

Abstract
The SecA protein is present in all bacteria, and it is a central component of the general Sec-dependent protein export pathway. An unusual property of Mycobacterium tuberculosis is the presence of two SecA proteins: SecA1, the essential “housekeeping” SecA, and SecA2, the accessory secretion factor. Here, we report that a ΔsecA2 mutant of M. tuberculosis was defective for growth in the early stages of low-dose aerosol infection of C57BL/6 mice, a time during which the bacillus is primarily replicating in macrophages. Consistent with this in vivo phenotype, we found that the ΔsecA2 mutant was defective for growth in macrophages from C57BL/6 mice. The ΔsecA2 mutant was also attenuated for growth in macrophages from phox−/− mice and from NOS2−/− mice. These mice are defective in the reactive oxygen intermediate (ROI)-generating phagocyte oxidase and the reactive nitrogen intermediate (RNI)-generating inducible nitric oxide synthase, respectively. This indicated a role for SecA2 in the intracellular growth of M. tuberculosis that is independent of protecting against these ROIs or RNIs. Macrophages infected with the ΔsecA2 mutant produced higher levels of tumor necrosis factor alpha, interleukin-6, RNI, and gamma interferon-induced major histocompatibility complex class II. This demonstrated a function for M. tuberculosis SecA2 in suppressing macrophage immune responses, which could explain the role of SecA2 in intracellular growth. Our results provide another example of a relationship between M. tuberculosis virulence and inhibition of the host immune response.
Mycobacterium tuberculosis, the causative agent of tuberculosis, is one of the most successful bacterial pathogens of all time. The global burden of tuberculosis is at its highest level ever and includes an increasing number of multiple-drug-resistant M. tuberculosis infections (74). Novel antituberculosis prevention and treatment measures are needed to control this health crisis, and a complete understanding of M. tuberculosis pathogenesis will help achieve this goal.
M. tuberculosis is inhaled and taken up by unactivated host macrophages, where it is able to replicate and survive. Much remains to be learned about how M. tuberculosis avoids host defenses in these macrophages. The processes involved are likely complex and multifactorial. Among the properties implicated in M. tuberculosis survival in macrophages are the abilities of the bacillus to block acidification of the phagocytic vacuole, to block phagosome-lysosome fusion, and to resist reactive oxygen intermediates (ROI) and reactive nitrogen intermediates (RNI) (34, 67).
The early innate immune response to M. tuberculosis includes Toll-like receptor (TLR) stimulation of macrophages (35, 55). TLR signaling leads to the secretion of RNI and proinflammatory cytokines that include tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). Later in infection, activation of host macrophages by an adaptive TH1 immune response occurs, leading to the inhibition of intracellular growth and control of M. tuberculosis infection. Gamma interferon (IFN-γ) is an important mediator of this response; it upregulates antimycobacterial processes and antigen presentation by macrophages (22). One of the antimycobacterial effectors induced by IFN-γ is RNI, which is produced by the inducible nitric oxide synthase (NOS2), although other IFN-γ-dependent effector mechanisms exist (13, 39, 40). TNF-α is another cytokine that plays an integral role in host control of M. tuberculosis infection (6, 8, 23). Among the many properties reported for TNF-α is that it can synergize with IFN-γ to induce potent antimycobacterial activity of macrophages (13, 21, 22).
It is increasingly apparent that M. tuberculosis can limit the host immune response and macrophage activation and that this inhibition is likely to be important for survival in macrophages (34). Although results are dependent on experimental conditions, there are several reports of macrophages infected with more-virulent M. tuberculosis producing lower levels or activities of TNF-α and/or RNI than macrophages infected with less virulent or attenuated mycobacteria (2, 7, 20, 58, 65). In a more direct fashion, M. tuberculosis has been shown to suppress expression of Escherichia coli-induced proinflammatory IL-12 (45). M. tuberculosis is also known to inhibit IFN-γ upregulation of genes, including the major histocompatibility complex (MHC) class II genes (25, 43, 50, 53). Inhibition of MHC class II reduces antigen presentation, which has implications for the development of an effective TH1 response in vivo.
A common theme in bacterial pathogenesis is the importance of protein export and secretion pathways of the pathogen to virulence. These pathways export proteins beyond the cytoplasm to the cell surface or secrete proteins out of the bacterium. Both surface and secreted proteins are exposed to the external environment. As a result, these proteins are ideally positioned to protect the bacterium from macrophage attack or to modify the host immune response to the bacillus (14, 36). Like all bacteria, M. tuberculosis has the conventional Sec pathway and Sec proteins for exporting proteins beyond the cytoplasm (36). More unusual is the presence of two SecA proteins (SecA1 and SecA2) in M. tuberculosis (9). We have shown that the SecA2 protein of M. tuberculosis is an accessory secretion factor that promotes secretion of a subset of proteins that include superoxide dismutase (SodA) and catalase peroxidase (KatG) (10). Both of these enzymes detoxify oxygen radicals: SodA converts superoxide to hydrogen peroxide and oxygen, and KatG converts hydrogen peroxide to water and oxygen. KatG is also able to break down peroxynitrite, which is a dangerous reaction product of superoxide and nitric oxide (72). SecA2 contributes to the virulence of M. tuberculosis, as shown previously using a ΔsecA2 mutant of M. tuberculosis in a high-dose murine model of tuberculosis (10).
Here, we describe our studies of the ΔsecA2 mutant of M. tuberculosis in an aerosol model of murine infection and in murine bone marrow-derived macrophages. Our results with the two models were consistent and revealed a role for SecA2 in promoting M. tuberculosis growth early in murine infection and in unactivated macrophages. Using macrophages from phox−/− and NOS2−/− mice, we further showed that the role of SecA2 is not simply explained by a failure to resist ROI produced by the phagocyte oxidase or RNI produced by the inducible nitric oxide synthase. In comparing macrophage responses to infection with the ΔsecA2 mutant and the parental M. tuberculosis strain H37Rv, we discovered that macrophages infected with the ΔsecA2 mutant were more activated on the basis of several criteria: increased TNF-α, IL-6, RNI, and IFN-γ-regulated MHC class II. This indicated a role for SecA2 in M. tuberculosis inhibition of the immune response, which is likely important for survival in the host and pathogenesis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The Mycobacterium tuberculosis strains used in this study, H37Rv, mc23112 (ΔsecA2 mutant, stock derived from a single colony), and mc23116 (ΔsecA2, attB::secA2) (10), were grown in Middlebrook 7H9 broth (Difco) with 0.2% glycerol, 1× albumin dextrose saline (ADS), and 0.05% Tween 80 (Tw). When appropriate, the medium was supplemented with the antibiotic kanamycin (20 μg/ml).
Animals.
Female C57BL/6 mice were purchased from Charles River Laboratories. p47phox−/− mice were generated and maintained as described previously (4). gp91phox−/− mice and NOS2−/− mice were purchased from Jackson Laboratories. All mice were housed in sterile microbarrier cages and were given autoclaved food and water ad libitum. Both strains of phox−/− mice were maintained on an antibiotic oral suspension of sulfamethoxazole (200 mg/5 ml) and trimethoprim (40 mg/5 ml) (Hi-Tech Pharmacal) in their water to prevent opportunistic infections.
Aerosol infection.
Female C57BL/6 mice aged 38 to 45 days were used for the aerosol studies. The M. tuberculosis strains were cultured to mid-log phase (optical density at 600 nm of ∼0.5 to 1.0). The cultures were washed one time and resuspended in phosphate-buffered saline (PBS) with 0.05% Tween 80 (PBS-Tw) to a concentration of 1 × 107 CFU/ml. The bacterial suspension was placed into the nebulizer jar of a whole-body exposure aerosol chamber (Mechanical Engineering Workshop, Madison, WI). Mice were exposed for 15 min with a chamber purge time of 20 min. At specific time points, four mice were sacrificed from each group by CO2 asphyxiation and the lungs, livers, and spleens were removed and homogenized in PBS-Tw with 100 ng/ml cycloheximide and 50 μg/ml carbenicillin. The homogenates were plated onto 7H10-ADS-glycerol plates with 10 mg/ml cycloheximide for CFU enumeration.
Macrophage infections.
Bone marrow-derived macrophages were prepared from C57BL/6, p47phox−/−, gp91phox−/−, and NOS2−/− mice as follows. Mice were sacrificed by CO2 asphyxiation, and the femurs were removed. The cells were flushed out of the femurs by using supplemented Dulbecco modified Eagle medium (DMEM; Sigma) containing 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, and 1× nonessential amino acids. The cells were washed twice and cultured in supplemented DMEM for 6 days in the presence of 20% L929-conditioned medium (LCM). After 6 days, the cells were removed using 5 mM EDTA (in PBS), washed twice with supplemented DMEM, and resuspended in supplemented DMEM with 10% LCM. The cells were then seeded into the wells of either an 8-well chamber slide (2 × 105 macrophages/well) or a 24-well plate (8 × 105 macrophages/well) and were allowed to adhere overnight before infection. In some experiments, the macrophages were pretreated for 24 h with 10 ng/ml recombinant murine IFN-γ (rmIFN-γ; Chemicon) before infection. M. tuberculosis strains were taken from mid-log growth phase and washed one time in PBS-Tw. Cells were resuspended in PBS-Tw and further diluted to the appropriate number of CFU/ml in supplemented DMEM. The bacteria were added at the appropriate concentration to the cell monolayers to achieve a multiplicity of infection (MOI) of 1 or 10. Macrophages were infected for 4 h, at which time the monolayers were washed three times with supplemented DMEM to remove any non-cell-associated bacteria, and then fresh medium with 10% LCM was added back. At various time points, the medium was removed from the wells and the cells were lysed with 0.1% Tween 80 and vigorous pipetting. Lysates were diluted in PBS-Tw and plated onto 7H10-ADS-glycerol-0.05% Tw plates for enumeration of CFU.
RNI measurements.
To measure RNI, we used Griess reagent (Molecular Probes) and followed the manufacturer's protocol. Briefly, supernatants from infected macrophage monolayers were filter sterilized twice using a 0.2-μm low-protein binding filter and mixed 1:1 with Griess reagent. Samples were measured at 548 nm, and the values were converted to μM nitrite by using a standard curve generated with sodium nitrite.
In vitro sensitivity assays.
Bacteria were grown to mid-log phase in 7H9-ADS-glycerol-Tw, washed, and diluted to a density of approximately 1 × 106 CFU/ml in PBS-Tw. Percent killing was determined for each strain by comparing the number of CFU from treated samples to that from untreated samples. The following treatments were employed: heat shock (53°C for 45 min) and acid pH (pH 4.0 for 24 h). In vitro ROI killing assays were performed using various compounds. We tested sensitivity to hypoxanthine-xanthine oxidase (0 and 3 h; 250 μΜ hypoxanthine [Sigma] and 0.1 U/ml xanthine oxidase [Roche]) in the presence or absence of catalase (1 U/ml; Sigma), added to detoxify the hydrogen peroxide generated (16, 54). Other ROI-generating compounds tested include hydrogen peroxide (0, 5, and 10 mM for 4 h), plumbagin (0.2 mM for 3.5, 7.5, and 10.5 h), pyrogallol (2 mM for 0, 1, and 4 h), and cumene hydroperoxide (0.5 mM for 0, 1, and 4 h) (Sigma). In vitro sensitivity to RNI was tested using spermidine NONOate (SPER/NO; 200 μM for 0 and 4 h; Alexis Biochemicals). All in vitro ROI and RNI assays were performed at 37°C.
CBA.
Infection of bone marrow-derived macrophages was performed as described above, with cells being infected at an MOI of 1. Supernatants were collected from the infected macrophages at 24 h postinfection, filtered twice through a 0.22-μm low-protein binding filter, and stored at −80°C. Cytometric bead array (CBA) was performed using a mouse inflammation CBA Kit (BD Biosciences) according to the manufacturer's protocol. Samples were analyzed on a BD Biosciences FACSCalibur flow cytometer. The triplicate samples for each strain were averaged, and the data are shown as means ± standard deviations.
Quantitative real-time PCR (qRT-PCR) to measure IFN-γ responses of infected macrophages.
Murine bone marrow-derived macrophages were prepared and infected as described above at an MOI of 10 for 3 h, at which time unincorporated bacteria were washed off and fresh medium was added back to the monolayers. At 21 h postinfection, the medium was removed from the monolayers and replaced with fresh medium containing 2 ng/ml rmIFN-γ. After 15 h of rmIFN-γ treatment, the supernatants were removed and the monolayers lysed with TRIzol reagent (Invitrogen). The human monocytic cell line THP-1 (ATCC TIB-202) was maintained in supplemented RPMI with 10% heat-inactivated FBS. To prepare the THP-1 cells for infection, we washed them twice in fresh RPMI with FBS, resuspended them in RPMI with FBS containing 50 ng/ml phorbol myristate acetate (Sigma), and seeded them into six-well tissue culture plates at 2 × 106 cells/well. After 24 h of phorbol myristate acetate treatment, the cells were infected at an MOI of 10 in the presence of 200 ng/ml recombinant human IFN-γ (rhIFN-γ; Pierce) for 4 h. After uptake, the cells were washed and fresh medium with rhIFN-γ was added. At 24 h postinfection, the supernatant was removed from the infected monolayers and the cells were lysed with TRIzol. TRIzol lysates were processed for RNA extraction. RNA samples were reverse transcribed, and real-time PCR was performed on cDNA using the TaqMan sequence detection system with ABsolute QPCR mix (Applied Biosystems). Values were calculated based on the standard curves generated for each gene. The normalization of samples was determined by dividing the number of copies of MHC class II mRNA by the number of copies of 18S rRNA. The following primers were used with the indicated probes: 18S probe (5′-tetrachloro-6-carboxyfluorescein-CAAATTACCCACTCCCGACCCG-6-carboxytetramethylrhodamine [TAMRA]-3′), primers F (5′-GCTGCTGGCACCAGACTT-3′) and R (5′-CGGCTACCACATCCAAGG-3′); murine MHC class II (I-Ab) probe (5′-6-carboxyfluorescein-CGCAGCGCATACGATATGTGACCA-TAMRA-3′), primers Rev (5′-CTGTCGTAGCGCACGTACT-3′) and For (5′-GGCGAGTGCTACTTCACCA-3′); human MHC class II (HLA-DRA) probe (5′-6-carboxyfluorescein-CTGGACCCTTTGCAAGAACCCTTCCC-TAMRA-3′), primers F (5′-TCCAATGAACGGAGTATCTTGTGT-3′) and R (5′-TGAGATGACGCATCTGTTGCT-3′) (28, 73).
RESULTS
The ΔsecA2 mutant is attenuated in the murine low-dose aerosol infection model.
Previously, we demonstrated that an in-frame and unmarked ΔsecA2 deletion mutant of M. tuberculosis is attenuated in mice following high-dose intravenous administration (10). To better characterize the phenotype of the ΔsecA2 mutant, we used a more natural low-dose aerosol exposure model, infecting C57BL/6 mice with approximately 200 bacilli to the lungs, and included earlier time points than previously analyzed. Following infection with the ΔsecA2 mutant or the virulent M. tuberculosis parent H37Rv, groups of mice were sacrificed at various times postinfection and the bacterial burden in the lungs, livers, and spleens was enumerated by plating homogenates for CFU. We observed a typical growth pattern for H37Rv in the lungs of the mice: significant growth during the early phase of infection, followed by a period of persistence in which the number of bacilli remained constant over time (Fig. 1A) (52). The transition between the growth and persistence phases correlates with the establishment of the TH1 response, a central feature of which is macrophage activation by IFN-γ (49, 52). The ΔsecA2 mutant was defective in the early growth phase of the infection compared to H37Rv. As early as 9 days postinfection, the ΔsecA2 mutant had 1 log fewer bacilli in the lungs than H37Rv (Fig. 1A). This difference was maintained throughout the experiment, although both strains continued to grow to day 16. We also observed fewer CFU for the ΔsecA2 mutant in livers and spleens (0.5 to 1.0 log less) than for H37Rv (data not shown). During the later phase of infection, the ΔsecA2 mutant resembled H37Rv in persistence, although at the final 80-day time point, the CFU burden in the lungs of the ΔsecA2 mutant-infected mice had dropped slightly. In addition to reduced bacillary load, mice infected with the ΔsecA2 mutant exhibited an increase in length of survival (372 ± 29 days) in comparison to mice infected with H37Rv (173 ± 16 days) (Fig. 1B).
FIG. 1.

The ΔsecA2 mutant of M. tuberculosis is attenuated in a low-dose aerosol infection of C57BL/6 mice. Immunocompetent C57BL/6 mice were exposed in a whole-body aerosol chamber to either M. tuberculosis H37Rv (▪) or the ΔsecA2 mutant (○), with each mouse receiving approximately 200 CFU per lung. (A) Mice were sacrificed at various time points postinfection, and the growth of the strains was assessed by plating for CFU from lung homogenates. The results are representative of two experiments. *, P values compared to the ΔsecA2 mutant are <0.005 by Student's t test. (B) Groups of four mice infected with either strain were monitored for survival.
The ΔsecA2 mutant has an attenuated phenotype in unactivated macrophages but not in activated macrophages.
The observed in vivo phenotype of the ΔsecA2 mutant indicated a role for SecA2 in the early phase of infection, during which M. tuberculosis is growing in macrophages. We hypothesized that the ΔsecA2 mutant is defective for growth in unactivated macrophages. We tested this hypothesis by comparing the abilities of the ΔsecA2 mutant and H37Rv to replicate within unactivated bone marrow-derived macrophages from C57BL/6 mice. The macrophages were infected ex vivo, and after 4 hours of uptake, the monolayers were washed and fresh medium was added back. The growth of each strain was evaluated over a 5-day period by plating macrophage lysates for viable CFU. The ΔsecA2 mutant showed diminished growth in the macrophages and displayed up to 1 log fewer CFU by day 5 (Fig. 2A). The intracellular growth defect of the ΔsecA2 mutant was complemented by introduction of a wild-type copy of secA2 on an integrating plasmid, which indicated that the mutant phenotype was due to the absence of secA2 (Fig. 2B).
FIG. 2.

The ΔsecA2 mutant of M. tuberculosis is attenuated in unactivated murine bone marrow-derived macrophages. Unactivated murine bone marrow-derived macrophages from C57BL/6 mice were infected at an MOI of 1 with (A) M. tuberculosis H37Rv (▪) or the ΔsecA2 mutant (○) and (B) the ΔsecA2 mutant (○) or the ΔsecA2-complemented mutant (ΔsecA2, attB::secA2) (▴). The numbers of CFU were determined by plating macrophage lysates at various time points postinfection. The infection was performed with triplicate wells for each strain per time point, and the error bars represent standard deviations for the triplicate wells. The data shown are representative of more than 20 experiments for the wild type and the ΔsecA2 mutant and 5 experiments for the complemented strain. *, P values compared to the ΔsecA2 mutant are <0.05 by Student's t test.
In contrast to unactivated macrophages, which are permissive for M. tuberculosis growth, activated macrophages inhibit M. tuberculosis growth due to enhanced antimycobacterial activities (13, 21, 22). We compared the ΔsecA2 mutant and H37Rv in murine bone marrow-derived macrophages activated by 24 h of pretreatment with rmIFN-γ. In the IFN-γ-treated macrophages, H37Rv failed to grow but survived over time, and the ΔsecA2 mutant behaved similarly (Fig. 3A). Infection of unactivated macrophages was performed in parallel and again revealed an intracellular growth defect of the ΔsecA2 mutant (Fig. 3B).
FIG. 3.

Survival of the ΔsecA2 mutant in IFN-γ-treated murine bone marrow-derived macrophages. In parallel experiments, we examined the survival and growth of the ΔsecA2 mutant and H37Rv in (A) activated and (B) unactivated murine bone marrow-derived macrophages from C57BL/6 mice. Macrophages were activated by 24 h pretreatment with 10 ng/ml rmIFN-γ. The macrophages were infected at an MOI of 1 with H37Rv (▪) or the ΔsecA2 mutant (○) as described above. Graphs are shown as the averages from five experiments ± standard errors of the means. *, P values compared to ΔsecA2 mutant are <0.05 by Student's t test.
These results revealed a role for SecA2 in promoting M. tuberculosis growth in unactivated macrophages but no apparent role for SecA2 in M. tuberculosis survival in activated macrophages. They are consistent with the pattern of growth in vivo, where the ΔsecA2 mutant exhibited reduced growth in the lungs of infected mice in the early growth phase of infection and then behaved similarly to H37Rv after the onset of the TH1 immune response and concomitant macrophage activation.
The ΔsecA2 mutant has an attenuated phenotype in oxidative burst-deficient macrophages.
We previously reported that the ΔsecA2 mutant of M. tuberculosis is defective in secreting the antioxidant enzymes SodA and KatG (10). M. tuberculosis is relatively resistant to ROI (13), which could be an important property for intracellular survival in the face of the macrophage oxidative burst, and the SecA2-dependent secreted SodA and KatG are among the M. tuberculosis molecules implicated in this resistance (67). We tested whether the sole role of SecA2 in macrophage growth is to protect the bacillus from the oxidative burst during intracellular infection. Bone marrow-derived macrophages from p47 phox−/−, gp91phox−/−, and C57BL/6 mice were prepared and infected in parallel with the ΔsecA2 mutant or H37Rv. These phox−/− mice lack different components of the phagocyte oxidase complex, which is responsible for the oxidative burst of macrophages (46). In these oxidative burst-deficient macrophages, the ΔsecA2 mutant showed the same growth defect as that observed in wild-type C57BL/6 macrophages (Fig. 4A and B). This indicated that SecA2 contributes to intracellular growth even in the absence of an oxidative burst. Consequently, SecA2 must have a function in intracellular growth other than just protecting the bacillus from ROI generated during the oxidative burst.
FIG. 4.

The ΔsecA2 mutant is attenuated in bone marrow-derived macrophages from mice defective in components of the phagocyte oxidase (phox−/−). Unactivated bone marrow-derived macrophages from C57BL/6 mice and from either (A) p47phox −/− or (B) gp91phox−/− mice were infected in parallel at an MOI of 1 with M. tuberculosis H37Rv (▪, in C57BL/6 macrophages; □, in phox−/− macrophages) and the ΔsecA2 mutant (•, in C57BL/6 macrophages; ○, in phox−/− macrophages). Graphs are shown as the averages from multiple experiments (six for p47phox−/− mice and three for gp91phox−/− mice) ± standard errors of the means. *, P values compared to the ΔsecA2 mutant are <0.05 by Student's t test.
As an independent assessment of the role of SecA2 in protecting against oxygen radicals, we compared in the vitro sensitivities of the ΔsecA2 mutant and H37Rv to those of a variety of ROI species. Among the compounds tested were pyrogallol and hypoxanthine-xanthine oxidase; both of these treatments generate extracellular superoxide. Plumbagin (cytoplasmic superoxide generator), hydrogen peroxide, and cumene hydroperoxide were also tested. In all cases, the ROI treatments showed equivalent degrees of killing for the ΔsecA2 mutant and H37Rv (data not shown). We further tested the strains for their sensitivity to other stresses, including acid pH and heat shock, and again saw no differences in killing. The unaltered in vitro sensitivities of the ΔsecA2 mutant in combination with the data for phox−/− macrophages suggested that the role of SecA2 in promoting growth in macrophages is not solely to protect the bacteria from damaging ROI but involves other mechanisms.
Macrophages infected with the ΔsecA2 mutant release higher levels of proinflammatory cytokines and RNI.
To determine whether the SecA2-dependent secretion pathway contributes to M. tuberculosis inhibition of host immune responses, we compared levels of cytokine production in macrophages infected with the ΔsecA2 mutant and H37Rv. Bone marrow-derived macrophages from C57BL/6 mice were infected as described before, and at 24 h postinfection, supernatants were assayed for cytokine production using the cytometric bead assay (BD Biosciences). Macrophages infected with the ΔsecA2 mutant produced more TNF-α and IL-6 than those infected with H37Rv (Fig. 5), with an average difference of 2.5-fold for TNF-α and 3.5-fold for IL-6. The introduction of an integrated plasmid expressing wild-type secA2 complemented the increased cytokine production phenotype of the ΔsecA2 mutant.
FIG. 5.
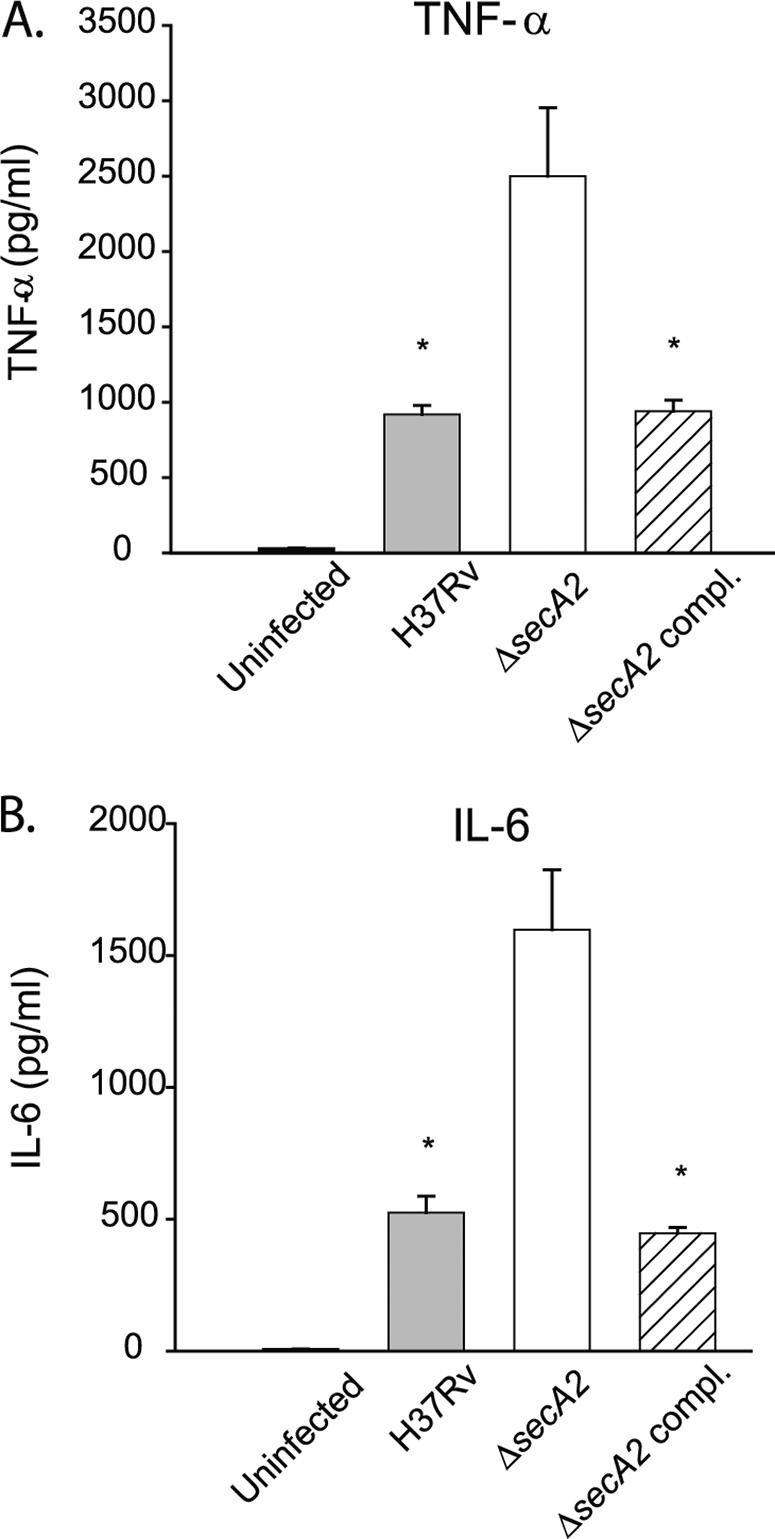
Cytokine release from infected bone marrow-derived macrophages. Cytokines were measured using the cytometric bead array kit (BD Biosciences). Bone marrow-derived macrophages from C57BL/6 mice were infected at an MOI of 1 in triplicate. Supernatants were collected at 24 h postinfection and assayed for cytokines. The graphs present levels of cytokine for (A) TNF-α or (B) IL-6 in uninfected macrophages (black bars) or those infected with H37Rv (gray bars), the ΔsecA2 mutant (white bars), or the ΔsecA2-complemented strain (hatched bars), with error bars depicting standard deviations for triplicate samples. The data shown are representative of eight experiments. *, P values compared to the ΔsecA2 mutant are <0.05 by Student's t test.
We also assayed the production of RNI in the supernatants of infected macrophages by using Griess reagent, which measures nitrite, the oxidation product of nitric oxide. Measurements were taken at 48, 72, and 96 h postinfection. The macrophages infected with the ΔsecA2 mutant produced more RNI at all time points than the macrophages infected with H37Rv (Fig. 6A), with an average difference of 1.5-fold. In the same experiments at 0, 4, and 24 h postinfection, we also measured the levels of ROI in the cells and saw no differences in levels of ROI between cells infected with the ΔsecA2 mutant and H37Rv (data not shown). We also tested the levels of RNI production in infected cells pretreated with IFN-γ 24 h prior to infection which upregulates RNI production. As expected, there was an overall increase in RNI production in cells pretreated with IFN-γ compared to that in untreated cells. At 24 h postinfection, macrophages infected with the ΔsecA2 mutant again exhibited higher levels of RNI in the supernatant (Fig. 6B). The increased RNI production observed following infection with the ΔsecA2 mutant in the absence and presence of IFN-γ was complemented by the introduction of a wild-type copy of secA2.
FIG. 6.

RNI produced by infected bone marrow-derived macrophages. RNI was measured using Griess reagent. Infections were performed at an MOI of 10, and samples were collected at 72 h postinfection for unactivated macrophages (A) and at 24 h postinfection for IFN-γ-activated macrophages (B). Bars show the averages of triplicate samples for uninfected cells (black bars) or those infected with H37Rv (gray bars), the ΔsecA2 mutant (white bars), or the ΔsecA2-complemented strain (hatched bars), with error bars depicting standard deviations for triplicate samples. The data shown are representative of three experiments. *, P values compared to the ΔsecA2 mutant are <0.05 by Student's t test.
It is important to note that in these experiments, the numbers of CFU in macrophage lysates following the 4-h infection period were equivalent for both strains. The above results suggest that, in comparison to H37Rv, the ΔsecA2 mutant is defective in the ability to inhibit macrophage production of immunostimulatory molecules, resulting in more highly activated macrophages upon infection with the ΔsecA2 mutant.
The ΔsecA2 mutant has an attenuated phenotype in NOS2−/− macrophages.
Given the antimycobacterial activity of RNI, we considered the possibility that the increased levels of RNI observed were alone responsible for the growth defect of the ΔsecA2 mutant in macrophages. To test this idea, we examined the growth of the ΔsecA2 mutant and H37Rv in unactivated macrophages from NOS2−/− mice, which lack the inducible nitric oxide synthase and are defective for the nitrosative burst (38). The ΔsecA2 mutant still exhibited a growth defect in NOS2−/− macrophages that was equivalent to the defect observed in macrophages from C57BL/6 mice (Fig. 7). These data indicated that the role of SecA2 in intracellular growth involves functions other than protecting against reactive nitrogen intermediates.
FIG. 7.

The ΔsecA2 mutant is attenuated in bone marrow-derived macrophages from mice defective in inducible nitric oxide synthase (NOS2). Unactivated bone marrow-derived macrophages from C57BL/6 mice and from NOS2−/− mice were infected in parallel at an MOI of 1 with M. tuberculosis H37Rv (▪, in C57BL/6 macrophages; □, in NOS2−/− macrophages) and the ΔsecA2 mutant (•, in C57BL/6 macrophages; ○, in NOS2−/− macrophages) as described above. Graphs are shown as the averages from four experiments ± standard errors of the means. *, P values compared to the ΔsecA2 mutant are <0.05 by Student's t test.
As was done for ROI, we also assayed in vitro sensitivity of the ΔsecA2 mutant and H37Rv to NO generated by SPER/NO. Treatment with SPER/NO led to equivalent degrees of killing of the ΔsecA2 mutant and H37Rv (data not shown). The in vitro sensitivity result taken together with the observed growth defect of the ΔsecA2 mutant in NOS2−/− macrophages suggests that the role of SecA2 in intracellular growth is not solely to protect against RNI.
The ΔsecA2 mutant is associated with higher IFN-γ-induced MHC class II expression.
Having demonstrated that macrophages infected with the ΔsecA2 mutant produced higher levels of cytokine and RNI in comparison to H37Rv-infected macrophages, we set out to see whether the inhibition of other macrophage responses was altered by the ΔsecA2 mutation. Multiple laboratories have demonstrated that M. tuberculosis inhibits the expression of several IFN-γ-regulated genes of the host. Among these genes are the IFN-γ-induced MHC class II genes required for antigen presentation (43, 50, 53). To test for a role of SecA2 in M. tuberculosis inhibition of IFN-γ responses, we employed two different systems previously used to study this IFN-γ inhibition: primary murine macrophages and the human monocytic cell line THP-1. Murine bone marrow-derived macrophages were preinfected with the ΔsecA2 mutant, H37Rv, or the complemented strain at an MOI of 10 for 24 h, followed by stimulation with rmIFN-γ. After 15 h of IFN-γ treatment, expression of MHC class II (IA-b) transcript was measured by qRT-PCR. As expected, IFN-γ treatment of uninfected cells increased MHC class II transcript and H37Rv inhibited the IFN-γ induction of MHC class II. In contrast, infection with the ΔsecA2 mutant did not reduce the level of MHC class II to the same degree as infection with H37Rv (Fig. 8A). This difference between the ΔsecA2 mutant and H37Rv in the level of IFN-γ-induced MHC class II was at least twofold greater with the ΔsecA2 mutant in five separate experiments (P = 0.008 by the Mann-Whitney U test). Similar experiments were performed with human THP-1 cells. In these experiments, rhIFN-γ was added to the cells at the time of infection with either the ΔsecA2 mutant, H37Rv, or the complemented strain. Expression of MHC class II (HLA-DR) transcript was measured by qRT-PCR of samples collected at 24 h postinfection. Again, infection with the ΔsecA2 mutant did not reduce the level of MHC class II to the same degree as infection with H37Rv. This difference between the ΔsecA2 mutant and H37Rv in the level of IFN-γ-induced MHC class II was reproducible with the ΔsecA2 mutant, exhibiting an average 1.5-fold difference in HLA-DR transcript level over five independent experiments (P = 0.00003 by Student's t test). The phenotypes of the ΔsecA2 mutant in both murine macrophages and THP-1 cells were complemented by the introduction of a wild-type copy of secA2. These data provided another example of SecA2 functioning in the process(es) of M. tuberculosis inhibition of host immune responses.
FIG. 8.

Inhibition of MHC class II expression measured by quantitative real-time PCR. Host mRNA expression was determined by qRT-PCR of cDNA made from murine bone marrow-derived macrophages or THP-1 cells. Cells were infected with M. tuberculosis strains at an MOI of 10 and stimulated with recombinant IFN-γ, and uninfected cells were similarly treated with recombinant IFN-γ. For murine macrophages, the cells were infected with M. tuberculosis strains and allowed to sit for 21 h prior to the addition of 2 ng/ml rmIFN-γ. RNA was sampled at 15 h post-recombinant IFN-γ addition. For THP-1 cells, the cells were simultaneously infected with M. tuberculosis and treated with 200 ng/ml rhIFN-γ. RNA was sampled at 24 h postinfection. The qRT-PCR samples were normalized to an internal 18S control. Bars represent the normalized MHC class II levels of triplicate infections performed on a single day, with error bars showing standard deviations. Data are representative of five experiments for (A) murine bone marrow-derived macrophages and (B) THP-1 cells. Samples are shown as uninfected cells (black bars) or cells infected with H37Rv (gray bars), the ΔsecA2 mutant (white bars), or the complemented (compl.) strain (hatched bars). *, P values compared to the wild-type and complemented strains are <0.003 by Student's t test. Attmol, attomoles.
DISCUSSION
The accessory secretion factor SecA2 contributes to M. tuberculosis virulence. Here, we showed with a natural low-dose aerosol model of infection that as early as 9 days postinfection, the ΔsecA2 mutant had a lower bacterial burden in the lungs of mice than the parental M. tuberculosis strain H37Rv. The attenuated phenotype of the mutant occurred during the early growth phase of infection, when M. tuberculosis is primarily growing in unactivated macrophages. Consistent with this result, we showed that the ΔsecA2 mutant had a growth defect in unactivated murine bone marrow macrophages in comparison to H37Rv. It should be noted that the ΔsecA2 mutant does not display any significant growth defect when grown in broth media (10). In contrast to the phenotype in unactivated murine macrophages, the ΔsecA2 mutant did not exhibit a phenotype in IFN-γ-activated macrophages. Our experiments in vitro with primary murine macrophages appear to reflect the phenotypes observed in vivo in the murine model of infection.
Since SecA2 is an accessory secretion factor, the role of SecA2 could be the proper secretion or surface localization of M. tuberculosis proteins required for intracellular growth. Previously, we identified SodA and KatG as proteins whose secretion into culture media depends on SecA2 (10). Since both of these proteins are antioxidants, we considered the simple hypothesis that SecA2 promotes detoxification of ROI and thereby protects the bacillus from the oxidative burst of macrophages to promote intracellular growth. Macrophage-generated ROI could have direct antimicrobial effects or indirect effects involving alterations in host cell signaling pathways (44, 69). There is a precedent for exported superoxide dismutases contributing to virulence in bacterial pathogens (5), and various experiments suggest roles for M. tuberculosis SodA, KatG, and a surface-localized superoxide dismutase, SodC, in pathogenesis and protection against the oxidative burst of macrophages (17, 18, 41, 47, 54). However, when we tested the ΔsecA2 mutant in phox−/− macrophages, it exhibited a growth defect equivalent to that observed in wild-type C57BL/6 macrophages. In addition, our efforts to detect altered in vitro sensitivity of the ΔsecA2 mutant to ROI species, including extracellularly generated superoxide, were unsuccessful. Taken together, these experiments do not reveal a role for SecA2 in protecting against the oxidative burst and they indicate an alternate function of SecA2 in intracellular growth.
Yet, we cannot entirely rule out the possibility that SecA2 contributes to ROI resistance. A role in protecting against ROI in macrophages and in vitro could have been masked by the another function(s) of SecA2 in intracellular growth and/or redundant ROI resistance mechanisms of M. tuberculosis. Furthermore, even in the absence of phagocyte oxidase, there are still reactive oxygen species generated in the cell through mitochondrial electron transport. Our experiments cannot rule out the possibility that mitochondrion-generated ROI contribute to the phenotypes of the ΔsecA2 mutant.
In our studies, we also assessed the role that SecA2 plays in modulating the immune response. It is becoming evident that virulent M. tuberculosis inhibits innate and adaptive immune responses. In regard to innate immune responses, M. tuberculosis has been shown to inhibit macrophage production of IL-12 in a coinfection assay (45). In addition, there is an emerging trend that virulent M. tuberculosis strains elicit reduced levels of proinflammatory cytokines (including TNF-α and IL-6) and RNI from macrophages compared to less virulent M. tuberculosis strains. This phenomenon has been described for the hypervirulent HN878 strain of M. tuberculosis and the hypervirulent mce1 mutant of M. tuberculosis (42, 58, 65). There are also reports of virulent M. tuberculosis eliciting lower levels or activities of cytokines and RNI from macrophages than attenuated and avirulent mycobacteria (2, 7, 20, 26). However, it must be noted that for TNF-α, a correlation between increased mycobacterial virulence and reduced cytokine production has not been a universal finding (12, 19, 57, 66). With regard to adaptive responses, M. tuberculosis inhibits macrophage responses to IFN-γ stimulation, such as the upregulation of MHC class II (25, 43, 50, 53). MHC class II and presentation of mycobacterial antigens to CD4+ T cells are essential components of the adaptive immune response to M. tuberculosis (22).
In this study, we found that macrophages infected with the ΔsecA2 mutant produced significantly greater levels of TNF-α, IL-6, and RNI than macrophages infected with H37Rv. This suggested a function for SecA2 in inhibiting the innate immune response. We were unable to detect IL-12 production by our infected macrophages. The increased level of IFN-γ-induced MHC class II message observed with the ΔsecA2 mutant versus H37Rv suggested an additional role for SecA2 in suppressing the adaptive immune response. To our knowledge, this is the first report of an M. tuberculosis mutant with a defect in the inhibition of IFN-γ responses.
These immunomodulatory activities of SecA2 are likely important for M. tuberculosis virulence. All of the host molecules that we identified to be regulated by SecA2 are demonstrated to contribute to the control of tuberculosis in the mouse model. Mice deficient in TNF-α, IL-6, NOS2, or CIITA (MHC class II transcriptional regulator) are all more permissive for M. tuberculosis infection, as shown by increased bacterial burden in organs and decreased length of survival, in comparison to wild-type mice (6, 23, 39, 60, 62). Immunosuppression by SecA2 may influence the course of M. tuberculosis infection in many ways, including by establishing a permissive environment for intracellular growth in macrophages and limiting antigen presentation in the host. By infecting NOS2−/− macrophages with the ΔsecA2 mutant, we demonstrated that suppression of RNI was not the only important factor regulated by SecA2 for promoting intracellular growth. We believe the attenuated phenotype of the ΔsecA2 mutant in macrophages and mice is a reflection of an imbalance in multiple cytokines and effector mechanisms, possibly including molecules that we have yet to identify.
Interestingly, all four of the molecules we found to be upregulated by macrophages infected with the ΔsecA2 mutant are associated with Toll-like receptor 2 (TLR2) and myeloid differentiation factor 88 (MyD88) signaling pathways in host cells. To various degrees, TNF-α, IL-6, and RNI are induced by mycobacterial stimulation of TLR2-dependent, MyD88-dependent pathways (11, 30, 32, 48, 59, 63, 64, 70). Thus, an increased ability of the ΔsecA2 mutant to signal through TLR2-MyD88-dependent pathways could account for the increased macrophage responses. However, there are alternate explanations given the complexities of cell signaling. Interestingly, there is also a role for TLR2 in the process of M. tuberculosis inhibition of IFN-γ-induced MHC class II (25, 27, 51). However, our observation that infection with the ΔsecA2 mutant led to higher IFN-γ-induced MHC class II message than infection with H37Rv is opposite to what would be predicted by increased TLR2 signaling. TLR2-independent pathways of M. tuberculosis inhibition of IFN-γ responses have also been identified in which SecA2 could be involved (25, 55). Alternatively, increased TNF-α levels generated by the ΔsecA2 mutant could synergize with IFN-γ to promote the observed increase in MHC class II (71).
Modification of the immune response is a strategy used by other bacterial pathogens to survive in the host, and the bacterial factors involved are often secreted and surface proteins localized by specialized secretion systems (14). Currently, there are two known specialized secretion systems in M. tuberculosis: the SecA2-dependent system and the ESAT-6/Snm system (36). Interestingly, the ESAT-6/Snm system, which localizes the small ESAT-6 and CFP-10 proteins, has also been reported to contribute to immunosuppression (37, 68). M. tuberculosis mutants lacking the snm4 (Rv3877) and snm9 (Rv3615c) components of the ESAT-6 system elicit increased levels of TNF-α, RNI, and IL-12 by macrophages. Further, M. tuberculosis snm mutants exhibit a growth defect in unactivated macrophages and an early growth phenotype in mice (24, 29, 31, 37, 68). There is no immediate explanation for the similarity in snm and ΔsecA2 mutant phenotypes, since the ΔsecA2 mutant secretes ESAT-6 (data not shown).
Because SecA2 is a secretion factor, its role in immunomodulation is most likely related to proper secretion or cell wall localization of an immunosuppressive factor. However, all of the proteins localized by SecA2, particularly cell wall proteins, are not yet known. The mycobacterial cell wall is a complex structure that contains many diverse molecules with reported immunomodulatory properties (33). These include several surface molecules reported to influence the production of inflammatory TNF-α, IL-6, and/or RNI, such as surface lipoproteins, lipoarabinomannan and its precursor, lipomannan, phthiocerol dimycocerosates, and modified trehalose dimycolate (1, 3, 11, 15, 35, 55-57, 61). The increased immune response elicited by the ΔsecA2 mutant could be explained by different amounts of immunoregulatory molecules in the cell wall or an altered cell wall structure with greater exposure of stimulatory molecules that interact with host receptors, such as TLR2. Thus, we are considering the possibility that the immunosuppressive effect of SecA2 is due to a role in localization of cell wall synthetic enzymes that influence cell wall architecture. A final possibility is that the ΔsecA2 mutant phenotypes actually represent increased release of a stimulatory molecule (10). Future efforts will be aimed at identifying the SecA2-dependent factors that suppress innate and adaptive immune responses.
Acknowledgments
We gratefully acknowledge Kristi Williams for her assistance with the qRT-PCR assays and Anthony Hickey for his advice on the aerosol infection experiments. We also thank Douglas Kernodle and the members of the Braunstein laboratory for critical review of the manuscript.
This work was supported by awards to M.B. from the American Lung Association (RG-049-N) and the NIAID (AI54540-01).
Editor: J. L. Flynn
Footnotes
Published ahead of print on 9 October 2006.
REFERENCES
- 1.Adams, L. B., Y. Fukutomi, and J. L. Krahenbuhl. 1993. Regulation of murine macrophage effector functions by lipoarabinomannan from mycobacterial strains with different degrees of virulence. Infect. Immun. 61:4173-4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balcewicz-Sablinska, M. K., J. Keane, H. Kornfeld, and H. G. Remold. 1998. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J. Immunol. 161:2636-2641. [PubMed] [Google Scholar]
- 3.Barnes, P. F., D. Chatterjee, J. S. Abrams, S. Lu, E. Wang, M. Yamamura, P. J. Brennan, and R. L. Modlin. 1992. Cytokine production induced by Mycobacterium tuberculosis lipoarabinomannan. Relationship to chemical structure. J. Immunol. 149:541-547. [PubMed] [Google Scholar]
- 4.Barry-Lane, P. A., C. Patterson, M. van der Merwe, Z. Hu, S. M. Holland, E. T. Yeh, and M. S. Runge. 2001. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J. Clin. Investig. 108:1513-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Battistoni, A. 2003. Role of prokaryotic Cu,Zn superoxide dismutase in pathogenesis. Biochem. Soc. Trans. 31:1326-1329. [DOI] [PubMed] [Google Scholar]
- 6.Bean, A. G., D. R. Roach, H. Briscoe, M. P. France, H. Korner, J. D. Sedgwick, and W. J. Britton. 1999. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162:3504-3511. [PubMed] [Google Scholar]
- 7.Beltan, E., L. Horgen, and N. Rastogi. 2000. Secretion of cytokines by human macrophages upon infection by pathogenic and non-pathogenic mycobacteria. Microb. Pathog. 28:313-318. [DOI] [PubMed] [Google Scholar]
- 8.Botha, T., and B. Ryffel. 2003. Reactivation of latent tuberculosis infection in TNF-deficient mice. J. Immunol. 171:3110-3118. [DOI] [PubMed] [Google Scholar]
- 9.Braunstein, M., A. M. Brown, S. Kurtz, and W. R. Jacobs, Jr. 2001. Two nonredundant SecA homologues function in mycobacteria. J. Bacteriol. 183:6979-6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braunstein, M., B. Espinosa, J. Chan, J. T. Belisle, and W. R. J. Jacobs. 2003. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol. Microbiol. 48:453-464. [DOI] [PubMed] [Google Scholar]
- 11.Brightbill, H. D., D. H. Libraty, S. R. Krutzik, R. B. Yang, J. T. Belisle, J. R. Bleharski, M. Maitland, M. V. Norgard, S. E. Plevy, S. T. Smale, P. J. Brennan, B. R. Bloom, P. J. Godowski, and R. L. Modlin. 1999. Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science 285:732-736. [DOI] [PubMed] [Google Scholar]
- 12.Byrd, T. F. 1997. Tumor necrosis factor alpha (TNFalpha) promotes growth of virulent Mycobacterium tuberculosis in human monocytes: iron-mediated growth suppression is correlated with decreased release of TNFalpha from iron-treated infected monocytes. J. Clin. Investig. 99:2518-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan, J., Y. Xing, R. S. Magliozzo, and B. R. Bloom. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 175:1111-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coombes, B. K., Y. Valdez, and B. B. Finlay. 2004. Evasive maneuvers by secreted bacterial proteins to avoid innate immune responses. Curr. Biol. 14:R856-R867. [DOI] [PubMed] [Google Scholar]
- 15.Dao, D. N., L. Kremer, Y. Guerardel, A. Molano, W. R. Jacobs, Jr., S. A. Porcelli, and V. Briken. 2004. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect. Immun. 72:2067-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Groote, M. A., U. A. Ochsner, M. U. Shiloh, C. Nathan, J. M. McCord, M. C. Dinauer, S. J. Libby, A. Vazquez-Torres, Y. Xu, and F. C. Fang. 1997. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc. Natl. Acad. Sci. USA 94:13997-14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dussurget, O., G. Stewart, O. Neyrolles, P. Pescher, D. Young, and G. Marchal. 2001. Role of Mycobacterium tuberculosis copper-zinc superoxide dismutase. Infect. Immun. 69:529-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards, K. M., M. H. Cynamon, R. K. Voladri, C. C. Hager, M. S. DeStefano, K. T. Tham, D. L. Lakey, M. R. Bochan, and D. S. Kernodle. 2001. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 164:2213-2219. [DOI] [PubMed] [Google Scholar]
- 19.Engele, M., E. Stossel, K. Castiglione, N. Schwerdtner, M. Wagner, P. Bolcskei, M. Rollinghoff, and S. Stenger. 2002. Induction of TNF in human alveolar macrophages as a potential evasion mechanism of virulent Mycobacterium tuberculosis. J. Immunol. 168:1328-1337. [DOI] [PubMed] [Google Scholar]
- 20.Falcone, V., E. B. Bassey, A. Toniolo, P. G. Conaldi, and F. M. Collins. 1994. Differential release of tumor necrosis factor-alpha from murine peritoneal macrophages stimulated with virulent and avirulent species of mycobacteria. FEMS Immunol. Med. Microbiol. 8:225-232. [DOI] [PubMed] [Google Scholar]
- 21.Flesch, I. E., and S. H. Kaufmann. 1990. Activation of tuberculostatic macrophage functions by gamma interferon, interleukin-4, and tumor necrosis factor. Infect. Immun. 58:2675-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flynn, J. L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93-129. [DOI] [PubMed] [Google Scholar]
- 23.Flynn, J. L., M. M. Goldstein, J. Chan, K. J. Triebold, K. Pfeffer, C. J. Lowenstein, R. Schreiber, T. W. Mak, and B. R. Bloom. 1995. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2:561-572. [DOI] [PubMed] [Google Scholar]
- 24.Fortune, S. M., A. Jaeger, D. A. Sarracino, M. R. Chase, C. M. Sassetti, D. R. Sherman, B. R. Bloom, and E. J. Rubin. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. USA 102:10676-10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fortune, S. M., A. Solache, A. Jaeger, P. J. Hill, J. T. Belisle, B. R. Bloom, E. J. Rubin, and J. D. Ernst. 2004. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J. Immunol. 172:6272-6280. [DOI] [PubMed] [Google Scholar]
- 26.Freeman, S., F. A. Post, L. G. Bekker, R. Harbacheuski, L. M. Steyn, B. Ryffel, N. D. Connell, B. N. Kreiswirth, and G. Kaplan. 2006. Mycobacterium tuberculosis H37Ra and H37Rv differential growth and cytokine/chemokine induction in murine macrophages in vitro. J. Interferon Cytokine Res. 26:27-33. [DOI] [PubMed] [Google Scholar]
- 27.Gehring, A. J., R. E. Rojas, D. H. Canaday, D. L. Lakey, C. V. Harding, and W. H. Boom. 2003. The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and FcγR1 on human macrophages through Toll-like receptor 2. Infect. Immun. 71:4487-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greer, S. F., E. Zika, B. Conti, X. S. Zhu, and J. P. Ting. 2003. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 4:1074-1082. [DOI] [PubMed] [Google Scholar]
- 29.Guinn, K. M., M. J. Hickey, S. K. Mathur, K. L. Zakel, J. E. Grotzke, D. M. Lewinsohn, S. Smith, and D. R. Sherman. 2004. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol. Microbiol. 51:359-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heldwein, K. A., M. D. Liang, T. K. Andresen, K. E. Thomas, A. M. Marty, N. Cuesta, S. N. Vogel, and M. J. Fenton. 2003. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J. Leukoc. Biol. 74:277-286. [DOI] [PubMed] [Google Scholar]
- 31.Hsu, T., S. M. Hingley-Wilson, B. Chen, M. Chen, A. Z. Dai, P. M. Morin, C. B. Marks, J. Padiyar, C. Goulding, M. Gingery, D. Eisenberg, R. G. Russell, S. C. Derrick, F. M. Collins, S. L. Morris, C. H. King, and W. R. Jacobs, Jr. 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. USA 100:12420-12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang, S., S. Uematsu, S. Akira, and P. Salgame. 2004. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J. Immunol. 173:3392-3397. [DOI] [PubMed] [Google Scholar]
- 33.Karakousis, P. C., W. R. Bishai, and S. E. Dorman. 2004. Mycobacterium tuberculosis cell envelope lipids and the host immune response. Cell. Microbiol. 6:105-116. [DOI] [PubMed] [Google Scholar]
- 34.Koul, A., T. Herget, B. Klebl, and A. Ullrich. 2004. Interplay between mycobacteria and host signalling pathways. Nat. Rev. Microbiol. 2:189-202. [DOI] [PubMed] [Google Scholar]
- 35.Krutzik, S. R., and R. L. Modlin. 2004. The role of Toll-like receptors in combating mycobacteria. Semin. Immunol. 16:35-41. [DOI] [PubMed] [Google Scholar]
- 36.Kurtz, S., and M. Braunstein. 2005. Protein secretion and export in Mycobacterium tuberculosis, p. 71-138. In T. Parish (ed.), Mycobacterium molecular microbiology. Horizon Bioscience, Norfolk, United Kingdom.
- 37.MacGurn, J. A., S. Raghavan, S. A. Stanley, and J. S. Cox. 2005. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol. Microbiol. 57:1653-1663. [DOI] [PubMed] [Google Scholar]
- 38.MacMicking, J., Q. W. Xie, and C. Nathan. 1997. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15:323-350. [DOI] [PubMed] [Google Scholar]
- 39.MacMicking, J. D., R. J. North, R. LaCourse, J. S. Mudgett, S. K. Shah, and C. F. Nathan. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 94:5243-5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacMicking, J. D., G. A. Taylor, and J. D. McKinney. 2003. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302:654-659. [DOI] [PubMed] [Google Scholar]
- 41.Manca, C., S. Paul, C. E. Barry, V. H. Freedman, and G. Kaplan. 1999. Mycobacterium tuberculosis catalase and peroxidase activities and resistance to oxidative killing in human monocytes in vitro. Infect. Immun. 67:74-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manca, C., M. B. Reed, S. Freeman, B. Mathema, B. Kreiswirth, C. E. Barry III, and G. Kaplan. 2004. Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infect. Immun. 72:5511-5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagabhushanam, V., A. Solache, L. M. Ting, C. J. Escaron, J. Y. Zhang, and J. D. Ernst. 2003. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. J. Immunol. 171:4750-4757. [DOI] [PubMed] [Google Scholar]
- 44.Nathan, C. 2003. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J. Clin. Investig. 111:769-778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nau, G. J., J. F. Richmond, A. Schlesinger, E. G. Jennings, E. S. Lander, and R. A. Young. 2002. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. USA 99:1503-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nauseef, W. M. 2004. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 122:277-291. [DOI] [PubMed] [Google Scholar]
- 47.Ng, V. H., J. S. Cox, A. O. Sousa, J. D. MacMicking, and J. D. McKinney. 2004. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52:1291-1302. [DOI] [PubMed] [Google Scholar]
- 48.Nicolle, D. M., X. Pichon, A. Bouchot, I. Maillet, F. Erard, S. Akira, B. Ryffel, and V. F. Quesniaux. 2004. Chronic pneumonia despite adaptive immune response to Mycobacterium bovis BCG in MyD88-deficient mice. Lab. Investig. 84:1305-1321. [DOI] [PubMed] [Google Scholar]
- 49.North, R. J., and Y. J. Jung. 2004. Immunity to tuberculosis. Annu. Rev. Immunol. 22:599-623. [DOI] [PubMed] [Google Scholar]
- 50.Noss, E. H., C. V. Harding, and W. H. Boom. 2000. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell. Immunol. 201:63-74. [DOI] [PubMed] [Google Scholar]
- 51.Noss, E. H., R. K. Pai, T. J. Sellati, J. D. Radolf, J. Belisle, D. T. Golenbock, W. H. Boom, and C. V. Harding. 2001. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J. Immunol. 167:910-918. [DOI] [PubMed] [Google Scholar]
- 52.Orme, I. M., and F. M. Collins. 1994. Mouse model of tuberculosis, p. 113-134. In B. R. Bloom (ed.), Tuberculosis: pathogenesis, protection, and control. ASM Press, Washington, D.C.
- 53.Pai, R. K., M. E. Pennini, A. A. Tobian, D. H. Canaday, W. H. Boom, and C. V. Harding. 2004. Prolonged Toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect. Immun. 72:6603-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Piddington, D. L., F. C. Fang, T. Laessig, A. M. Cooper, I. M. Orme, and N. A. Buchmeier. 2001. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect. Immun. 69:4980-4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quesniaux, V., C. Fremond, M. Jacobs, S. Parida, D. Nicolle, V. Yeremeev, F. Bihl, F. Erard, T. Botha, M. Drennan, M. N. Soler, M. Le Bert, B. Schnyder, and B. Ryffel. 2004. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6:946-959. [DOI] [PubMed] [Google Scholar]
- 56.Quesniaux, V. J., D. M. Nicolle, D. Torres, L. Kremer, Y. Guerardel, J. Nigou, G. Puzo, F. Erard, and B. Ryffel. 2004. Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 172:4425-4434. [DOI] [PubMed] [Google Scholar]
- 57.Rao, V., N. Fujiwara, S. A. Porcelli, and M. S. Glickman. 2005. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J. Exp. Med. 201:535-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reed, M. B., P. Domenech, C. Manca, H. Su, A. K. Barczak, B. N. Kreiswirth, G. Kaplan, and C. E. Barry III. 2004. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431:84-87. [DOI] [PubMed] [Google Scholar]
- 59.Reiling, N., C. Holscher, A. Fehrenbach, S. Kroger, C. J. Kirschning, S. Goyert, and S. Ehlers. 2002. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J. Immunol. 169:3480-3484. [DOI] [PubMed] [Google Scholar]
- 60.Repique, C. J., A. Li, W. J. Brickey, J. P. Ting, F. M. Collins, and S. L. Morris. 2003. Susceptibility of mice deficient in the MHC class II transactivator to infection with Mycobacterium tuberculosis. Scand. J. Immunol. 58:15-22. [DOI] [PubMed] [Google Scholar]
- 61.Rousseau, C., N. Winter, E. Pivert, Y. Bordat, O. Neyrolles, P. Ave, M. Huerre, B. Gicquel, and M. Jackson. 2004. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell. Microbiol. 6:277-287. [DOI] [PubMed] [Google Scholar]
- 62.Saunders, B. M., A. A. Frank, I. M. Orme, and A. M. Cooper. 2000. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect. Immun. 68:3322-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi, S., A. Blumenthal, C. M. Hickey, S. Gandotra, D. Levy, and S. Ehrt. 2005. Expression of many immunologically important genes in Mycobacterium tuberculosis-infected macrophages is independent of both TLR2 and TLR4 but dependent on IFN-alphabeta receptor and STAT1. J. Immunol. 175:3318-3328. [DOI] [PubMed] [Google Scholar]
- 64.Shi, S., C. Nathan, D. Schnappinger, J. Drenkow, M. Fuortes, E. Block, A. Ding, T. R. Gingeras, G. Schoolnik, S. Akira, K. Takeda, and S. Ehrt. 2003. MyD88 primes macrophages for full-scale activation by interferon-gamma yet mediates few responses to Mycobacterium tuberculosis. J. Exp. Med. 198:987-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shimono, N., L. Morici, N. Casali, S. Cantrell, B. Sidders, S. Ehrt, and L. W. Riley. 2003. Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc. Natl. Acad. Sci. USA 100:15918-15923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silver, R. F., Q. Li, and J. J. Ellner. 1998. Expression of virulence of Mycobacterium tuberculosis within human monocytes: virulence correlates with intracellular growth and induction of tumor necrosis factor alpha but not with evasion of lymphocyte-dependent monocyte effector functions. Infect. Immun. 66:1190-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith, I. 2003. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 16:463-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stanley, S. A., S. Raghavan, W. W. Hwang, and J. S. Cox. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. USA 100:13001-13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thannickal, V. J., and B. L. Fanburg. 2000. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L1005-L1028. [DOI] [PubMed] [Google Scholar]
- 70.Underhill, D. M., A. Ozinsky, K. D. Smith, and A. Aderem. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA 96:14459-14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watanabe, Y., and C. O. Jacob. 1991. Regulation of MHC class II antigen expression. Opposing effects of tumor necrosis factor-alpha on IFN-gamma-induced HLA-DR and Ia expression depends on the maturation and differentiation stage of the cell. J. Immunol. 146:899-905. [PubMed] [Google Scholar]
- 72.Wengenack, N. L., M. P. Jensen, F. Rusnak, and M. K. Stern. 1999. Mycobacterium tuberculosis KatG is a peroxynitritase. Biochem. Biophys. Res. Commun. 256:485-487. [DOI] [PubMed] [Google Scholar]
- 73.Williams, K. L., D. J. Taxman, M. W. Linhoff, W. Reed, and J. P. Ting. 2003. Cutting edge: Monarch-1: a pyrin/nucleotide-binding domain/leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J. Immunol. 170:5354-5358. [DOI] [PubMed] [Google Scholar]
- 74.World Health Organization. 2005. Tuberculosis fact sheet no. 104. [Online.] http://www.who.int/mediacentre/factsheets/fs104/en/#global.