

Abstract
Gamma interferon (IFN-γ)-induced indoleamine dioxygenase (IDO), which inhibits chlamydial replication by reducing the availability of tryptophan, is up-regulated by interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α). The mechanisms by which this occurs include an increase in the synthesis of interferon regulatory factor-1 as well as a nuclear factor-κB (NF-κB)-dependent increase in the expression of IFN-γ receptors (IFN-γR). Although Chlamydia is susceptible to IDO, it up-regulates IFN-γR expression to a greater degree than either IL-1β or TNF-α, perhaps through interaction with Toll-like receptors (TLR). The purpose of this study was to determine the mechanism by which Chlamydia psittaci up-regulates IFN-γR expression and evaluate this effect on IDO induction. Infection of HEK 293 cells with C. psittaci increased IFN-γR expression only in cells expressing either TLR2 or TLR4 and the adaptor protein MD-2. In addition, up-regulation of IFN-γR expression in Chlamydia-infected HeLa cells could be blocked either by neutralizing TLRs with anti-TLR2 and/or anti-TLR4 or by inhibiting NF-κB transactivation with a proteasome inhibitor. Although the newly expressed IFN-γR in Chlamydia-infected cells were capable of binding IFN-γ, they did not enhance IFN-γ-induced IDO activity in a manner similar to those observed for IL-1β and TNF-α. Instead, IDO activation in Chlamydia-infected cells was no different than that induced in uninfected cells, despite the increase in IFN-γR expression. Furthermore, the amount of IFN-γ-induced signal transducer and activator of transcription 1 (STAT-1) activation in infected cells paralleled that observed in uninfected cells, suggesting that STAT-1 activation by these newly expressed receptors was impaired.
The Toll-like receptors (TLRs) belong to a family of innate immune receptors found on both immune and nonimmune cells (8, 45). They are essential for the activation of subsequent immune responses to various conserved microbial components (13, 15, 18, 37), including lipopolysaccharide (LPS), peptidoglycan, fimbriae, and heat shock proteins (10, 21, 29). The TLRs are type I transmembrane receptors whose cytoplasmic domains have strong homologies to those of interleukin receptors (1). Interleukin 1 (IL-1) receptors and TLRs have a common signaling pathway, and the stimulation by IL-1 or bacterial components of the TLRs triggers activation and nuclear translocation of the transcription factor nuclear factor-κB (NF-κB). The subsequent binding of NF-κB to DNA results in the transcription of a number of genes, including those inducing chemokines, adhesion molecules, proinflammatory cytokines, and cytokine receptors (9, 40, 42). Up-regulation of gamma interferon receptor (IFN-γR) expression following NF-κB transactivation renders the cell more sensitive to IFN, thereby requiring less IFN for subsequent gene activation (23, 40).
Among the genes activated by IFN-γ is indoleamine 2,3-dioxygenase (IDO), which converts tryptophan to N-formylkynurenine, resulting in the depletion of tryptophan (38). The IFN-induced loss of tryptophan has been shown to restrict the growth of several pathogens, including Toxoplasma, group B streptococci, and Chlamydia species (2, 25, 30). Furthermore, IDO regulation can be influenced by IL-1, tumor necrosis factor alpha (TNF-α), and LPS, each of which enhances the amount of IDO activity, and thereby the antimicrobial effect of IFN-γ, in a dose-related manner (3, 6, 16, 19). Activation of NF-κB in response to the binding of IL-1, TNF-α, and LPS to their appropriate receptors plays a central role in this synergistic up-regulation of IDO activity. In addition to the NF-κB-dependent increase in IFN-γR expression, induction of IDO requires synthesis of interferon regulatory factor-1 (5, 12), and its efficient transcriptional activation is dependent on increased nuclear NF-κB (33).
The interaction of Chlamydia, an obligate intracellular pathogen, with surface receptors of epithelial cells is also capable of up-regulating IFN-γR expression. A TLR4 antagonist, diphosphoryl lipid A, partially inhibits this up-regulation, suggesting that Chlamydia may be interacting with more than one receptor. While intact chlamydiae have been shown to interact predominantly with TLR2 (31), purified chlamydial macromolecules may interact with both TLR2 and TLR4 (7, 14, 35). In addition, Chlamydia psittaci has been shown to enhance the expression of IFN-γRs more effectively than cytokines (39), yet the effect of this increase in receptor expression on IDO induction has not been explored.
The purpose of this study was twofold. First, the mechanism by which Chlamydia psittaci is able to up-regulate IFN-γR expression on epithelial cells was examined. HEK 293 cells transfected to express either TLR2 or TLR4 and specific TLR-neutralizing antibodies against TLR2 or TLR4 were used to identify TLRs used by Chlamydia to up-regulate IFN-γR expression. Second, the capacities of these newly expressed cytokine receptors to elicit a synergistic induction of IDO activity in a manner similar to that observed with IL-1 (40) was assessed. The results indicate that although the up-regulated cytokine receptors were able to bind IFN-γ, there was not an enhancement in signal transducer and activator of transcription 1 (STAT-1) signaling or IDO induction in the infected epithelial cells.
MATERIALS AND METHODS
Cytokines and immunoreagents.
Human recombinant IL-1β (specific activity = 1 × 107 U/mg; <0.1 ng LPS/mg) was purchased from Peprotech (Rocky Hill, NJ). Human recombinant IFN-γ (specific activity = 108 U/mg; <0.4 ng LPS/mg) was obtained from Biogen (Cambridge, MA). Rabbit polyclonal anti-human IFN-γ receptor (alpha chain) was purchased from Fitzgerald (Dublin, Ireland). Genus-specific mouse monoclonal anti-chlamydial LPS immunoglobulin G2a (IgG2a) was purchased from Chemicon (MAB8321; Temecula, CA). Fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse IgG F(ab′)2, phycoerythrin (PE)-conjugated sheep anti-mouse IgG F(ab′)2, PE-conjugated goat anti-rabbit IgG F(ab′)2, rabbit IgG, goat IgG, and mouse IgG2a were purchased from Sigma (St. Louis, MO). Mouse monoclonal neutralizing TLR2 (TL2.1) IgG1 and TLR4 (HTA125) IgG2a antibodies were purchased from eBioscience (San Diego, CA). Mouse monoclonal anti-human NF-κB IgG1 (sc8008), goat-polyclonal anti-human p-Stat1 (Tyr701), and goat polyclonal anti-human PCNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Photoprobe was purchased from Vector Laboratories (Burlingame, CA). FITC-conjugated streptavidin was purchased from Southern Biotechnology (Birmingham, AL). Proteasome inhibitor II (PSI) was purchased from Calbiochem (San Diego, CA).
Cell cultivation.
HeLa 229 cells, obtained from the American Type Culture Collection (Rockville, MD), were cultivated in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (vol/vol), gentamicin sulfate (10 μg/ml), and 100 μg/ml of streptomycin sulfate (complete MEM). Cells were maintained at 106 cells/ml at 37°C in 5% CO2 in air. HeLa cells transfected with pGASinsIDO/EGFP-C1, encoding the enhanced green fluorescent protein (GFP) under the regulation of the IDO promoter (34), were cultivated in complete medium containing G418 (100 μg/ml) for maintenance of transfected cells. HEK 293 cells transfected with either TLR2, TLR4 and MD-2 (an adaptor protein required for the binding of LPS to TLR4), or the empty pcDNA expression vector were a gift from Douglas Golenbock (University of Massachusetts) and were cultivated in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (vol/vol), 10 μg/ml of gentamicin sulfate, and 100 μg/ml of streptomycin sulfate (complete Dulbecco's modified Eagle's medium) as described above, and transfection was maintained with G418 (500 μg/ml).
Chlamydia cultivation.
C. psittaci strain 6BC was propagated in HeLa 229 cell monolayers in complete MEM containing cycloheximide (2 μg/ml). To prepare chlamydial stocks, HeLa cells were infected at a multiplicity of infection (MOI) of 2 inclusion-forming units (IFUs) per cell and incubated for 2 days; infected cells were then removed from the tissue culture plate with trypsin and disrupted by sonication. Elementary bodies were purified by a series of differential centrifugations; frozen in sucrose, l-glutamic acid, and sodium phosphate (SPG) buffer; and stored at −80°C (32). Chlamydial stocks were standardized with HeLa 229 cells by the IFU method. Following 30 h of infection, cells were stained with fluorescent anti-chlamydial LPS, and fluorescent chlamydial inclusions were counted. Numbers of IFU/ml were determined using the following formula:
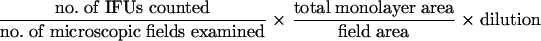 |
(1) |
IFN-γR expression assay.
Cells were seeded into six-well plates at a density of 2 × 105 cells/ml in complete medium and cultured for 12 h. Monolayers were washed twice with warm Hanks balanced salt solution (HBSS) before addition of fresh complete medium, IL-1β (100 ng/ml), or 6BC (multiplicity of infection, 2 IFUs), in triplicate. Plates were incubated at 37°C with centrifugation at 2,975 × g for 1 h. The inoculum was aspirated, the monolayer was washed twice with warm HBSS, and complete medium was added to each well before culturing the plates for 24 h to permit maximal increases in receptor expression. After the cells were collected by gently scraping them with cell lifters, they were incubated with anti-IFN-γR or anti-chlamydial antibodies for 1 h and FITC- or PE-conjugated secondary antibodies for 1 h on ice and washed with phosphate-buffered saline (PBS). Two-color flow cytometric analyses were performed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA) with an argon laser tuned to 488 nm. Red fluorescence for receptor expression was recorded in the FL2 emission channel containing a 580-nm filter. Green fluorescence for chlamydial infection was recorded in the FL1 emission channel containing a 530-nm filter. CELLQuest software (Becton Dickinson) was used for analysis of the data.
Since HEK 293 cells do not express TLR2 or TLR4, they were used to assess the effect of individual TLRs in transfectants expressing either TLR2 or TLR4 plus MD-2 (17, 24). HEK 293 cells transfected with the empty expression vector served as a negative control, and HeLa 229 cells, which naturally express TLRs (8, 28), served as positive controls. Each was treated with IL-1β or infected with Chlamydia as described above before changes in IFN-γR expression were assessed by flow cytometric analysis using anti-IFN-γR antibody. Statistical analysis of changes in TLR expression was by one-way analysis of variance (ANOVA).
To determine whether the increases in IFN-γR expression on HeLa 229 cells in response to Chlamydia infection were TLR mediated, they were treated with IL-1β or infected with Chlamydia as described above. Some wells also received mouse neutralizing anti-human TLR2 (100 μg/ml), anti-human TLR4 (100 μg/ml), or both antibodies. Changes in IFN-γR expression were assessed by flow cytometric analysis using anti-IFN-γR antibody. Statistical analysis of antibody neutralization used a 3 by 2 by 2 factorial design ANOVA.
NF-κB requirement for IFN-γR increase.
NF-κB has been shown to be important in cytokine-induced receptor increase as well as in IDO transcriptional activation (33, 40). To assess the role of NF-κB transactivation in the Chlamydia-induced increase in IFN-γR expression, HeLa 229 cells were cultivated in triplicate in medium alone, treated with IL-1β, or infected with C. psittaci as described above. Some wells also received PSI (3 mM) for 12 h, since concentrations of PSI in this range have been shown to maintain nuclear NF-κB concentrations at steady-state levels in TNF-α- or IL-1β-treated cells (33, 40). The cells were harvested, and changes in IFN-γR expression were assessed by flow cytometric analysis using anti-IFN-γR antibody. Statistical analysis of changes in IFN-γR expression was by one-way ANOVA.
Cytokine binding assay.
To quantify changes in the capacities of cells to bind IFN-γ following IFN-γR up-regulation, HeLa 229 cells (106 cells/ml) were cultivated in medium alone, treated with IL-1β, or infected with C. psittaci for 24 h, in triplicate, to allow for maximum receptor expression. Parallel sets of cells were then collected by scraping them with cell lifters and then incubated with biotinylated-IFN-γ or with anti-IFN-γR antibody for 1 h on ice. FITC-conjugated streptavidin was added to the cells that received biotinylated IFN-γ at a streptavidin-to-biotinylated-cytokine ratio of 10:1, whereas PE-conjugated antibodies were added to cells receiving anti-receptor antibodies. After an additional 1 h incubation on ice, during which the streptavidin and the secondary antibody bind to biotin and primary antibody, respectively, the cells were washed twice with PBS and assessed by flow cytometric analysis. Statistical analysis of IFN-γ binding was by one-way ANOVA.
IFN-γ was biotinylated by diluting the cytokine in 10 mM Tris, pH 8.0, 1 mM EDTA (TE) to a final concentration of 100 ng/ml. Biotin photoprobe was added to IFN-γ at a 20:1 biotin-to-IFN-γ ratio and UV photocoupled for 30 min at a 2-cm distance from the UV lamp (30W) according to the manufacturer's instructions.
Assessment of nuclear transcription factor translocation.
Nuclei were isolated as described previously (40). Briefly, cells (106 cells/ml) were incubated on ice in cold PBS for 1 h and collected by gently scraping them with cell lifters. The cells were washed with PBS by centrifugation. Nuclei were harvested by suspending the cell pellets in cold nuclear extraction buffer (230 mM sucrose, 5 mM MgCl2, 10 mM HEPES, 1% Triton X-100) and incubating them on ice for 10 min. The nuclei were pelleted by centrifugation and washed with nuclear wash buffer (320 mM sucrose, 5 mM MgCl2, and 10 mM HEPES). Nuclear yield was determined by microscopic examination using DAPI (4′,6′-diamidino-2-phenylindole) stain. To assess whether NF-κB was activated by infection, HeLa 229 cells were cultivated with medium alone, treated with IL-1β, or infected with C. psittaci for 1 h. After the nuclei were harvested, they were incubated in a 1:50 dilution of PE-conjugated anti-NF-κB p65 antibody in nuclear labeling buffer (320 mM sucrose, 5 mM MgCl2, 10 mM HEPES, 1% bovine serum albumin, and 0.1% sodium azide) overnight before the amount of NF-κB present was assessed by flow cytometric analysis.
To determine the amount of phosphorylated STAT-1 translocated into the nucleus upon IFN-γ stimulation, HeLa 229 cells were cultured with complete medium, treated with IL-1β, or infected with C. psittaci for 12 h. Then, the cells were washed and incubated for an additional 4 h, at which point they were treated with IFN-γ (20 ng/ml) for 4 h. Nuclei were harvested and held in a 1:150 dilution of anti-p-STAT-1 IgG specific for the tyrosine 701 residue overnight. The nuclei were washed twice with nuclear labeling buffer and incubated for 4 h with a 1:50 dilution of sheep FITC-conjugated anti-mouse IgG secondary antibody. The nuclei were washed once again with labeling buffer before flow cytometric analysis for the presence of p-STAT-1 tyrosine 701. Statistical analysis of changes in STAT-1 phosphorylation was by one-way ANOVA with Dunnett's test for multiple comparisons.
IDO quantitation.
To assess the effect of cytokine stimulation by IL-1β on IDO induction, HeLa 229 cells (105 cells/ml) were cultivated with IL-1β, cultivated with complete medium alone, or infected with C. psittaci and incubated for 12 h. At that time, the cells were washed with PBS and incubated for an additional 4 h in complete medium. IFN-γ was then added in increasing concentrations (up to 100 ng/ml), and the cells were incubated for an additional 24 h. For determination of IDO activity, the medium was replaced with 0.4 ml of HBSS containing [5-3H]tryptophan (1 μCi/ml; specific activity = 20 Ci/mmol; MP Biomedicals, Irvine, CA) and 25 μM tryptophan carrier. After an additional 4 h of incubation, supernatants were collected and analyzed for tryptophan metabolites by reversed-phase high-performance liquid chromatography (HPLC) (44). Briefly, aliquots (50 μl) were injected into a μBondapak C18 column (Millipore) and eluted with 1 mM KH2PO4 buffer, pH 4.0, containing 10% MeOH, at a flow rate of 1.6 ml/min. Radioactivity in tryptophan and metabolite fractions was quantified by flowthrough scintillation spectroscopy using an HPLC (Varian, Palo Alto, CA) equipped with a radioisotope detector (Radiomatic instruments, Tampa, FL). The percent specific tryptophan catabolism was calculated by the following equation:
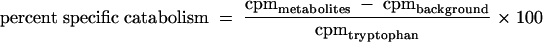 |
(2) |
where cpmmetabolites corresponds to the counts per minute (cpm) present in metabolite fractions, cpmbackground represents the cpm resulting from nonspecific breakdown of tryptophan to metabolites, and cpmtryptophan equals the total number of cpm for all fractions. Statistical analysis of IDO activity was by analysis of covariance with several regression lines.
RESULTS
Requirement of TLRs for increased IFN-γR expression.
To assess the involvement of TLRs in the Chlamydia-induced increase in IFN-γR expression, HEK 293 cells transfected with TLR2, TLR4 plus MD-2, or the empty expression vector (24, 36, 41, 43) were cultivated with complete medium alone, treated with IL-1β, or infected with C. psittaci for 24 h. IFN-γR expression and chlamydial infection were assessed by flow cytometric analysis using anti-IFN-γR and anti-chlamydial antibodies. As described previously (39), flow cytometry revealed that the uninfected as well as the infected cell populations in C. psittaci-infected cultures exhibited increased IFN-γR expression relative to that in cells cultured in medium alone. When infected with C. psittaci, HEK 293 cells expressing either TLR2 or TLR4/MD-2 displayed significantly increased (P < 0.001) IFN-γR expression (Fig. 1), supporting a role for TLRs in up-regulation of IFN-γR expression following chlamydial stimulation. HEK 293 cells transfected with the empty vector did not increase IFN-γR expression during infection, indicating that an intact TLR was required for up-regulation of IFN-γR expression by Chlamydia.
FIG. 1.

Up-regulation of IFN-γR expression by C. psittaci is TLR dependent. HEK 293 cells stably transfected with TLR2, TLR4/MD-2, or the empty pcDNA expression vector were cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci for 24 h and stained with antibody to IFN-γR and PE-conjugated secondary antibody. The mean fluorescent intensities (MFIs) were determined using flow cytometry. The results are representative of several independent experiments with similar results (MFIs ± standard deviations).
To confirm the involvement of the TLRs in up-regulation of IFN-γR expression during chlamydial infection, neutralizing antibodies to either TLR2 or TLR4 were used. HeLa 229 cells were treated with medium alone, treated with IL-1β, or infected with C. psittaci for 24 h. The cells were simultaneously treated with anti-TLR2, anti-TLR4, or combined TLR-neutralizing antibodies, and IFN-γR expression was assessed by flow-cytometric analysis. As described previously (39), both IL-1β and C. psittaci stimulated increased IFN-γR expression in the absence of neutralizing antibody (Fig. 2), and Chlamydia induced more IFN-γR expression than did IL-1β. However, only infected cultures receiving either TLR2- or TLR4-neutralizing antibodies had significantly lower increases in IFN-γR expression (P < 0.0001); the neutralizing antibodies had no effect on IFN-γR expression in either uninfected or IL-1β-treated cells. While neither neutralizing antibody alone was sufficient to fully block up-regulation of IFN-γR expression in infected cultures, the combination of both TLR-neutralizing antibodies completely inhibited the increase in IFN-γR expression, indicating that only TLR2 and TLR4 were stimulated by C. psittaci to up-regulate IFN-γR expression.
FIG. 2.

Neutralization of TLRs inhibits up-regulation or IFN-γR expression. HeLa 229 cells were cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci. Some cultures also received neutralizing anti-TLR2, anti-TLR4, both antibodies, or normal IgG. After 24 h, the cells were collected and receptor expression was assessed using flow cytometric analysis. Visual confirmation was by fluorescent microscopy. The results for one of three similar experiments (mean fluorescent intensities [MFIs] ± standard deviations) are presented.
Role of NF-κB in IFN-γR up-regulation.
IL-1β-mediated up-regulation of IFN-γR expression requires activation of NF-κB (40). Because IL-1R and TLRs are in the same receptor family, they may utilize the same mechanism for receptor up-regulation. To assess whether TLRs require NF-κB transactivation to increase IFN-γR expression, NF-κB translocation to the nucleus was quantified with HeLa 229 cells treated with medium, treated with IL-1β, or infected with C. psittaci for 1 h. At this time, maximal nuclear NF-κB can be detected following IL-1β stimulation (40). Nuclei were harvested from the cells, and the amount of intranuclear NF-κB was determined by flow cytometric analysis using an anti-NF-κB p65 antibody. Cells either treated with IL-1β or infected with C. psittaci were found to transactivate NF-κB to the same extent (Fig. 3, top panel) relative to basal levels detected in unstimulated cells. To confirm that the up-regulation of IFN-γR expression was due to NF-κB activation, PSI, which prevents NF-κB activation and nuclear translocation by inhibiting degradation of IκB (4, 11), was used at a concentration previously shown to maintain nuclear NF-κB at constitutive levels (33, 40). HeLa cells were cultivated in medium alone, treated with IL-1β, or infected with C. psittaci, with or without PSI. After 12 h, the cells were harvested and analyzed for IFN-γR expression by flow cytometry. In cultures treated with IL-1β or infected with C. psittaci, PSI completely inhibited IFN-γR up-regulation (P < 0.001), such that IFN-γR expression was no different from that in cells cultivated in medium alone (Fig. 3, bottom panel). This suggests that in the absence of NF-κB transactivation, C. psittaci is unable to up-regulate IFN-γR expression in a manner similar to that described for IL-1β (40).
FIG. 3.

Dependency of IFN-γR expression up-regulation on NF-κB transactivation. (Top) HeLa 229 cells were cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci for 1 h. Nuclei were harvested and subjected to flow cytometric analysis of intranuclear NF-κB present with an antibody to the p65 subunit of NF-κB. (Bottom) HeLa 229 cells were cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci, alone or in combination with PSI (3 μM) for 12 h. The cells were harvested, and IFN-γR expression was determined by flow cytometric analysis using an anti-receptor antibody. The results from one of several experiments with similar results are presented (mean fluorescent intensities [MFIs] ± standard deviations).
C. psittaci-stimulated HeLa cells do not increase IDO activity.
In a previous study, IL-1β was shown to increase the expression of IFN-γRs, resulting in a synergistic induction of IDO activity when the cells were induced with IFN-γ (40). Therefore, if increased receptor expression influences IDO induction, up-regulation of IFN-γR expression in response to chlamydiae should also result in enhanced IDO activity. To test this hypothesis, HeLa 229 cells were cultivated with complete medium alone, treated with IL-1β, or infected with C. psittaci for 12 h, followed by cultivation with complete medium alone for 4 h. Under these conditions, IFN-γR expression is maximal and nuclear NF-κB concentration has returned to basal levels (40). Cells were then incubated with increasing concentrations of IFN-γ (0 to 100 ng/ml) for 24 h before IDO reporter activity was assessed using flow cytometric analysis. As shown in Fig. 4 (top panel), cells treated first with IL-1β before induction of IDO had significantly increased reporter activities (P < 0.01) compared to cells induced with only IFN-γ, requiring 1/10 the amount of IFN-γ to induce the same amount of IDO reporter activity as that measured in untreated cells. However, no difference was seen between untreated cells and cells infected first with C. psittaci to up-regulate IFN-γR expression. Parallel cultures of HeLa 229 cells treated under the same conditions and analyzed for IDO enzyme activity by HPLC showed the same trends (Fig. 4, bottom panel). Only up-regulation of IFN-γR expression by IL-1β, and not by C. psittaci, resulted in enhancement of IDO activity. Taken together, these data suggest that in C. psittaci-infected cultures, newly expressed receptors are not contributing to transcriptional activation of the IDO gene.
FIG. 4.

IFN-γR expression up-regulated in response to Chlamydia does not contribute to increased IDO activity. (Top) HeLa 229 cells, stably transfected with the IDO reporter plasmid pGASinsIDO/EGFP-C1, were cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci for 12 h and then incubated in medium alone for 4 h before being induced with increasing concentrations of IFN-γ for an additional 12 h. The cells were harvested, and GFP expression was assessed using flow cytometric analysis (top panel), or the cells were analyzed for IDO enzymatic activity by HPLC (bottom panel). The results for one of three similar experiments (means ± standard deviations) are presented. MFI, mean fluorescent intensity.
Binding of IFN-γ to cells exhibiting increased IFN-γR expression.
Identification of cytokine receptors by flow cytometry indicates the existence of specific epitopes on the receptor molecules but does not assess whether the receptors retain their ability to bind cytokine. The inability of IFN-γRs expressed in response to Chlamydia to bind IFN-γ could explain why the increase in IFN-γR expression does not increase the sensitivities of those cells to IFN-γ treatment. To address this possibility, biotinylated IFN-γ was used to assess the ability of IFN-γRs to bind IFN-γ. HeLa 229 cells were cultivated in complete medium, treated with IL-1β, or infected with C. psittaci for 24 h. IFN-γ binding was assessed by first incubating the cells with biotinylated IFN-γ followed by FITC-conjugated streptavidin to bind to the biotin. Binding of biotinylated IFN-γ was compared to binding of anti-IFN-γR antibody in parallel cultures (Fig. 5). Flow cytometric analysis revealed that increases in IFN-γ binding were similar to increases in binding of anti-IFN-γR antibodies. This indicated that the IFN-γRs expressed in response to C. psittaci were fully capable of binding IFN-γ. Therefore, the inability of newly expressed IFN-γRs to up-regulate IFN-γ-induced IDO activity could not be explained by an inability to bind IFN-γ.
FIG. 5.

Up-regulation of cytokine binding to Chlamydia-infected cells. Parallel sets of HeLa 229 cells were cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci for 24 h. One set was then incubated with biotinylated-IFN-γ followed by FITC-conjugated streptavidin. The other set was stained with anti-IFN-γR antibody and PE-conjugated secondary antibody. The mean fluorescent intensity (MFI) of each was determined using flow cytometric analysis. The results for one of three similar experiments (MFIs ± standard deviations) are presented.
Chlamydia-induced IFN-γRs do not increase STAT-1 phosphorylation.
Since IFN-γRs expressed in response to Chlamydia bind IFN-γ normally, the lack of increased responsiveness by the cells to IFN-γ may be due to changes in receptor signaling. After the binding of IFN-γ to its receptor, STAT-1 is activated by phosphorylation. Upon activation, STAT-1 dimerizes and translocates to the nucleus, where it binds to control elements in IFN-γ-responsive genes. To determine whether the infected cells still retained the ability to induce STAT-1 phosphorylation and translocation to the nucleus following IFN-γ treatment, HeLa 229 cells were cultivated in complete medium, treated with IL-1β, or infected with C. psittaci for 12 h and then induced with IFN-γ. Nuclei from the cells were isolated, stained using anti-phosphorylated STAT-1 antibody, and analyzed by flow cytometry. Cells treated with IL-1β and then induced with IFN-γ showed significant (P < 0.001) increases in amounts of STAT-1 phosphorylation; however, cells that were infected showed no difference in phosphorylated-STAT-1 levels compared with uninfected cells (Fig. 6).
FIG. 6.

Up-regulated IFN-γR expression does not increase STAT-1 activation. HeLa 229 cells cultivated in complete medium, treated with IL-1β (100 ng/ml), or infected with C. psittaci for 12 h were induced with IFN-γ (20 ng/ml) for 4 h. Nuclei were isolated, and phosphorylated STAT-1 levels were assessed by flow cytometric analysis using an anti-STAT-1P Tyr 701 antibody. The results are representative of several independent experiments with similar results (mean fluorescent intensities [MFI] ± standard deviations).
DISCUSSION
That Chlamydia is able to modulate expression of IFN-γRs is not unique. Increases and decreases in cytokine receptor expression have been demonstrated with a variety of other pathogens. Mycobacterium and Trypanosoma cruzi have been reported to down-regulate IFN-γR expression on murine macrophages and B cells, respectively, while Legionella has been shown to increase expression of both IL-1RI and TNF receptor on macrophages (20, 22, 26). Changes in cytokine responsiveness by modulation of the expression of cytokine-specific receptors are one means by which a pathogen could manipulate cell-mediated immune responses. Chlamydia up-regulates the expression of IL-1β, TNF-α, and IFN-γ receptors, each of which is involved in synergistic IDO induction. Furthermore, these increases in cytokine receptor expression are significantly higher than increases stimulated by cytokines (39). The increases in cytokine receptor expression induced by Chlamydia are facilitated by the TLR system. Although a TLR4 antagonist partially blocks up-regulation of IFN-γR expression, other TLRs might be involved. Intact chlamydiae have been shown to interact predominantly with TLR2 (31), yet purified chlamydial components interact with TLR differentially; chlamydial LPS activates TLR4 (14), whereas chlamydial HSP60 may activate both TLR2 and TLR4 (7, 35). The binding of a chlamydial component to a TLR would initiate a signal transduction cascade, resulting in an NF-κB-dependent increase in cytokine receptor expression.
To identify the TLRs involved in up-regulation of IFN-γR expression, transfected cells expressing either TLR2 or TLR4 and its required adaptor protein MD-2 were tested. Stimulation of either TLR by C. psittaci was capable of up-regulating IFN-γR expression, indicating that chlamydial components were capable of using either TLR. Furthermore, neutralization of either TLR with antibody only partially abrogated the effect of Chlamydia; complete inhibition of receptor up-regulation required the neutralization of both receptors, indicating that both TLR2 and TLR4 are important in the regulation of IFN-γR expression. That the signaling cascade induced by TLR stimulation and leading to up-regulation of IFN-γR expression required the activation of NF-κB was demonstrated by inhibiting proteasomal degradation of IκB. In the absence of an increase in NF-κB activation, IFN-γR expression was not changed.
Given the magnitude of increase in IFN-γR expression in Chlamydia-infected cultures, one would expect increases in IDO induction induced by IFN-γ to be greater than that seen following IL-1β treatment (40). However, infection with C. psittaci did not affect IDO induction; instead, IFN-γ induced the same amount of IDO activity in C. psittaci-infected cells as it did in uninfected cells. This would suggest that the signaling pathway used by the newly synthesized receptors in Chlamydia-infected cells was incomplete, the basis of which could be an inability to bind cytokine or to generate a signal. To ensure that the receptors detected by flow cytometry retained their ability to bind their respective cytokines, their binding of biotinylated cytokines was determined in parallel to detection by antibody to receptor subunits. In each case, the amount of cytokine bound was proportional to the amount of anti-cytokine receptor antibody bound. Since these newly expressed receptors could bind cytokine, the endpoint of the JAK/STAT signaling pathway was examined. To determine whether this STAT-1 was being activated and translocated to the nucleus, changes in nuclear phosphorylated STAT-1 following IFN-γ treatment were measured. While cells expressing increased IFN-γRs in response to IL-1β treatment exhibited increased nuclear p-STAT-1 following IFN-γ exposure, cells from infected cultures failed to respond similarly, despite the expression of a greater number of IFN-γRs. In fact, the amount of nuclear p-STAT-1 was the same as that in cells cultivated in medium alone. The same trend was observed in HEK 293 cells transfected to express either TLR2 or TLR4/MD-2 (data not shown). Clearly, although the increases in IFN-γR expression in response to Chlamydia are TLR dependent, these receptors do not contribute to the IFN-γ responsiveness of these cells.
Although these experiments were performed with epithelial cells, C. psittaci is capable of infecting a variety of cell types (27), including macrophages that are capable of producing proinflammatory cytokines such as IL-1β and TNF-α upon stimulation of their TLRs (1). These proinflammatory cytokines have been shown to be important in the up-regulation of IDO activity, in an NF-κB-dependent manner. Activation of NF-κB is required for both the up-regulation of IFN-γR expression (40) and the synthesis of interferon regulatory factor-1 (33), a transcription factor required for IDO transcriptional activation (5, 12). However, production of IL-1β or TNF-α may not be sufficient to overcome the effect of chlamydial infection on the STAT-1 signaling pathway. Although infection of macrophages can elicit production of IL-1, which can up-regulate IDO induction, it will do so only in combination with IFN-γ (3). Induction of IDO activity is dependent on IFN-γ-mediated STAT-1 phosphorylation and nuclear translocation. Thus, while IL-1 can increase the amount of IDO activity induced by IFN-γ, IL-1 alone does not induce either STAT-1 signaling or IDO activity. In the absence of a functional JAK/STAT signaling pathway, C. psittaci might up-regulate a localized inflammatory response without inducing the protective benefits of IDO induction.
It is tempting to speculate about the role that these receptors play during infection with Chlamydia. One possibility is that these receptors could function as decoys. IFN-γ would bind to these receptors but not activate signal transduction, thereby reducing the local concentration of IFN-γ. By reducing the effecting concentration of IFN-γ, induction of IDO would be diminished, resulting in prolonged survival of Chlamydia in the face of an otherwise effective immune response.
Acknowledgments
This work was supported by Public Health Service grant no. AI45836 from the National Institute of Allergy and Infectious Diseases (J.M.C.).
We thank the following people for their contributions: Douglas Golenbock and Eicke Latz from the University of Massachusetts for providing us with the HEK 293 cells expressing TLR2 and TLR4 plus MD-2 and Michael Hughes in the Department of Mathematics and Statistics at Miami University for his help in all statistical data analyses.
Editor: J. L. Flynn
Footnotes
Published ahead of print on 9 October 2006.
REFERENCES
- 1.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675-680. [DOI] [PubMed] [Google Scholar]
- 2.Byrne, G. I., L. K. Lehmann, and G. J. Landry. 1986. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect. Immun. 53:347-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlin, J. M., and J. B. Weller. 1995. Potentiation of interferon-mediated inhibition of Chlamydia infection by interleukin-1 in human macrophage cultures. Infect. Immun. 63:1870-1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, Z., J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard, and T. Manatis. 1995. Signal-induced site-specific phosphorylation targets Iκ Bα to the ubiquitin-proteasome pathway. Genes Dev. 9:1586-1597. [DOI] [PubMed] [Google Scholar]
- 5.Chon, S. Y., H. H. Hassanain, and S. L. Gupta. 1996. Cooperative role of interferon regulatory factor 1 and p91 (STAT-1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J. Biol. Chem. 271:17247-17452. [DOI] [PubMed] [Google Scholar]
- 6.Currier, A. R., M. H. Ziegler, M. M. Riley, T. A. Babcock, V. P. Telbis, and J. M. Carlin. 2000. Tumor necrosis factor-α and lipopolysaccharide enhance interferon-induced antichlamydial indoleamine dioxygenase activity independently. J. Interferon Cytokine Res. 20:369-376. [DOI] [PubMed] [Google Scholar]
- 7.DaCosta, C., N. Wantia, C. J. Kirschning, D. H. Busch, N. Rodriguez, H. Wagner, and T. Miethke. 2004. Heat shock protein 60 from Chlamydia pneumoniae elicits an unusual set of inflammatory responses via Toll-like receptor 2 and 4 in vivo. Eur. J. Immunol. 34:2874-2884. [DOI] [PubMed] [Google Scholar]
- 8.Deva, R., P. Shankaranarayanan, R. Ciccoli, and S. Nagam. 2003. Candida albicans induces selectively transcriptional activation of cyclooxygenase-2 in HeLa cells: pivotal roles of Toll-like receptors, p38 mitogen-activated protein kinase, and NF-κ B. J. Immunol. 171:3047-3055. [DOI] [PubMed] [Google Scholar]
- 9.Duffey, D. C., Z. Chen, G. Dong, F. G. Ondrey, J. S. Wolfe, K. Brown, U. Siebenlist, and C. Van Waes. 1999. Expression of a dominant-negative mutant inhibitor-κ Bα of nuclear factor-κ Β in human head and neck squamous cell carcinoma inhibitis survival, proinflammatory cytokine expression, and tumor growth in vivo. Cancer Res. 59:3468-3474. [PubMed] [Google Scholar]
- 10.Frendéus, B., C. Wachtler, M. Hedlund, H. Fischer, P. Samuelsson, M. Svensson, and C. Svanborg. 2001. Escherichia coli P fimbriae utilize the Toll-like receptor 4 pathway for cell activation. Mol. Microbiol. 40:37-51. [DOI] [PubMed] [Google Scholar]
- 11.Griscavage, J., S. Wilk, and L. J. Ignarro. 1996. Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-κ B. Proc. Natl. Acad. Sci. USA 93:3308-3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta, S., D. Xia, M. Jiang, S. Lee, and A. B. Pernis. 1998. Signaling pathways mediated by the TNF- and cytokine-receptor families target a common cis-element of the IFN regulatory factor 1 promoter. J. Immunol. 161:5997-6004. [PubMed] [Google Scholar]
- 13.Hajishengallis, G., M. Martin, H. T. Sojar, A. Sharma, R. E. Schifferle, E. DeNardin, M. W. Russell, and R. J. Genco. 2002. Dependence of bacterial protein adhesions on Toll-like receptors for proinflammatory cytokine induction. Clin. Diagn. Lab. Immunol. 9:403-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heine, H., S. Müller-Loennies, L. Brade, B. Lindner, and H. Brade. 2003. Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur. J. Biochem. 270:440-450. [DOI] [PubMed] [Google Scholar]
- 15.Hirschfeld, M., C. J. Kirschning, R. Schwandner, H. Wesche, J. H. Weis, R. M. Wooten, and J. J. Weis. 1999. Inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by Toll-like receptor 2. J. Immunol. 163:2382-2386. [PubMed] [Google Scholar]
- 16.Hissong, B. D., G. I. Byrne, M. L. Padilla, and J. M. Carlin. 1995. Upregulation of interferon-induced indoleamine 2,3-dioxygenase in human macrophage cultures by lipopolysaccharide, muramyl tripeptide, and interleukin-1. Cell. Immunol. 160:264-269. [DOI] [PubMed] [Google Scholar]
- 17.Hornung, V., S. Rothenfusser, S. Britsch, A. Krug, B. Jahrsdorfer, T. Giese, S. Endres, and G. Hartmann. 2002. Quantitative expression of Toll-like receptor 1-10 mRNA cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168:4531-4537. [DOI] [PubMed] [Google Scholar]
- 18.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749-3752. [PubMed] [Google Scholar]
- 19.Hu, B., B. D. Hissong, and J. M. Carlin. 1995. Interleukin-1 enhances indoleamine 2,3-dioxygenase activity by increasing specific mRNA expression in human mononuclear phagocytes. J. Interferon Cytokine Res. 15:1870-1875. [DOI] [PubMed] [Google Scholar]
- 20.Hussain, S., B. S. Zwilling, and W. P. Lafuse. 1999. Mycobacterium avium infection of mouse macrophages inhibits IFN-γ janus kinase-STAT signaling and gene induction by down-regulation of the IFN-γ receptor. J. Immunol. 163:2041-2048. [PubMed] [Google Scholar]
- 21.Kadowaki, N., S. Ho, S. Antonenko, R. de Waal Malefyt, R. A. Kastelein, F. Bazan, and Y.-J. Liu. 2001. Subsets of human dendritic cell precursors express different Toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194:863-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kierszenbaum, F., H. Mejia Lopez, M. K. Tanner, and M. B. Sztein. 1995. Trypanosoma cruzi-induced decrease in the level of interferon-gamma receptor expression by resting and activated human blood lymphocytes. Parasite Immunol. 17:207-214. [DOI] [PubMed] [Google Scholar]
- 23.Krakauer, T., and J. J. Oppenheim. 1993. IL-1 and tumor necrosis factor-α each up-regulate both the expression of IFN-γ receptors and enhance IFN-γ-induced HLA-DR expression on human monocytes and a human monocytic cell line (THP-1). J. Immunol. 150:1205-1211. [PubMed] [Google Scholar]
- 24.Latz, E., A. Visintin, E. Lien, K. A. Fitzgerald, B. G. Monks, E. A. Kurt-Jones, D. T. Golenbock, and T. Espevik. 2002. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the Toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 277:47834-47843. [DOI] [PubMed] [Google Scholar]
- 25.Mackenzie, C. R., U. Hadding, and W. Däubner. 1998. Interferon-gamma-induced activation of the indoleamine 2,3-dioxygenase in cord blood monocyte-derived macrophages inhibits the growth of group B Streptococci. J. Infect. Dis. 178:875-878. [DOI] [PubMed] [Google Scholar]
- 26.McHugh, S., Y. Yamamoto, T. W. Klein, and H. Friedman. 2000. Differential expression of IL-1 and TNF receptors in murine macrophages infected with virulent vs. avirulent Legionella pneumophila. Can. J. Microbiol. 46:885-890. [PubMed] [Google Scholar]
- 27.Moulder, J. W. 1991. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 55:143-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimura, M., and S. Naito. 2005. Tissue-specific mRNA expression profiles of human Toll-like receptors and related genes. Biol. Pharm. Bull. 28:886-892. [DOI] [PubMed] [Google Scholar]
- 29.Njoroge, T., R. J. Genco, H. T. Sojar, N. Hamada, and C. A. Genco. 1997. A role for fimbriae in Porphyromonas gingivalis invasion of oral epithelial cells. Infect. Immun. 65:1980-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfefferkorn, E. R. 1982. Interferon-γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 81:908-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prebeck, S., C. Kirschning, S. Durr, C. da Costa, B. Donath, K. Brand, V. Redecke, H. Wagner, and T. Miethke. 2001. Predominant role of Toll-like receptor 2 versus 4 in Chlamydia pneumoniae-induced activation of dendritic cells. J. Immunol. 167:3316-3323. [DOI] [PubMed] [Google Scholar]
- 32.Ripa, K. T., and P. Mårdh. 1977. Cultivation of Chlamydia trachomatis in cycloheximide-treated McCoy cells. J. Clin. Microbiol. 6:328-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson, C. M., P. T. Hale, and J. M. Carlin. 2006. NF-κB contributes to indoleamine dioxygenase transcriptional synergy by IFN-γ and tumor necrosis factor-alpha. Cytokine 35:53-61. [DOI] [PubMed]
- 34.Robinson, C. M., K. Shirey, and J. M. Carlin. 2003. Synergistic transcriptional activation of indoleamine dioxygenase by IFN-γ and tumor necrosis factor-α. J. Interferon Cytokine Res. 23:413-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasu, S., D. LaVerda, N. Qureshi, D. T. Golenbock, and D. Beasley. 2001. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via Toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ. Res. 89:244-250. [DOI] [PubMed] [Google Scholar]
- 36.Schromm, A. B., E. Lein, P. Henneke, J. C. Chow, A. Yoshimura, H. Heine, E. Latz, B. G. Monks, D. A. Schwartz, K. Miyake, and D. T. Golenbock. 2001. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: a point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J. Exp. Med. 194:79-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C. J. Kirschning. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J. Biol. Chem. 274:17406-17409. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu, T., S. Nomiyama, F. Hirata, and O. Hayaishi. 1978. Indoleamine 2,3-dioxygenase. Purification and some properties. J. Biol. Chem. 253:4700-4706. [PubMed] [Google Scholar]
- 39.Shirey, K., and J. M. Carlin. 2006. Chlamydiae modulate gamma interferon, interleukin-1β-, and tumor necrosis factor alpha receptor expression in HeLa cells. Infect. Immun. 74:2482-2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirey, K., J. Jung, G. S. Maeder, and J. M. Carlin. 2006. Up-regulation of interferon-γ receptor expression by proinflammatory cytokines influences IDO activation in epithelial cells. J. Interferon Cytokine Res. 26:53-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visintin, A., A. Mazzoni, J. A. Spitzer, and D. M. Segal. 2001. Secreted MD-2 is a large polymeric protein that efficiently confers lipopolysaccharide sensitivity to Toll-like receptor 4. Proc. Natl. Acad. Sci. USA 98:12156-12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weih, F., G. Warr, H. Yang, and R. Bravo. 1997. Mutlifocal defects inimmune responses in RelB-deficient mice. J. Immunol. 158:5211-5218. [PubMed] [Google Scholar]
- 43.Yang, H., D. W. Young, F. Gusovsky, and J. C. Chow. 2000. Cellular events mediated by lipopolysaccharide-stimulated Toll-like receptor 4: MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J. Biol. Chem. 275:20861-20866. [DOI] [PubMed] [Google Scholar]
- 44.Yong, S., and S. Lau. 1979. Rapid separation of tryptophan, kynurenine, and indoles using reversed phase high performance liquid chromatography. J. Chromatogr. 175:343-347. [DOI] [PubMed] [Google Scholar]
- 45.Zarember, K. A., and P. J. Godowski. 2002. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168:554-561. [DOI] [PubMed] [Google Scholar]