

Abstract
Gliotoxin is a nonribosomal peptide produced by Aspergillus fumigatus. This compound has been proposed as an A. fumigatus virulence factor due to its cytotoxic, genotoxic, and apoptotic properties. Recent identification of the gliotoxin gene cluster identified several genes (gli genes) likely involved in gliotoxin production, including gliZ, encoding a putative Zn2Cys6 binuclear transcription factor. Replacement of gliZ with a marker gene (ΔgliZ) resulted in no detectable gliotoxin production and loss of gene expression of other gli cluster genes. Placement of multiple copies of gliZ in the genome increased gliotoxin production. Using endpoint survival data, the ΔgliZ and a multiple-copy gliZ strain were not statistically different from the wild type in a murine pulmonary model; however, both the wild-type and the multiple-copy gliZ strain were more virulent than ΔlaeA (a mutant reduced in production of gliotoxin and other toxins). A flow-cytometric analysis of polymorphonuclear leukocytes (PMNs) exposed to supernatants from wild-type, ΔgliZ, complemented ΔgliZ, and ΔlaeA strains supported a role for gliotoxin in apoptotic but not necrotic PMN cell death. This may indicate that several secondary metabolites are involved in A. fumigatus virulence.
Aspergillus fumigatus, a saprophytic and opportunistic pathogenic filamentous fungus, causes mycotoxicosis, allergic reactions, and a life-threatening systemic disease called “invasive aspergillosis” (IA) in immunocompromised individuals. Insufficient defense mechanisms by the innate and acquired immune systems result in high IA mortality rates in neutropenic and immunosuppressed patients. The incidence of IA has increased in recent decades, largely due to an increased population of immunosuppressed patients at risk after organ transplantation or therapy for cancer. In spite of advances in early diagnosis and new antifungal therapy, IA continues to be a leading cause of death in these patients, with mortality rates reported to be as high as 80% to 95% (11).
A. fumigatus virulence attributes contributing to IA are not likely to be due to a single factor but rather a combination of interactions of various molecules and biological properties of the fungus (22, 32, 40). Growth characteristics such as its high spore concentration in the air and its faster growth relative to any other airborne fungi at 40°C are thought to contribute to its virulence (22, 39). However, identification of unique, single-molecule, virulence factors has been elusive in this system. One molecule hypothesized as a unique virulence factor is the secondary metabolite gliotoxin.
Gliotoxin is a well-studied nonribosomal peptide toxin (14) and has long been fingered as a putative factor contributing to IA due to its cytotoxic (15), genotoxic (26), and apoptotic properties (21, 29, 38). A potential role for gliotoxin in IA was recently supported by genetic studies of an A. fumigatus secondary metabolite mutant, ΔlaeA, which was shown to be crippled in gliotoxin biosynthesis (5, 6). The loss of laeA in A. fumigatus results in reduced virulence in a murine model, increased conidial susceptibility to macrophage phagocytosis, and decreased hyphal killing of neutrophils (5). This latter trait was hypothesized to be due to lack of gliotoxin production. However, along with the decrease in gliotoxin production, the ΔlaeA strain is decreased in the production of several other secondary metabolites implicated as virulence factors, including fumagillin, fumagatin, and helvolic acid, among others (5; http://www.aspergillus.man.ac.uk/indexhome.htm and references therein).
Recently, a predicted gliotoxin biosynthetic gene cluster was identified in A. fumigatus (14). In an attempt to assess the contributions of gliotoxin to the role of LaeA in virulence, we have created a null mutant in gliZ encoding a putative Zn2Cys6 binuclear finger transcription factor. Here we show that gliZ is required for gliotoxin biosynthesis and expression of other genes in the gli gene cluster and that placement of two or more copies of gliZ in the genome results in increased gliotoxin synthesis. Although statistical examination of the results of a murine pulmonary model did not support a difference in virulence in the wild type compared to either ΔgliZ or multiple-copy gliZ, there was a tendency towards a decrease and increase in mice death associated with these strains, respectively. Two concurrent studies, where gliP encoding a nonribosomal peptide synthase required for gliotoxin synthesis was deleted from the A. fumigatus genome, yielded similar results where the authors report no difference in mouse survival (10, 20). However, in both studies loss of gliotoxin resulted in decreased toxicity as measured either by mast cell degranulation (10) or macrophage/T-cell viability (20), thus leading to speculation that this metabolite can play a role in disease development. Here, cytotoxicity assays with polymorphonuclear leukocytes (PMNs) support a role for gliotoxin in apoptotic but not necrotic cell death. Taken together, we posit that gliotoxin is one factor that can be involved in disease development and that its effects may not be readily measured by the current animal model systems. We suggest that other LaeA-regulated metabolites or attributes also contribute to A. fumigatus virulence.
MATERIALS AND METHODS
Strains.
All fungal strains used in this study (Table 1) were maintained as glycerol stocks and were routinely cultured at 25°C or 37°C on glucose minimal medium (GMM) (34).
TABLE 1.
A. fumigatus strains used for this study
| Strain(s) | Genotypea |
|---|---|
| AF293 | Wild type |
| AF293.1 | pyrG1 |
| TDWC5.6 and TDWC5.33 | ΔgliZ::pyrG pyrG1 |
| TDWC8.3 and TDWC8.10 | gliZ hygB ΔgliZ::pyrG pyrG1 |
| TJW54.2 | ΔlaeA::pyrG pyrG1 |
| TJW55.1 | pyrG pyrG1 |
| TJW68.6 | laeA hygB ΔlaeA::pyrG pyrG1 |
pyrG is from A. parasiticus.
gliZ deletion and complementation.
Aspergillus fumigatus gliZ was disrupted in wild-type strain AF293.1 (a pyrG auxotroph [46]) by replacement of gliZ with the A. parasiticus pyrG marker gene obtained from pBZ5 (35). An A. fumigatus gliZ gene disruption vector, pJW74.3, was constructed by insertion of a 1.2-kb DNA fragment upstream of the gliZ start codon (primers GZ5F and GZ5R) and a 0.9-kb DNA fragment downstream of the gliZ stop codon (primers GZ3F and GZ3R) on either side of the A. parasiticus pyrG marker gene. Fungal protoplasts were transformed by the polyethylene glycol method as previously described (5). Homologous single-gene replacement of gliZ was confirmed by Southern blot analysis and PCR.
pJW78.3 was constructed to complement the ΔgliZ strain TDWC5.6. The plasmid contained a 3.2-kb wild-type gliZ gene including a 1.2-kb promoter. The 3.2-kb gliZ gene was amplified by primers GZCOMF and GZCOMR. The PCR product was subcloned in the Zero Blunt TOPO vector (Invitrogen Co.) to produce pJW75.1. pJW78.3 was created by inserting the 3.2-kb HindIII-XbaI gliZ fragment from pJW75.1 into a HindIII-XbaI site of pUCH2-8 (2), which contains the selectable marker hygromycin B phosphotransferase.
Extraction of fungal DNA, restriction enzyme digestion, gel electrophoresis, Southern and Northern blotting, hybridization, and probe preparation were performed using standard methods (33). RNA blots were hybridized with a 1.2-kb PCR fragment of gliZ amplified with primers GZINTF and GZINTR and a 0.7-kb gliI PCR product using primers GIINTF and GIINTR. Primers for PCR and probes are listed in Table 2.
TABLE 2.
Primers used in this study
| Primer | Sequence |
|---|---|
| GZ5F | 5′AGTGACCGTCCAAGAACCGTAG3′ |
| GZ5R | 5′GGAGAGAATTCGCGTTTAACCTTCTATCGCAG3′ |
| GZ3F | 5′GCGGTAAGCTTGCAGATCTGGGATCTGCTCCG3′ |
| GZ3R | 5′TGCCAGTCCAAGCTGTAAAATCC3′ |
| GZCOMF | 5′AGCAGCCGAACTCTCTAGAAGAAATCTG3′ |
| GZCOMR | 5′TGCCAGTCCAAGCTTTAAAATCCAGTTGAC3′ |
| GZINTF | 5′AAGGGCCGGTAGTCTACCTCTTC3′ |
| GZINTR | 5′CGATCTGGTAGCTGCCCAGCTGGAAG3′ |
| GIINTF | 5′TGTTGATCGAGACGCCGTTCTG3′ |
| GIINTR | 5′CAGAGCGGCTCGATTCTGGTG3′ |
| gliIRTF | 5′TCGTTGCTGGAGAATCTGCTGTATGA3′ |
| gliIRTR | 5′ACAAAGGGAATCTGGTGGAAGGCGA3′ |
| nestedgliIF | 5′TGTTGATCGAGACGCCGTTCTG3′ |
| nestedgliIR | 5′CAGAGCGGCTCGATTCTGGTG3′ |
Detection of gliI expression in A. fumigatus-infected lung by reverse transcriptase PCR (RT-PCR).
Total RNA was extracted from lungs of 10 mice of two groups; one of them was infected intranasally by A. fumigatus as described previously (5) with 108 spores/ml, and the other group, not infected by the fungus, served as a negative control. These mice were separate from the mice used for the virulence test as described below. At day 3, all mice were sacrificed and all lungs from each group were combined per treatment and homogenized with 10 ml of phosphate-buffered saline. Nonhomogenizable tissue was removed using a pipette. The homogenate was spun down in 50-ml Oakridge tubes at 5,000 rpm, and then supernatant (SN) was removed. The pellet was resuspended with 4 ml of 1% Triton X-100 and homogenized for 1 min. The homogenate was spun down in 50-ml Oakridge tubes at 5,000 rpm, and supernatant was removed. These last two steps were repeated one more time. The pellet was washed twice with sterile distilled water, and then homogenate was spun down at 5,000 rpm to obtain the final pellet. This pellet was lyophilized overnight. The freeze-dried pellet was mixed with TRIzol (Invitrogen Co.) to extract total RNA according to the manufacturer's instructions. The RNA pellet was dissolved in diethyl pyrocarbonate-treated water prior to cDNA synthesis using a SuperScript III kit (Invitrogen Co.) according to the manufacturer's directions. The following primers were used to amplify the A. fumigatus-specific gene, gliI (a biosynthetic gene of gliotoxin [14]), from the cDNA pool. gliIRTF and gliIRTR were used to amplify the primary PCR product. Two microliters of this PCR product was used as template for secondary PCR amplification with nested primers, nestedgliIF and nestedgliIR. These primers are described in Table 2.
Phenotypic characterization and secondary metabolite analysis.
Colony growth radius and spore production were measured as previously described (17). Gliotoxin production was assessed by thin-layer chromatography (TLC) and by high-pressure liquid chromatography-photo diode array detection-high-resolution mass spectrometry (HPLC-HR-MS) using previously published methods (5, 8, 40). For TLC, fungi were grown in liquid glucose minimal medium (GMM) for 3 days at 25 or 37°C and at 280 rpm, and organic extracts were measured for secondary metabolites. Gliotoxin was also measured by HPLC-diode array detection and HPLC-HR-MS from extracts of cultures grown on Czapek yeast extract agar (CYA), malt extract agar (MEA), oatmeal agar (OAT), and yeast extract-sucrose agar (YES) media at either 25 or 37°C (13). Here, two agar plugs of the center of colonies of 11-day-old cultures (three point inoculations) grown in the dark on each of the CYA, MEA, OAT, and YES (13) media were pooled and extracted according to Smedsgaard (36). The wild type and mutants were also grown on GMM agar in the dark for 1 week at 25°C and 37°C, and the culture extracts were examined for gliotoxin, desmethyl-(dimethylthio)-gliotoxin, helvolic acid, fumigaclavines, pseurotins, trypacidin, mono-methylsulochrin, chloroanthraquionones, fumiquinazolines, tryptoquivalins, and fumagillin by analyzing them using HPLC-diode array detection and HPLC-HR-MS. The monitoring wavelength was 210 nm. Gliotoxin and other metabolites were identified by their retention time, UV spectra, and positive electrospray (ESI+) spectra, which should match both the relative intensities of ions and their respective accurate masses (±<0.01-Da deviation).
Gliotoxin was also measured from homogenates of murine lung as previously described (5).
Animal model of Aspergillus infection.
Virulence of near-isogenic strains of A. fumigatus differing only in the presence of laeA or gliZ gene (e.g., wild-type, ΔlaeA, ΔgliZ, and ΔgliZ-complemented strains; Table 1) was assessed twice in a lung infection model, as described previously (5). Briefly, 6-week-old, outbred Swiss ICR mice (Harlan Sprague Dawley) weighing 24 to 27 g were immunosuppressed by intraperitoneal injection of cyclophosphamide (150 mg/kg of body weight) on days 4 and 1 preinfection and day 3 postinfection, with a single dose of cortisone acetate (200 mg/kg) on the morning of infection. Anesthetized mice (10 mice/fungal strain in the first test and 15 mice/fungal strain in the second test) were infected by nasal instillation of 50 μl of 5 × 106 conidia/ml (day 1) and monitored three times daily for 7 days postinfection. All surviving mice were sacrificed at day 7. Homogenates of lung tissue were examined for gliotoxin by HPLC-HR-MS as previously described (5).
Polymorphonuclear leukocytes and fungal cytotoxicity.
Whole blood of healthy adult human donors was collected after consent, using a protocol approved by the Fred Hutchinson Cancer Research Center Institutional Review Board. Polymorphonuclear leukocytes (PMNs) were purified by Ficoll centrifugation per the leukocyte separation protocol (Sigma). Red blood cells were lysed in hypotonic solution, and PMNs were separated by centrifugation (800 × g, 10 min) and resuspended in RPMI 1640-HEPES. Fungal supernatants were prepared as follows: 30 ml of GMM was inoculated with 107 conidia/ml and incubated for 72 h at 37°C (with shaking at 220 rpm). The fungal mat was separated from the culture supernatant by passing through sterile gauze and filtered through a 0.2-μm filter; the resultant supernatant was vacuum dried for 36 h. The dried precipitate was resuspended in 4 ml phosphate-buffered saline (concentrated SN) and frozen at −70°C for later use. PMNs (2 × 106 cells) were exposed to 200 μl of SN (test conditions), medium (live controls), or Triton X-100 (dead controls) for 60 min. Cells were centrifuged (1,200 rpm, 10 min) and resuspended in 100 μl binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2; pH 7.4) containing either Annexin V (5 μl; BD Biochemicals, San Jose, CA), propidium iodide (PI; 10 μl; Sigma Chemical Co., St. Louis, MO), or both Annexin V and PI. Cells were incubated for 15 min in the dark, and 400 μl of 1× binding buffer was added. Four separate experiments were conducted. Propidium iodide and Annexin V positivity were measured by a FACScan flow cytometer (BD Biosciences) with CELLQuest software (BD Biosciences).
Statistical analyses.
Wilcoxon rank-sum tests (44) were performed to detect differences in PMN death and apoptosis after exposure to supernatant from wild-type and mutant strains. In animal experiments, Fisher's exact tests (12) were used to compare proportions surviving at day 7. All P values presented were adjusted for multiple comparisons using the false discovery rate method (4).
RESULTS
gliZ is required for gliotoxin production.
gliZ was deleted from the A. fumigatus genome by replacing it with the A. parasiticus pyrG gene. Six gliZ null mutants (ΔgliZ) were obtained from 55 transformants. Southern blot and PCR analyses were carried out to confirm single-gene replacement events in the transformants (Fig. 1; unpublished data). Two transformants, TDWC5.6 and TDWC5.33, were selected for physiological and virulence studies. The ΔgliZ mutant TDWC5.6 was complemented with the gliZ gene. Two complemented strains, TDWC8.3 and TDWC8.10, were confirmed by PCR and Southern blot hybridization (data not shown). Both of these strains carried two or more copies of gliZ. Wild-type, ΔgliZ, and ΔgliZ-complemented strains were not statistically different in growth rate or conidial production (data not shown).
FIG. 1.

Disruption of a transcription factor of gliotoxin biosynthesis, gliZ, in A. fumigatus. A. Schematic diagram of the gliZ open reading frame and how it was replaced with the pyrG selectable marker from pJW74.3 by homologous recombination to generate the gliZ mutant TDWC5. B. Southern blot analysis of the wild-type (WT) strain and the gliZ mutant strain. Genomic DNA was digested by EcoRI. A 3.2-kb PCR product containing gliZ was obtained with the primers GZ5F and GZ3R and was used as a gliZ probe (indicated by a bar in panel A). Expected hybridization band patterns: wild-type strain, 7-kb band; ΔgliZ strain, 6- and 3.5-kb bands. The arrowheads at the top of the figure indicate correct knockout mutants. The ∼5-kb bands in the other three lanes indicate ectopic copies of knockout cassettes, and thus these strains were discarded.
A thin-layer chromatography (TLC) examination of chloroform extracts from liquid shake GMM cultures of A. fumigatus ΔgliZ and the ΔgliZ-complemented strains grown at 25°C showed loss of or increased production of gliotoxin, respectively (Fig. 2). Production of a few other unknown metabolites was also affected by loss or gain of gliZ in this condition (Fig. 2). HPLC data of the ΔgliZ and ΔgliZ-complemented strains grown on solid GMM supported the TLC gliotoxin results and further showed that strains produced levels of fumigaclavine C, mono-methylsulochrin, trypacidin, and fumiquinazolins similar to those of the wild type. However, at 37°C on solid GMM, the ΔgliZ-complemented strains produced helvolic acid, which was not detected in the wild type or the ΔgliZ mutant (Table 3) .
FIG. 2.

Thin-layer chromatography profiles of metabolite production by A. fumigatus ΔlaeA (TJW54.2), wild-type (AF293), ΔgliZ (TDWC5.6), and two complemented ΔgliZ (TDWC8.3 and TDWC8.10, designated ΔgliZcom8.3 and ΔgliZcom8.10, respectively) strains. Metabolites were extracted by chloroform from 50 ml GMM liquid shaking culture grown for 3 days at 25°C and 280 rpm. Dried chloroform extracts were resuspended in 100 μl of methanol, and 10 μl was used for separation on TLC. All experiments were triplicated. Solvent condition was the following: 7:3 chloroform:acetone. Gt, gliotoxin standard. Solid black arrows indicate gliotoxin, and dotted black arrows indicate possible changes in production of other metabolites in ΔgliZ and/or complemented gliZ mutants. Note the near-complete absence of metabolites in ΔlaeA. WT, wild type.
TABLE 3.
Metabolite production in strains grown on GMM agar for 1 week at 25 and 37°C in the dark
| Metabolite | Growth of strain:
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
ΔgliZ TWDC5.6
|
ΔgliZ- complemented TWDC8.3
|
ΔgliZ- complemented TWDC8.10
|
WT
|
ΔlaeA
|
||||||
| 25°C | 37°C | 25°C | 37°C | 25°C | 37°C | 25°C | 37°C | 25°C | 37°C | |
| Helvolic acid | 1.57 | ND | 1.88 | 1.61 | 1.88 | 1.44 | 1.82 | ND | ND | ND |
| Gliotoxin | ND | ND | 2.46 | ND | 2.53 | ND | 2.41 | ND | ND | ND |
| Bisdethio-(bisdithiomethyl)- gliotoxin | ND | ND | 2.38 | 1.71 | 2.46 | ND | 2.23 | ND | ND | ND |
Data are recorded as log(peak area in milli-absorbance units [mAu] per second) measured at 210 nm using a diode array detector. The detection limit was 10 mAu/s. ND, not detected. Metabolites were extracted from 10 agar plugs. WT, wild type.
As the purported role of GliZ as a Zn(II)2Cys2 transcription factor would be to activate expression of biosynthetic genes in the gli gene cluster (14, 49), we assessed the expression of gliI in wild-type, ΔlaeA, ΔgliZ, and ΔgliZ-complemented strains grown in liquid GMM. gliI encodes a putative aminocyclopropane carboxylate synthase, a function required for gliotoxin synthesis (14). As predicted, gliI expression was abolished in both the ΔlaeA and ΔgliZ strains and increased in the ΔgliZ-complemented strains in comparison to the wild type (Fig. 3A). Early expression of both gliI and gliZ was observed in the complemented strain under these conditions.
FIG. 3.

gliI gene expression in liquid shaking cultures and in infected mice. A. gliZ and gliI expression in A. fumigatus (AF293) wild-type, ΔlaeA (TJW54.2), ΔgliZ (TDWC5.6), and ΔgliZ-complemented (TDWC8.3) strains. Two lanes of TDWC8.3 are shown (designated ΔgliZcom). All strains were grown in 50 ml GMM liquid shaking culture grown at 25°C and 280 rpm. Total RNA was extracted from mycelia grown for 36 h and 72 h and probed with gliZ and gliI. B. Amplification of gliI from wild-type A. fumigatus-infected mouse lung by reverse transcriptase PCR (RT-PCR) using the secondary PCR nested primers (see Materials and Methods). Lane 1, 1-kb size marker; lane 2, PCR from genomic DNA; lane 3, PCR from RT-PCR product from noninfected lungs; lane 4, PCR from RT-PCR product from infected lungs. The size difference between lanes 2 and 4 is due to an intron (63 bp) in the gliI gene which is only present in the genomic DNA lane. WT, wild type.
Infection with GliZ mutants is correlated with gliotoxin concentrations in murine lung.
Relative virulence of wild type, ΔlaeA, ΔgliZ, and ΔgliZ-complemented strains of A. fumigatus was evaluated in murine pulmonary infection models by comparing the proportion surviving at day 7 after infection. As shown in Fig. 4, whereas a significant difference was observed between all four groups (P = 0.002), pairwise comparisons indicated a significant difference between groups of animals infected with the wild-type and ΔlaeA strains (P = 0.05). However, animals infected with ΔgliZ and complemented ΔgliZ did not demonstrate significant differences in survival compared to that of the wild type (P = 0.45 for both). Animals infected with the ΔgliZ-complemented and ΔlaeA strains also demonstrated differences in survival (P = 0.0004).
FIG. 4.

Virulence in a murine lung infection model. Fifteen outbred Swiss ICR mice immunosuppressed by intraperitoneal injection of cyclophosphamide (150 mg/kg) and cortisone acetate (200 mg/kg) were inoculated intranasally with A. fumigatus wild-type (AF293), ΔlaeA (TJW54.2), ΔgliZ (TDWC5.6), and ΔgliZ-complemented (TDWC8.3; designated ΔgliZcom8.3) strains (50 μl of 5 × 106 conidia/ml). Results presented are from one experiment that is representative of the two performed. Using Fisher's exact test at day 7, the overall P value was 0.002. Pairwise comparisons only indicated a significant difference between wild-type and ΔlaeA strains (P = 0.05) and the ΔgliZ-complemented and ΔlaeA strains (P = 0.0004). WT, wild type.
Lungs of five mice of each group were homogenized and extracted to assess metabolite content. Results of liquid chromatography high-resolution mass spectrometry showed that gliotoxin was present in lungs infected by wild-type and ΔgliZ-complemented strains but not in ΔgliZ or noninfected lungs (Fig. 5). The ΔlaeA-infected lungs showed significant amounts of gliotoxin in this test, in contrast to prior tests (5), and the ΔgliZ-complemented strains showed the most gliotoxin (Fig. 5). RT-PCR analysis of noninfected lung and lung from mice infected with wild-type A. fumigatus showed gliI expression only in the infected lung (Fig. 3B). As previously reported (5), no trace of other known secondary metabolites normally produced during in vitro culture of A. fumigatus (e.g., helvolic acids, fumigaclavines A to C, tryptoquivalines A to H, fumiquinazolines A to E, pseurotins A to F2, fumagillin, fumitremorgins A to C, verruculogen, or TR-2) could be detected in the lung tissue infected with any strain, even though these metabolites ionize significantly better than gliotoxin and thus have lower detections limits (25).
FIG. 5.

HPLC-electrospray-positive (ESI+)-MS-extracted ion chromatograms (m/z, 263.01 ± 0.04) of lung chloroform extracts, cleaned up by solid-phase extraction, showing the major fragment ion of gliotoxin. The extracts were from five mice lungs/strain infected by A. fumigatus ΔlaeA (TJW54.2), wild type (AF293), ΔgliZ (TDWC5.6), and complemented ΔgliZ (TDWC8.3). ΔgliZ and noninfected mice lung extracts just show noise. Inset A shows the ESI+ spectrum of gliotoxin in TDWC8.3, and inset B shows the spectrum of the reference standard. The y axis is adjusted to show the maximum peak level for each graph. WT, wild type.
PMN death.
Previously we have demonstrated that ΔlaeA supernatants (SN) were less cytotoxic to PMNs than SN from wild-type A. fumigatus (4). To test the hypothesis that gliotoxin mediates the perceived differences in PMN cytotoxicity, we exposed PMNs to ΔgliZ, ΔgliZ-complemented, and wild-type SN and quantified propidium iodide (PI)-positive cells after 60-min incubations. Results of four different experiments with PMNs from four different donors showed equivalent cell death after exposure to the SNs from ΔgliZ and ΔgliZ-complemented strains (Fig. 6A). Although there was a trend for decreased cell death from the ΔlaeA SN, this decrease was not significant at P ≤ 0.05 in this study.
FIG. 6.
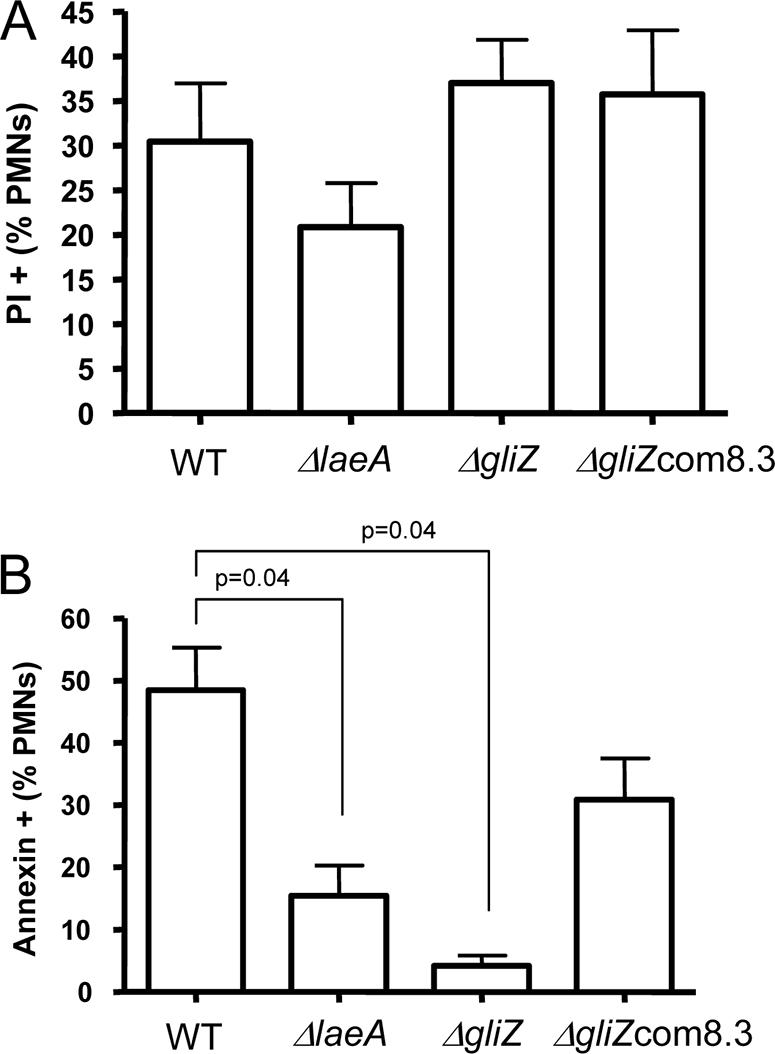
A. PMN PI positivity after exposure to concentrated culture supernatants from wild-type (AF293), ΔlaeA (TJW54.2), ΔgliZ (TDWC5.6), and complemented ΔgliZ (TDWC8.3; designated ΔgliZcom8.3) strains. Percentages of total cells that were PI positive are shown. Control is untreated PMN, which was <10% dead under assay conditions. Data represent means (±standard deviations) calculated from triplicate measurements in four different experiments. Wilcoxon rank-sum P values adjusted for multiple comparisons showed no difference between any treatment at P ≤ 0.05. B. PMN apoptosis after exposure to concentrated culture supernatants from wild-type (AF293), ΔlaeA (TJW54.2), ΔgliZ (TDWC5.6), and complemented ΔgliZ (TDWC8.3) strains. Percentages of total cells that were Annexin V positive (apoptotic) are shown. Data represent means (±standard deviations) calculated from triplicate measurements in four different experiments. Wilcoxon-rank sum P values adjusted for multiple comparisons are shown only for treatments showing significant differences. WT, wild type.
As a more sensitive indicator of the potential impact of gliotoxin in inducing cellular apoptosis, Annexin V uptake was measured after exposure to the different supernatants. Results of four different experiments from four different donors demonstrated that PMN apoptosis was significantly decreased after exposure to SNs from ΔgliZ and ΔlaeA compared to the wild type (Fig. 6B). Complementation of gliZ appeared to restore approximately wild-type levels of PMN apoptosis (for complemented ΔgliZ versus the wild type, P = 0.12).
DISCUSSION
Secondary metabolite production in fungi is a complex process yielding compounds with obscure or unknown functions in the producing organism. However, many of these metabolites have tremendous importance to humankind, in that they display a broad range of useful antibiotic and pharmaceutical activities as well as less desirable immunosuppressant and toxic activities (49). Although several fungal toxins have been shown to be involved in plant pathogenesis (1, 24, 45, 48), an unambiguous role of fungal toxins in human disease has yet to be established, with the exception of mycotoxin and mushroom ingestion. The present study is the first to demonstrate that A. fumigatus gliotoxin plays a role in mediating human neutrophil apoptosis ex vivo.
Gliotoxin is an epipolythiodioxopiperazine cyclic dipeptide produced by several fungi, including A. fumigatus; in the past it has been implicated in the establishment of severe fungal infections in both animals and humans (28). Although the mechanism by which gliotoxin exhibits its toxic effects is not clear, a number of studies using commercially produced gliotoxin or crude culture filtrates have shown that exposure can have profound immunosuppressive effects on different mammalian cell types: for example, gliotoxin induces death by apoptosis in neutrophils (42), macrophages (43), and human hepatocytes (16). Gliotoxin also appears to have immunomodulatory effects on human monocytes (18), mast cells (27), and T lymphocytes (37). Further, it has been recently proposed that gliotoxin may provide a mechanism by which A. fumigatus can selectively suppress antigen-presenting cells (37).
In support of a possible role for gliotoxin in mediating development of invasive aspergillosis, we have recently described an A. fumigatus gliotoxin mutant that showed less virulence in a murine mouse model (5). Deletion of laeA, a developmentally regulated transcriptional regulator of multiple secondary metabolites, resulted not only in reduced mortality and growth in a pulmonary murine model but also in increased susceptibility to macrophage phagocytosis and decreased ability to kill neutrophils ex vivo. Coupled with this phenotype was the near loss of gliotoxin production. However, the laeA mutant was also crippled in the production of other known metabolites that could play a role in IA (5), thus leaving open the question of exactly what, if any, altered host-fungus interactions could be attributed to reduction in gliotoxin synthesis.
To genetically elucidate the contribution of loss of gliotoxin to the ΔlaeA phenotype, we have deleted gliZ, a putative Zn2Cys6 transcription factor located in the gliotoxin gene cluster. Deletion of gliZ resulted in loss of gliotoxin production in vivo and in vitro, as assessed by examination of murine lung homogenates and culture extracts from several media. This loss was associated with absence of transcription of a biosynthetic gene in the gliotoxin gene cluster. Complementation of ΔgliZ with multiple copies of gliZ resulted in strains with increased production of gliotoxin compared to the wild type (Fig. 2). Metabolite profiles were notably different in cultures grown at 25 or 37°C or in liquid shake versus solid agar medium. In general, secondary metabolite production was enhanced at 25°C. We saw evidence that gliZ regulated the production of metabolites other than simply gliotoxin (Fig. 2) when the strains were grown at 25°C in GMM liquid shake medium. This was not observed in other growth conditions, with the exception of the unexpected production of helvolic acid in the gliZ-complemented strains grown on solid GMM at 37°C (Table 3). However, examination of lung extracts from the gliZ mutant and wild-type-infected mice only showed differences in gliotoxin production.
Virulence of the gliZ mutants was assessed by the murine model and an ex vivo assay with human PMNs. The murine model system used in our laboratory did not support a role for gliotoxin in virulence using P = 0.05 as a cutoff score. This was similar to a recent report of two gliP disruption strains that did not show a decrease in virulence in their mouse models (10, 20). However, there was a significant difference in virulence between the ΔlaeA strain compared to the wild-type and the gliZ-complemented strain in our model. Examination of lungs from infected mice showed an expected profile of highest gliotoxin concentration in the gliZ-complemented infection, followed by the wild type (Fig. 5). No gliotoxin was measured in the ΔgliZ strain, and in contrast to our earlier report (5), this time we found considerable production in the ΔlaeA mutant, although it was well below that of the wild type. Production of secondary metabolites varies in both A. fumigatus and A. nidulans ΔlaeA mutants in any given experiment, likely due to the mechanism of LaeA regulation of secondary metabolite gene clusters (7 and unpublished data). However, production is always less than that of the wild type. Our murine pulmonary results suggest that some LaeA-regulated factors in addition to gliotoxin may be important in IA. We suggest, however, that the current murine model systems used here and as reported in the gliP works (10, 20) may not be sensitive enough to detect subtle differences exerted by A. fumigatus gliotoxin levels and that gliotoxin does contribute to A. fumigatus pathogenicity, although its predominant role in A. fumigatus biology may not lie in animal pathogenesis.
Considering that our earlier studies had shown a loss of PMN sensitivity to A. fumigatus ΔlaeA SNs ex vivo (5), we were most interested in the response of PMNs to SNs of the gliZ mutants. While there was no statistical difference in PMN PI positivity after exposure to the SNs, although a trend was noted (Fig. 6A), exposures to ΔgliZ SN and ΔlaeA SN were associated with less PMN apoptosis compared to exposure to WT SN (Fig. 6B). This may be an important observation given the fact that neutrophils constitute the primary cellular defense against invasive Aspergillus hyphae. Many microorganisms including bacteria and viruses evade destruction by neutrophils by triggering neutrophil apoptosis, thereby resulting in subacute to chronic infections (19, 41, 47). Increased PMN apoptosis has been observed in the past after exposure to A. fumigatus hyphae (3); it is also known that the purified commercially available gliotoxin is a powerful inducer of neutrophil apoptosis, possibly exerting this effect by inhibiting NF-κB (42). In keeping with these studies, results of the PMN-SN interaction experiments emphasize a role for gliotoxin in mediating apoptosis of neutrophils. Recently, it was suggested that A. fumigatus elaborates gliotoxin as an immunoevasive strategy by specifically targeting antigen-presenting cells such as monocytes and monocyte-derived dendritic cells (37). Induction of neutrophil apoptosis by gliotoxin production by A. fumigatus may be yet another way for the organism to escape killing by immune cells. Given that some A. fumigatus isolates produce varying levels of gliotoxin while some isolates do not produce any gliotoxin (23), it remains to be seen if this correlates with the extent of pathogenicity or persistence of infection caused by A. fumigatus.
Apart from indicating a role for gliotoxin in A. fumigatus pathogenesis, the present study also strongly implicates other factors regulated by LaeA in the possible development of IA. Previously we demonstrated that LaeA, a nuclear protein, regulated several putative fungal virulence factors, including synthesis of gliotoxin and other secondary metabolites as well as conidial morphology (5). A. fumigatus secretes uncharacterized and characterized secondary metabolites, including verruculogen, fumagillin, fumitremorgin C, and helvolic acid. Recently an ergot biosynthetic cluster was identified in A. fumigatus (9), and it has been demonstrated that A. fumigatus can produce the alkaloid festuclavine and its derivatives fumigaclavine A and fumigaclavine B2 (30), compounds with known toxic properties. Reeves et al. (31) also reported the identity of a nonribosomal peptide synthetase gene, pes1, which when deleted altered conidial morphology and exhibited a loss of virulence in the Galleria mellonella model system. Interestingly, the genes for both the ergot alkaloids and pes1 are regulated by laeA (R. Perrin, J. W. Bok, N. Federova, J. Wortman, H. Kim, W. Nierman, and N. P. Keller, unpublished data). Thus, it appears likely that metabolites other than gliotoxin are involved in mediating cellular interactions ex vivo and potentially development or outcomes of invasive infection.
Acknowledgments
This research was funded by NIH R01AI051468 to K.A.M., NSF MCB-0236393 to N.P.K., and NIH 1 R01 AI065728-01A1 to N.P.K. and D.A.
Editor: A. Casadevall
Footnotes
Published ahead of print on 9 October 2006.
REFERENCES
- 1.Ahn, J. H., and J. D. Walton. 1996. Chromosomal organization of TOX2, a complex locus controlling host-selective toxin biosynthesis in Cochliobolus carbonum. Plant Cell 8:887-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, N. J., T. H. Hohn, and S. P. McCormick. 1998. The TRI11 gene of Fusarium sporotrichioides encodes a cytochrome P-450 monooxygenase required for C-15 hydroxylation in trichothecene biosynthesis. Appl. Environ. Microbiol. 64:221-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellocchio, S., S. Moretti, K. Perruccio, F. Fallarino, S. Bozza, C. Montagnoli, P. Mosci, G. B. Lipford, L. Pitzurra, and L. Romani. 2004. TLRs govern neutrophil activity in aspergillosis. J. Immunol. 173:7406-7415. [DOI] [PubMed] [Google Scholar]
- 4.Benjamini, Y., and Y. Hochberg. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 57:289-300. [Google Scholar]
- 5.Bok, J. W., S. A. Balajee, K. A. Marr, D. Andes, K. F. Nielsen, J. C. Frisvad, and N. P. Keller. 2005. LaeA, a regulator of morphogenetic fungal virulence factors. Eukaryot. Cell 4:1574-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bok, J. W., and N. P. Keller. 2004. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3:527-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bok, J. W., D. Noodermeer, S. P. Kale, and N. P. Keller. 2006. Secondary metabolic gene cluster silencing in Aspergillus nidulans. Mol. Microbiol., 61:1636-1645. [DOI] [PubMed] [Google Scholar]
- 8.Bruhn, J. B., A. B. Christensen, L. R. Flodgaard, K. F. Nielsen, T. O. Larsen, M. Givskov, and L. Gram. 2004. Presence of acylated homoserine lactones (AHLs) and AHL-producing bacteria in meat and potential role of AHL in spoilage of meat. Appl. Environ. Microbiol. 70:4293-4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coyle, C. M., and D. G. Panaccione. 2005. An ergot alkaloid biosynthesis gene and clustered hypothetical genes from Aspergillus fumigatus. Appl. Environ. Microbiol. 71:3112-3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cramer, R. A., Jr., M. P. Gamcsik, R. M. Brooking, L. K. Najvar, W. R. Kirkpatrick, T. F. Patterson, C. J. Balibar, J. R. Graybill, J. R. Perfect, S. N. Abraham, and W. J. Steinbach. 2006. Disruption of a nonribosomal peptide synthetase in Aspergillus fumigatus eliminates gliotoxin production. Eukaryot. Cell 5:972-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denning, D. W. 1998. Invasive aspergillosis. Clin. Infect. Dis. 26:781-803. [DOI] [PubMed] [Google Scholar]
- 12.Fisher, R. A. 1956. Statistical methods for scientific inference. Oliver and Boyd, Edinburgh, United Kingdom.
- 13.Frisvad, J. C., and R. A. Samson. 2004. Polyphasic taxonomy of Penicillium subgenus Penicillium. A guide to identification of the food and air-borne terverticillate penicillia and their mycotoxins. Studies Mycol. 49:1-174. [Google Scholar]
- 14.Gardiner, D. M., and B. J. Howlett. 2005. Bioinformatic and expression analysis of the putative gliotoxin biosynthetic gene cluster of Aspergillus fumigatus. FEMS Microbiol. Lett. 248:241-248. [DOI] [PubMed] [Google Scholar]
- 15.Grovel, O., Y. F. Pouchus, T. Robiou du Pont, M. Montagu, Z. Amzil, and J. Verbist. 2002. Ion trap MS(n) for identification of gliotoxin as the cytotoxic factor of a marine strain of Aspergillus fumigatus Fresenius. J. Microbiol. Methods 48:171-179. [DOI] [PubMed] [Google Scholar]
- 16.Hagens, W. I., P. Olinga, D. K. Meijer, G. M. Groothuis, L. Beljaars, and K. Poelstra. 2006. Gliotoxin non-selectively induces apoptosis in fibrotic and normal livers. Liver Int. 26:232-239. [DOI] [PubMed] [Google Scholar]
- 17.Jin, Y., J. W. Bok, D. Guzman-de-Pena, and N. P. Keller. 2002. Requirement of spermidine for developmental transitions in Aspergillus nidulans. Mol. Microbiol. 46:801-812. [DOI] [PubMed] [Google Scholar]
- 18.Johannessen, L. N., A. M. Nilsen, and M. Lovik. 2005. The mycotoxins citrinin and gliotoxin differentially affect production of the pro-inflammatory cytokines tumour necrosis factor-alpha and interleukin-6, and the anti-inflammatory cytokine interleukin-10. Clin. Exp. Allergy 35:782-789. [DOI] [PubMed] [Google Scholar]
- 19.Kasahara, K., I. Sato, K. Ogura, H. Takeuchi, K. Kobayashi, and M. Adachi. 1998. Expression of chemokines and induction of rapid cell death in human blood neutrophils by Mycobacterium tuberculosis. J. Infect. Dis. 178:127-137. [DOI] [PubMed] [Google Scholar]
- 20.Kupfahl, C., T. Heinekamp, G. Geginat, T. Ruppert, A. Härtl, H. Hof, and A. A. Brakhage. 2006. Deletion of the gliP gene of Aspergillus fumigatus results in loss of gliotoxin production but has no effect on virulence of the fungus in a low-dose mouse infection model. Mol. Microbiol. 62:292-302. [DOI] [PubMed] [Google Scholar]
- 21.Kweon, Y. O., Y. H. Paik, B. Schnabl, T. Qian, J. J. Lemasters, and D. A. Brenner. 2003. Gliotoxin-mediated apoptosis of activated human hepatic stellate cells. J. Hepatol. 39:38-46. [DOI] [PubMed] [Google Scholar]
- 22.Latge, J. P. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 12:310-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis, R. E., N. P. Wiederhold, M. S. Lionakis, R. A. Prince, and D. P. Kontoyiannis. 2005. Frequency and species distribution of gliotoxin-producing Aspergillus isolates recovered from patients at a tertiary-care cancer center. J. Clin. Microbiol. 43:6120-6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez, J. P., N. W. Oesch, and L. M. Ciuffetti. 2004. Characterization of the multiple-copy host-selective toxin gene, ToxB, in pathogenic and nonpathogenic isolates of Pyrenophora tritici-repentis. Mol. Plant Microbe Interact. 17:467-474. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen, K. F., and J. Smedsgaard. 2003. Fungal metabolite screening: database of 474 mycotoxins and fungal metabolites for dereplication by standardised liquid chromatography-UV-mass spectrometry methodology. J. Chromatogr. A 1002:111-136. [DOI] [PubMed] [Google Scholar]
- 26.Nieminen, S. M., J. Maki-Paakkanen, M. R. Hirvonen, M. Roponen, and A. von Wright. 2002. Genotoxicity of gliotoxin, a secondary metabolite of Aspergillus fumigatus, in a battery of short-term test systems. Mutat. Res. 520:161-170. [DOI] [PubMed] [Google Scholar]
- 27.Niide, O., Y. Suzuki, T. Yoshimaru, T. Inoue, T. Takayama, and C. Ra. 2006. Fungal metabolite gliotoxin blocks mast cell activation by a calcium- and superoxide-dependent mechanism: implications for immunosuppressive activities. Clin. Immunol. 118:108-116. [DOI] [PubMed] [Google Scholar]
- 28.Nishida, S., L. S. Yoshida, T. Shimoyama, H. Nunoi, T. Kobayashi, and S. Tsunawaki. 2005. Fungal metabolite gliotoxin targets flavocytochrome b558 in the activation of the human neutrophil NADPH oxidase. Infect. Immun. 73:235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pahl, H. L., B. Krauss, K. Schulze-Osthoff, T. Decker, E. B. Traenckner, M. Vogt, C. Myers, T. Parks, P. Warring, A. Muhlbacher, A. P. Czernilofsky, and P. A. Baeuerle. 1996. The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-κB. J. Exp. Med. 183:1829-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panaccione, D. G., and C. M. Coyle. 2005. Abundant respirable ergot alkaloids from the common airborne fungus Aspergillus fumigatus. Appl. Environ. Microbiol. 71:3106-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reeves, E. P., K. Reiber, C. Neville, O. Scheibner, K. Kavanagh, and S. Doyle. 2006. A nonribosomal peptide synthetase (Pes1) confers protection against oxidative stress in Aspergillus fumigatus. FEBS J. 273:3038-3053. [DOI] [PubMed] [Google Scholar]
- 32.Rementeria, A., N. Lopez-Molina, A. Ludwig, A. B. Vivanco, J. Bikandi, J. Ponton, and J. Garaizar. 2005. Genes and molecules involved in Aspergillus fumigatus virulence. Rev. Iberoam. Micol. 22:1-23. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook, J., E. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 34.Shimizu, K., and N. P. Keller. 2001. Genetic involvement of a cAMP-dependent protein kinase in a G protein signaling pathway regulating morphological and chemical transitions in Aspergillus nidulans. Genetics 157:591-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skory, C. D., P. K. Chang, J. Cary, and J. E. Linz. 1992. Isolation and characterization of a gene from Aspergillus parasiticus associated with the conversion of versicolorin A to sterigmatocystin in aflatoxin biosynthesis. Appl. Environ. Microbiol. 58:3527-3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smedsgaard, J. 1997. Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J. Chromatogr. A 760:264-270. [DOI] [PubMed] [Google Scholar]
- 37.Stanzani, M., E. Orciuolo, R. Lewis, D. P. Kontoyiannis, S. L. Martins, L. S. St. John, and K. V. Komanduri. 2005. Aspergillus fumigatus suppresses the human cellular immune response via gliotoxin-mediated apoptosis of monocytes. Blood 105:2258-2265. [DOI] [PubMed] [Google Scholar]
- 38.Suen, Y. K., K. P. Fung, C. Y. Lee, and S. K. Kong. 2001. Gliotoxin induces apoptosis in cultured macrophages via production of reactive oxygen species and cytochrome c release without mitochondrial depolarization. Free Radic. Res. 35:1-10. [DOI] [PubMed] [Google Scholar]
- 39.Tekaia, F., and J. P. Latge. 2005. Aspergillus fumigatus: saprophyte or pathogen? Curr. Opin. Microbiol. 8:385-392. [DOI] [PubMed] [Google Scholar]
- 40.Tsitsigiannis, D. I., J.-W. Bok, D. Andes, N. K. Fog, J. C. Frisvad, and N. P. Keller. 2005. Aspergillus cyclooxygenase-like enzymes are associated with prostaglandin production and virulence. Infect. Immunol. 73:4548-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang, S. Z., P. K. Smith, M. Lovejoy, J. J. Bowden, J. H. Alpers, and K. D. Forsyth. 1998. The apoptosis of neutrophils is accelerated in respiratory syncytial virus (RSV)-induced bronchiolitis. Clin. Exp. Immunol. 114:49-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward, C., E. R. Chilvers, M. F. Lawson, J. G. Pryde, S. Fujihara, S. N. Farrow, C. Haslett, and A. G. Rossi. 1999. NF-κB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 274:4309-4318. [DOI] [PubMed] [Google Scholar]
- 43.Waring, P., R. D. Eichner, A. Mullbacher, and A. Sjaarda. 1988. Gliotoxin induces apoptosis in macrophages unrelated to its antiphagocytic properties. J. Biol. Chem. 263:18493-18499. [PubMed] [Google Scholar]
- 44.Wilcoxon, F. 1945. Individual comparisons by ranking methods. Biomet. Bull. 1:80-83. [Google Scholar]
- 45.Wolpert, T. J., L. D. Dunkle, and L. M. Ciuffetti. 2002. Host-selective toxins and avirulence determinants: what's in a name? Annu. Rev. Phytopathol. 40:251-285. [DOI] [PubMed] [Google Scholar]
- 46.Xue, T., C. K. Nguyen, A. Romans, D. P. Kontoyiannis, and G. S. May. 2004. Isogenic auxotrophic mutant strains in the Aspergillus fumigatus genome reference strain AF293. Arch. Microbiol. 182:346-353. [DOI] [PubMed] [Google Scholar]
- 47.Yang, Y. F., M. J. Sylte, and C. J. Czuprynski. 1998. Apoptosis: a possible tactic of Haemophilus somnus for evasion of killing by bovine neutrophils? Microb. Pathog. 24:351-359. [DOI] [PubMed] [Google Scholar]
- 48.Yoder, O. C., and B. G. Turgeon. 1996. Molecular-genetic evaluation of fungal molecules for roles in pathogenesis to plants. J. Genet. 75:425-440. [Google Scholar]
- 49.Zhang, Y.-Q., H. Wilkinson, N. P. Keller, and D. Tsitsigiannis. 2005. Secondary metabolite gene clusters, p. 355-386. In Z. An, Handbook of industrial microbiology. Marcel Dekker, New York, N.Y.