water‡) | 0.12(1) | 0.29(9) | – | – | – | 0.29(9) | – | 3 |
λex = 480 nm, T ≈ 20°C. The fitting parameters represent an average over a number of cells (n) with the standard deviation in the last digit shown in parentheses to reflect the cell-to-cell variation of the individual fit parameters.
p < 0.05, as determined from unpaired, one-tailed Student's t-tests, indicating a statistically significant difference in the means as compared to cells in the absence of receptor cross-linking.
The time constant associated with the third exponential term is ∼40–100 ns with a preexponential factor that is shown in this case as residual anisotropy (r∞). The uncertainty in this time constant is due to the rather small amplitude and the relatively long rotational time as compared with the excited state lifetime of A488-IgE.
The pH of water is ∼5–5.5, and the pH of PBS is 7.4.
To probe possible changes in protein-protein interactions upon α-IgE binding, similar time-resolved anisotropy measurements were conducted on A488-IgE. In the cell environment, A488-IgE anisotropy decays as a biexponential with a residual (r∞) anisotropy component (Fig. 4 and Table 2) with and without IgE receptor cross-linking. The rotational times of A488-IgE bound to the cell surface were slower when IgE-FcɛRI was cross-linked with α-IgE (φ1 = 0.16 ± 0.03 ns (p = 0.045), β1 = 0.07 ± 0.01, φ2 = 1.83 ± 0.05 ns (p = 0.010), and β2 = 0.07 ± 0.01; Table 2) as compared to cells in the absence of cross-linking (φ1 = 0.13 ± 0.02 ns, β1 = 0.10 ± 0.03, φ2 = 1.56 ± 0.02 ns, and β2 = 0.07 ± 0.02; Table 2). In agreement with the observed increase in lifetime, the enhanced rotational times of A488-IgE-FcɛRI support conformational changes of membrane order (i.e., a constraining environment) and the likelihood of the recruitment of other proteins upon α-IgE-IgE-FcɛRI cross-linking. The corresponding total initial anisotropies of A488-IgE-FcɛRI in cells with (r0 = 0.19 ± 0.04) and without cross-linking (r0 = 0.18 ± 0.05) are not statistically different (see Fig. 4 B for representative decays); however, the degrees of orientational constraint are significantly different between the two, with r∞/r0 = 0.59 ± 0.01 (p = 0.034) for A488-IgE in cells with IgE cross-linking and 0.45 ± 0.09 in cells in the absence of cross-linking (Table 2). Because of the short timescale of our time-resolved anisotropy experiments (picoseconds-nanoseconds), the anisotropy reported by A488-IgE may reflect rapid tumbling of covalently attached A488 or wagging motions of the IgE Fab segments (43) that are somewhat slowed by the binding of a secondary antibody, and not motions of the much larger IgE (∼180 kDa). This explanation is further supported by the significant segmental mobility associated with the rotational diffusion of free A488-IgE in solution (Table 2). The anisotropy of A488-IgE (in PBS) decays as a biexponential with a residual anisotropy component (φ1 = 0.17 ± 0.02 ns, β1 = 0.12 ± 0.03, φ2 = 2.01 ± 0.02 ns (p = 0.023), β2 = 0.06 ± 0.01 and r∞ = 0.03 ± 0.01; Table 2), where the slowest time constant, associated with such a small r∞ amplitude fraction, is uncertain (∼40–100 ns, as compared to the relatively shorter excited-state lifetime, i.e., the observation time window for rotational diffusion).
To the best of our knowledge, this is the first time that the ultrafast (picoseconds-nanoseconds) excited-state dynamics and rotational diffusion of fluorescently labeled IgE have been monitored for elucidating the structure-function relationship of IgE receptor cross-linking in intact, live mast cells. Previously, time-resolved fluorescence (43) and phosphorescence measurements (44,45) of IgE-FcɛRI on mast cells in solution have been carried out with nanosecond-microsecond resolution. In one of these studies, the rotational correlation time of IgE-FcɛRI on both RBL plasma membrane blebs and cells was found to increase significantly for cells that had been treated with α-IgE, and the degree of orientational constraint, r∞/r0, also increased from 0.515 in uncross-linked cells to 1.00 in α-IgE-treated cells (44). These results agree well with the trend observed in this report with ∼20 ps time resolution (Table 2). In addition, the improved time resolution here enables us to monitor ultrafast segmental mobility of the A488-IgE-FcɛRI complex in real time, which is beyond the resolution of previous phosphorescence experiments (46). Although the differences in time resolution between our techniques and those of Myers et al. (44) do not permit a true comparison of dynamics and order parameters, the observed enhancement of environmental constraints on the segmental mobility of A488-IgE under cross-linking conditions agree well with former FRAP studies on the lateral diffusion of Cy3-IgE-FcɛRI (42). In those studies, a cross-linking induced reduction in lateral mobility of Cy3-IgE-FcɛRI was reported in mast cells (∼40% decrease in mobile fraction) as the complex moved into a more ordered environment from the uncross-linked state (42). Overall, our single-point time-resolved anisotropy results for diI-C18 and A488-IgE, together with the lifetime results, suggest that fluorophore dynamics become more restricted and local membrane structure increases upon IgE-FcɛRI cross-linking.
CONCLUSION
Short-range sampling of the immediate local environment of a fluorescent probe, whether membrane-incorporated or protein-associated, allows for the real-time molecular-level interrogation and visualization of specialized cholesterol-rich microdomains and associated signaling proteins, as well as the monitoring of their short-range fluorescence dynamics. In general, the changes in anisotropy and lifetime of both diI-C18 and A488-IgE in individual RBL-2H3 mast cells can be correlated with cross-linking induced changes in local membrane structure, which are indicative of the degree of membrane heterogeneity, organization, and composition. Here, we have shown that upon extensive IgE-FcɛRI cross-linking, diI-C18 undergoes a change in environment on the nanoscale as it coredistributes in the plasma membrane to well-defined patches of A488-IgE-FcɛRI (Fig. 5 B), resulting in longer lifetimes (Figs. 2 and 3; Table 1) and higher anisotropy (Figs. 1 and 4; Table 2), as compared to uncross-linked cells (Fig. 5 A). We have also shown that, upon cross-linking with α-IgE, the environment and dynamics of A488-IgE are affected by increased protein-protein interactions, which results in distinguishably longer lifetimes and higher anisotropy as compared with A488-IgE bound to uncross-linked cells. Based on these results, we hypothesize that protein-protein interaction (i.e., IgE-FcɛRI cross-linking) recruits essential signaling proteins and lipid molecules into more ordered domains that serve as a platform for signaling. The results presented here will provide a valuable frame of reference for the interpretation of studies on mast cells that are stimulated under more physiological conditions (e.g., 37°C), where domains are much smaller in size and exist for shorter periods of time, although the exact spatial and temporal scale of these domains remains to be determined. Moreover, these studies offer an opportunity to follow the spatial and temporal relationships linking plasma membrane nanostructure to the dynamics of FcɛRI and other signaling molecules, both in living cells and in model membrane systems.
FIGURE 5.

Model of A488-IgE and diI-C18 orientation and location within the plasma membrane in the absence (A) and presence (B) of IgE receptor cross-linking. Ordered regions of the membrane are indicated by high concentrations of cholesterol, sphingomyelin, glycosphingolipids, and glycerophospholipids. Upon receptor cross-linking, these domains cluster to nucleate a larger, functional domain.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Minjoung Kyoung and Florly Ariola of Penn State University for helpful discussion and David Holowka of Cornell University for providing the purified mouse α-2,4-dinitrophenyl IgE and rabbit α-IgE.
This work was supported, in part, by The Pennsylvania State University (E.D.S. and A.A.H.), the Penn State Materials Research Institute (E.D.S. and A.A.H.), the Penn State Materials Research Science and Engineering Center (under National Science Foundation grant DMR 0213623) (E.D.S. and A.A.H.), and the Lehigh-Penn State Center for Optical Technologies (E.D.S. and A.A.H.), which is supported by the Commonwealth of Pennsylvania. Acknowledgment is also made to the Donors of the American Chemical Society Petroleum Research Fund (E.D.S.) and to the Johnson & Johnson/Penn State Innovative Technology Research Seed Grant (A.A.H.) for partial support of this research. A.A.H. is also grateful for the generosity of Coherent Lasers for the loan of a pulse picker (Mira 9200, Coherent).
References
- 1.Mukherjee, S., and F. R. Maxfield. 2004. Membrane domains. Annu. Rev. Cell Dev. Biol. 20:839–866. [DOI] [PubMed] [Google Scholar]
- 2.Edidin, M. 2003. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32:257–283. [DOI] [PubMed] [Google Scholar]
- 3.Simons, K., and W. L. C. Vaz. 2004. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 33:269–295. [DOI] [PubMed] [Google Scholar]
- 4.Vereb, G., J. Szollosi, J. Matko, P. Nagy, T. Farkas, L. Vigh, L. Matyus, T. A. Waldmann, and S. Damjanovich. 2003. Dynamic, yet structured: the cell membrane three decades after the Singer-Nicolson model. Proc. Natl. Acad. Sci. USA. 100:8053–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas, J. L., D. Holowka, B. Baird, and W. W. Webb. 1994. Large-scale co-aggregation of fluorescent lipid probes with cell surface proteins. J. Cell Biol. 125:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holowka, D., E. D. Sheets, and B. Baird. 2000. Interactions between FcɛRI and lipid raft components are regulated by the actin cytoskeleton. J. Cell Sci. 113:1009–1019. [DOI] [PubMed] [Google Scholar]
- 7.Sheets, E. D., D. Holowka, and B. Baird. 1999. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcɛRI and their association with detergent-resistant membranes. J. Cell Biol. 145:877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feigenson, G. W., and J. T. Buboltz. 2001. Ternary phase diagram of dipalmitoyl-PC/dilauroyl-PC/cholesterol: nanoscopic domain formation driven by cholesterol. Biophys. J. 80:2775–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veatch, S. L., and S. L. Keller. 2002. Organization in lipid membranes containing cholesterol. Phys. Rev. Lett. 89:268101. [DOI] [PubMed] [Google Scholar]
- 10.Crane, J. M., and L. K. Tamm. 2004. Role of cholesterol in the formation and nature of lipid rafts in planar and spherical model membranes. Biophys. J. 86:2965–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dietrich, C., Z. N. Volovyk, M. Levi, N. L. Thompson, and K. Jacobson. 2001. Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl. Acad. Sci. USA. 98:10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stottrup, B. L., S. L. Veatch, and S. L. Keller. 2004. Nonequilibrium behavior in supported lipid membranes containing cholesterol. Biophys. J. 86:2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gidwani, A., D. Holowka, and B. Baird. 2001. Fluorescence anisotropy measurements of lipid order in plasma membranes and lipid rafts from RBL-2H3 mast cells. Biochemistry. 40:12422–12429. [DOI] [PubMed] [Google Scholar]
- 15.Sinha, M., S. Mishra, and P. G. Joshi. 2003. Liquid-ordered microdomains in lipid rafts and plasma membrane of U-87 MG cells: a time-resolved fluorescence study. Eur. Biophys. J. 32:381–391. [DOI] [PubMed] [Google Scholar]
- 16.Ge, M., K. A. Field, R. Aneja, D. Holowka, B. Baird, and J. Freed. 1999. ESR characterization of liquid ordered phase of detergent resistant membranes from RBL-2H3 cells. Biophys. J. 77:925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge, M., A. Gidwani, H. A. Brown, D. Holowka, B. Baird, and J. H. Freed. 2003. Ordered and disordered phases coexist in plasma membrane vesicles of RBL-2H3 mast cells. An ESR study. Biophys. J. 85:1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veatch, S. L., I. V. Polozov, K. Gawrisch, and S. L. Keller. 2004. Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophys. J. 86:2910–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacia, K., D. Scherfeld, N. Kahya, and P. Schwille. 2004. Fluorescence correlation spectroscopy relates rafts in model and native membranes. Biophys. J. 87:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietrich, C., B. Yang, T. Fujiwara, A. Kusumi, and K. Jacobson. 2002. Relationship of lipid rafts to transient confinement zones detected by single particle tracking. Biophys. J. 82:274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheets, E. D., G. M. Lee, R. Simson, and K. Jacobson. 1997. Transient confinement of a glycosylphosphatidylinositol-anchored protein in the plasma membrane. Biochemistry. 36:12449–12458. [DOI] [PubMed] [Google Scholar]
- 22.Harms, G. S., M. Sonnleitner, G. J. Schutz, H. J. Gruber, and T. Schmidt. 1999. Single-molecule anisotropy imaging. Biophys. J. 77:2864–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum Publishers, New York.
- 24.Holowka, D., J. A. Gosse, A. T. Hammond, X. Han, P. Sengupta, N. L. Smith, A. Wagenknecht-Wiesner, M. Wu, R. M. Young, and B. Baird. 2005. Lipid segregation and IgE receptor signaling: a decade of progress. Biochim. Biophys. Acta. 1743:252–259. [DOI] [PubMed] [Google Scholar]
- 25.Pierini, L., D. Holowka, and B. Baird. 1996. FcɛRI-mediated association of 6-μm beads with RBL-2H3 mast cells results in exclusion of signaling proteins from the forming phagosome and abrogation of normal downstream signaling. J. Cell Biol. 134:1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ariola, F. S., D. J. Mudaliar, R. P. Walvick, and A. Heikal. 2006. Dynamics imaging of lipid-marker interactions in model biomembranes. Phys. Chem. Chem. Phys. 8:4517–4529. [DOI] [PubMed] [Google Scholar]
- 27.Xu, C., W. Zipfel, J. B. Shear, R. M. Williams, and W. W. Webb. 1996. Multiphoton fluorescence excitation: new spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. USA. 93:10763–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hess, S. T., E. D. Sheets, A. Wagenknecht-Wiesner, and A. Heikal. 2003. Quantitative analysis of the fluorescence properties of intrinsically fluorescent proteins in living cells. Biophys. J. 85:2566–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owen, D. M., P. M. P. Lanigan, C. Dunsby, I. Munro, D. Grant, M. A. A. Neil, P. M. W. French, and A. I. Magee. 2006. Fluorescence lifetime imaging provides enhanced contrast when imaging the phase-sensitive dye di-4-ANEPPDHQ in model membranes and live cells. Biophys. J. 90:L80–L82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinosita, K. J., A. Ikegami, and S. Kawato. 1982. On the wobbling-in-cone analysis of fluorescence anisotropy decay. Biophys. J. 37:461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinosita, K. J., S. Kawato, and A. Ikegami. 1977. A theory of fluorescence polarization decay in membranes. Biophys. J. 20:289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rocheleau, J., M. Edidin, and D. W. Piston. 2003. Intrasequence GFP in class I MHC molecules, a rigid probe for fluorescence anisotropy measurements of the membrane environment. Biophys. J. 84:4078–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gryczynski, I., H. Malak, and J. R. Lakowicz. 1995. Three-photon induced fluorescence of 2,5-diphenyloxazole with a femtosecond Ti:sapphire laser. Chem. Phys. Lett. 245:30–35. [Google Scholar]
- 34.Volkmer, A., V. Subramaniam, D. J. S. Birch, and T. M. Jovin. 2000. One- and two-photon excited fluorescence lifetimes and anisotropy decays of green fluorescent proteins. Biophys. J. 78:1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Axelrod, D. 1979. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 26:557–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rusinova, E., V. Tretyachenko-Ladokhina, O. E. Vele, D. F. Senear, and J. B. A. Ross. 2002. Alexa and Oregon Green dyes as fluorescence anisotropy probes for measuring protein-protein and protein-nucleic acid interactions. Anal. Biochem. 308:18–25. [DOI] [PubMed] [Google Scholar]
- 37.Young, R. M., D. Holowka, and B. Baird. 2003. A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. J. Biol. Chem. 278:20746–20752. [DOI] [PubMed] [Google Scholar]
- 38.Packard, B. S., and D. E. Wolf. 1985. Fluorescence lifetimes of carbocyanine lipid analogues in phospholipid bilayers. Biochemistry. 24:5176–5181. [DOI] [PubMed] [Google Scholar]
- 39.Rahman, N. A., I. Pecht, D. A. Roess, and B. G. Barisas. 1992. Rotational dynamics of type I Fc epsilon receptors on individually-selected rat mast cells studied by polarized fluorescence depletion. Biophys. J. 61:334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Windengren, J., and P. Schwille. 2000. Characterization of photo-induced isomerization and back-isomerization of the cyanine dye Cy5 by fluorescence correlation spectroscopy. J. Phys. Chem. 104:6416–6428. [Google Scholar]
- 41.Agronskaia, A. V., L. Tertoolen, and H. C. Gerritsen. 2003. High frame rate fluorescence lifetime imaging. J. Phys. D: Appl. Phys. 36:1655–1662. [Google Scholar]
- 42.Pyenta, P. S., P. Schwille, W. W. Webb, D. Holowka, and B. Baird. 2003. Lateral diffusion of membrane lipid-anchored probes before and after aggregation of cell surface IgE-receptors. J. Phys. Chem. A. 107:8310–8318. [Google Scholar]
- 43.Holowka, D., T. Wensel, and B. Baird. 1990. A nanosecond fluorescence depolarization study on the segmental flexibility of receptor-bound immunoglobulin E. Biochemistry. 29:4607–4612. [DOI] [PubMed] [Google Scholar]
- 44.Myers, J. N., D. Holowka, and B. Baird. 1992. Rotational motion of monomeric and dimeric immunoglobulin E-receptor complexes. Biochemistry. 31:567–575. [DOI] [PubMed] [Google Scholar]
- 45.Song, J., G. M. Hagen, D. A. Roess, I. Pecht, and B. G. Barisas. 2002. The mast cell function-associated antigen and its interactions with the type I Fcɛ receptor. Biochemistry. 41:881–889. [DOI] [PubMed] [Google Scholar]
- 46.Zidovetski, R., M. Bartholdi, D. Arndt-Jovin, and T. M. Jovin. 1986. Rotational dynamics of the Fc receptor for immunoglobulin E on histamine-releasing rat basophilic leukemia cells. Biochemistry. 25:4397–4401. [DOI] [PubMed] [Google Scholar]
- 47.Vishwasrao, H. D., A. A. Heikal, K. A. Kasischke, and W. W. Webb. 2005. Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. J. Biol. Chem. 280:25119–25126. [DOI] [PubMed] [Google Scholar]