

Abstract
Antimicrobial peptides are present in most living species and constitute important effector molecules of innate immunity. Recently, we and others have detected antimicrobial peptides in the brain. This is an organ that is rarely infected, which has mainly been ascribed to the protective functions of the blood-brain barrier (BBB) and meninges. Since the bactericidal properties of the BBB and meninges are not known, we hypothesized that antimicrobial peptides could play a role in these barriers. We addressed this hypothesis by infecting mice with the neuropathogenic bacterium Neisseria meningitidis. Brains were analyzed for expression of the antimicrobial peptide CRAMP by immunohistochemistry in combination with confocal microscopy. After infection, we observed induction of CRAMP in endothelial cells of the BBB and in cells of the meninges. To explore the functional role of CRAMP in meningococcal disease, we infected mice deficient of the CRAMP gene. Even though CRAMP did not appear to protect the brain from invasion of meningococci, CRAMP knockout mice were more susceptible to meningococcal infection than wild-type mice and exhibited increased meningococcal growth in blood, liver, and spleen. Moreover, we could demonstrate that carbonate, a compound that accumulates in the circulation during metabolic acidosis, makes meningococci more susceptible to CRAMP.
Bacterial infection of the brain is a rare clinical condition, partly because of the protective properties of the blood-brain barrier (BBB). The BBB is mainly formed by the microvascular endothelium and exhibits intercellular tight junctions together with a low rate of pinocytosis, making it different from other endothelia in the body (31, 34). The BBB is essentially impermeable for large and hydrophilic molecules as well as for most bacteria and viruses. However, certain bacterial species have developed strategies to translocate across the BBB and enter the subarachnoid space, leading to meningitis. The meninges surrounding the subarachnoid space consist of the arachnoid and the pia mater, collectively named the “leptomeninges.” The innermost layer, pia mater, consists of a thin layer of epithelial-like cells (meningothelial cells) which cover the surface of the brain.
Neisseria meningitidis is a serious pathogen with the capacity to cross the BBB and colonizes the nasopharynx of 5 to 10% of the population. In susceptible individuals, the bacterium can cross the epithelial layer, reach the bloodstream, and cause massive bacteremia (33). The level of bacteremia has been suggested to be an important determinant for the ability of bacteria to cross the BBB, although the exact mechanism is not known (31). In the subarachnoid space, Neisseria meningitidis initiates a massive inflammatory response involving the release of cytokines and chemokines, ultimately leading to meningitis. It has been proposed that cells of the BBB and the meninges possess intrinsic bactericidal properties, but the nature of this defense system has not been determined.
Antimicrobial peptides are widespread in nature and constitute important effector molecules of the innate immune system (44). In mammals there are two main families of antimicrobial peptides, the defensins (17) and the cathelicidins (43). Alpha-defensins are expressed mainly in neutrophils and in Paneth cells of the small intestine, while β-defensins are found in various epithelia (17). The single human cathelicidin-derived peptide gene, named CAMP, is expressed by both circulating immune cells and epithelia. This gene encodes the precursor of the antimicrobial peptide LL-37 (designated hCAP18) from which the mature peptide is cleaved. A single cathelicidin gene is also found in mice and rats, and the active peptides are designated CRAMP (16) and rCRAMP (38), respectively.
In addition to their antimicrobial effects, both defensins and cathelicidins display a panel of other biological functions (17, 18). β-Defensins have the capacity to be chemotactic for T cells and dendritic cells (41) but can also induce maturation of dendritic cells (5). The human cathelicidin peptide LL-37 has been associated with a number of biological effects, such as immunomodulation (6), chemotaxis (11), wound healing (21), and angiogenesis (26).
The in vivo relevance of antimicrobial peptides is becoming firmly established, which is demonstrated by cathelicidin-related antimicrobial peptide knockout (CRAMP-KO) mice being more susceptible to skin infection with group A streptococcus (32). A further “proof of concept” is provided by a transgenic mouse model expressing the human α-defensin HD-5 in the small intestine. This “humanized” mouse was found to be resistant to Salmonella enterica serovar Typhimurium, which normally causes severe disease in mice (35). However, since several antimicrobial peptides are expressed simultaneously at sites of infection, compensatory functions can be expected, as exemplified by studies in β-defensin-1 KO mice (29, 30).
Apart from epithelial expression, antimicrobial peptides have also been detected in organs that are rarely exposed to bacteria, such as testis (2) and brain (20). Recently, we have performed a detailed study showing that rCRAMP is expressed in the central nervous system of the rat with a distinct regional distribution (4). In addition, β-defensins have previously been detected in the brain of mouse (24), rat (22), and cow (37). In humans, HBD-1 and HBD-2 have been shown to be expressed in astrocytes but not in neurons (20). The transcript of the human cathelicidin LL-37 has been detected in the brain with mRNA dot blot hybridization (3).
In the present study, we have investigated the expression of the antimicrobial peptide CRAMP in mice infected with Neisseria meningitidis. CRAMP was found to be induced in endothelial cells of the BBB and in cells of the leptomeninges after meningococcal infection. Moreover, results from these in vivo experiments in mice demonstrated that CRAMP inhibits bacterial growth in blood, liver, and spleen early in meningococcal sepsis. In vitro experiments could further demonstrate that carbonate, a compound that accumulates in the circulation during metabolic acidosis (associated with sepsis), makes meningococci more susceptible to CRAMP. Combined, our results suggest that CRAMP contributes to innate immunity in meningococcal disease.
MATERIALS AND METHODS
Synthetic peptides and antisera.
CRAMP, the extended-form CRAMP(1-39), and LL-37 were synthesized (Innovagen, Lund, Sweden) and used in the present study. The sequence of CRAMP is NH2-GLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPEQ-COOH, and for CRAMP(1-39) it is NH2-ISRLAGLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPEQ-COOH. The human homologue of CRAMP, LL-37, was used as a control peptide in the antibacterial assay (19). Antiserum against CRAMP was prepared according to previous guidelines (16), and the CRAMP(1-39) antiserum was produced by Innovagen. In control experiments, the antiserum against CRAMP was blocked with its corresponding antigen (underlined in the CRAMP sequence above) according to previous guidelines (13).
Bacterial strains and growth conditions.
Neisseria meningitidis serogroups A (Z6244), B (NMB), C (FAM20), and W-135 (JB515) were utilized in the antimicrobial assays, while all animal experiments were performed with serogroup C (FAM20) (14). The meningococci were grown on GCB agar containing Kellogg's supplement (25) at 37°C in 5% CO2 atmosphere and passaged every 18 to 20 h. When used in the experiments, the meningococci were resuspended in GC liquid (1.5% [wt/vol] proteose peptone no. 3 [Beckton Dickinson], 3 mM soluble starch [Sigma], 23 mM K2HPO4 [Merck], 7 mM KH2PO4 [Merck], 50 mM NaCl [Merck]). In some experiments, the bacteria were grown in GC liquid where the phosphate was replaced by carbonate (50 mM NaHCO3).
Mouse strains.
The mouse strains 129/SVJ (The Jackson Laboratory, Bar Harbor, Maine) and C57BL/6 as well as CRAMP-KO mice were bred in the animal facility at the Microbiology and Tumor Biology Center (MTC), Karolinska Institutet (Stockholm, Sweden). CRAMP-deficient mice were generated by targeted disruption of the gene encoding CRAMP (Cnlp) by homologous recombination. Embryonic stem cells of 129/SvJ mice were injected into C57Bl/6 blastocysts and transferred into C57Bl/6 foster mothers (32). Chimeric offspring were crossed with C57Bl/6 females. Afterwards, backcrosses into 129/SvJ were performed for seven generations. The contribution of the C57Bl/6 background was therefore minimized (less than 1%). Hence, 129/SvJ mice (CRAMP+/+) were used as controls in all experiments. All mice were 5 to 8 weeks old when challenged with bacteria. The experiments have been approved by the Stockholm North Ethical Committee on Animal Experiments (N230/03). In addition, all animal procedures were performed in accordance with institutional protocol guidelines at MTC.
Animal model.
N. meningitidis cells were suspended in GC liquid to 6 × 109 CFU per ml (initial experiment; n = 1). A dose of 100 μl bacterial suspension injected intraperitoneally (i.p.) causes meningococcal sepsis but not meningitis (defined as bacterial growth in cerebrospinal fluid [CSF]) in immunocompetent mice. However, to reduce the discomfort of the mice, a lower dose was chosen for all other experiments (1 × 109 to 2 × 109 bacteria per ml). Five- to 8-week-old mice were randomly redistributed in groups and injected i.p. with 100 μl of the bacterial suspension, equal to 1 × 108 to 2 × 108 or 6 × 108 CFU per mouse. The actual number of viable bacteria in the challenge dose was determined by viable count the following day. The clinical status of the mice was closely monitored according to a standardized protocol during the experiments (MTC protocol guidelines). Blood samples were obtained from the tail vein at 6 h, 24 h, and 48 h after bacterial challenge. The blood was diluted in phosphate-buffered saline (PBS), and serial dilutions were plated on GCB agar plates and incubated over night at 37°C in a 5% CO2 atmosphere. The number of CFU was counted, and the meningococcal identity of the bacteria was verified by gram staining, microscopy, and oxidase test.
Bacterial counts in liver and spleen.
Mice were euthanized by cervical dislocation. Liver and spleen were removed 6 h postchallenge and stored in PBS. Mean liver weights were 1.55 g (wild-type [wt] mouse; n = 6) and 1.61 g (KO mouse; n = 6), and mean spleen weights were 1.51 g (wt mouse; n = 6) and 1.50 g (KO mouse; n = 6). The tissues were homogenized in PBS, and serial dilutions of the homogenates were plated directly on GCB agar plates. The number of CFU was counted the following day.
Sampling of cerebrospinal fluid.
Mice were anesthetized by isoflurane before the skin of the neck was disinfected with ethanol. The cisterna magna was punctured as previously described (8). Approximately 1 to 2 μl of CSF was obtained from each mouse. The CSF was immediately suspended in 100 μl of GC liquid. Samples with signs of blood were discarded. The GC liquid mixtures were plated directly on GC plates, and the number of CFU was counted the following day. After the procedure the animals were euthanized by cervical dislocation.
Whole blood and serum bactericidal assay.
Whole blood (0.5 ml) was taken from the orbital vein of the mice using heparinized microcapillary tubes and placed on ice. Bacterial suspensions containing 102 CFU of N. meningitidis (35 μl) were mixed with blood (315 μl) to a final volume of 350 μl. Serum was prepared by storing whole blood (without heparin) at 4°C overnight. Approximately 100 to 150 μl of serum was isolated from a total volume of 0.5 ml blood. A bacterial suspension containing 102 CFU of N. meningitidis (35 μl) was mixed with the serum samples of 100 μl. The number of surviving colonies was counted after an overnight incubation.
Immunofluorescence.
Mice were anesthetized and transcardially perfused with 10% formalin in PBS at different time points after meningococcal challenge (3, 6, 12, 24, and 30 h). The brain and liver were removed, postfixed, rinsed, frozen, and processed as previously described (7). Briefly, the tissues were cut to 14-μm slices in a cryostat before being processed for the tyramide signal amplification (TSA) method (1). Tissue sections were incubated with a rabbit polyclonal CRAMP antiserum (1:1,000) (16) and incubated with a secondary anti-rabbit antibody conjugated with horseradish peroxidase (HRP) (1:200; DAKO) and processed with reagents from a commercial TSA kit (DuPont/NEN), including fluorescein isothiocyanate-conjugated tyramide. An indirect immunofluorescence technique (9) was employed to detect neutrophils, utilizing a rat monoclonal anti-neutrophil antibody (1:50; clone 7/4; Serotec). To detect endothelial cells, a rat anti-mouse CD31 antibody (1:100; PharMingen, San Diego, CA) was utilized. The secondary antibodies that visualized the presence of neutrophils and endothelial cells were Cy5-conjugated donkey anti-rat antibody (1:40; Jackson ImmunoResearch Europe, Cambridgeshire, United Kingdom) and RRX-conjugated donkey anti-rat antibody (1:80; Jackson ImmunoResearch Europe), respectively. The tissue sections were analyzed in a Nikon fluorescence microscope or in a Bio-Rad confocal microscope (7).
Peptide/protein extraction of spleen.
CRAMP-KO mice (n = 20) and C57BL/6 mice (n = 20) were euthanized by cervical dislocation. Spleens were removed and placed in ice-cold PBS. The tissue was homogenized in PBS, utilizing a 40-μm nylon cell strainer (BD Falcon). The homogenate was centrifuged, and the cells were counted in a Bürkner chamber. Approximately 5 × 107 cells from each mouse strain were degranulated with phorbol-ester (TPA) for 1 h at 37°C. After centrifugation, the supernatant was collected in a separate tube and trifluoroacetic acid (TFA) was added to a final concentration of 0.1%. The mixture was applied to OASIS cartridges (Waters, Milford, MA) in order to enrich for peptides and proteins. Before the material was applied, the cartridges were activated with acetonitrile and equilibrated in 0.1% TFA. Unbound material was washed away with 0.1% TFA, and proteins/peptides were eluted with 80% acetonitrile in 0.1% TFA. Finally, the eluate was frozen at −70°C, lyophilized, and redissolved in 0.1% TFA to a concentration of 10 μg/μl.
Dot blot analysis.
Sera from CRAMP-KO mice and 129/SVJ mice were prepared as described above for whole blood and serum bactericidal assay, and 20 μl was frozen, lyophilized, and redissolved in 10 μl 0.1% TFA. One microliter of the redissolved sera was applied to a Hybond Super C membrane (Amersham Pharmacia Biotech, Uppsala, Sweden). The membrane was blocked in 5% fat-free milk and then incubated with a rabbit polyclonal CRAMP(1-39) antibody (0.25 μg/ml) in PBS with 5% fat-free milk for 1 h. Incubation with the secondary antibody, a monkey horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (Amersham Pharmacia Biotech) diluted 1:1,000 in PBS with 5% fat-free milk, was performed for 1 h. Immunoreactivity was visualized using an ECL detection system (Amersham Pharmacia Biotech) according to the manufacturer's instructions, and the light was recorded on Hyperfilm ECL (Amersham Pharmacia Biotech). All incubations were performed at room temperature.
Western blot analysis.
Peptide/protein extracts and lyophilized serum samples were mixed with sample buffer (Invitrogen, Carlsbad, CA), followed by incubation at 70°C for 10 min. The samples were then loaded onto Nu-polyacrylamide gel electrophoresis 4-to-12% gradient gels (Invitrogen) and subjected to electrophoresis (200 V). After separation, peptides/proteins in the gel were transferred (30 V) to polyvinylidene difluoride membranes (Invitrogen). The membranes were then treated as described above for dot blot analysis, except that the primary antibody was incubated overnight at room temperature.
Antibacterial assay (microtiter plate).
The antibacterial activity of the synthetic peptides CRAMP, CRAMP(1-39), and LL-37 were analyzed against all N. meningitidis strains. Bacteria were grown on GCB agar plates and resuspended in GC liquid to 105 bacteria per ml. In some experiments, carbonate-based GC liquid was utilized (50 mM NaHCO3). Bacteria were incubated in the absence (controls) or presence of different amounts of peptide in a 96-well microtiter plate (90 μl bacterial suspension plus 10 μl of the peptide solution). Different concentrations of the peptides were achieved by twofold serial dilutions in 0.1% TFA. The final concentration of the peptides in each well ranged from 2 μM to 32 μM. The peptide/bacterial mixtures were incubated at 37°C with 5% CO2 for 1 h. The killing efficacies of CRAMP, CRAMP(1-39), and LL-37 were analyzed by plating serial dilutions of the incubation mixture in triplicate on GCB agar plates, and the number of CFU was enumerated the following day. The percentage of killed bacteria was expressed with the following formula: (CFU after peptide incubation)/(CFU after control incubation) × 100. The 50% effective concentration (EC50) was defined as the concentration of peptide that decreased CFU counts by 50% relative to the peptide-free control.
Peptide/protein extraction of whole brain.
Brains were removed from infected and noninfected wt mice (129/SVJ) and CRAMP-KO mice. The brains were immediately frozen on dry ice, kept at −70°C, and later ground into small pieces in liquid nitrogen using a mortar and pestle. The material was then homogenized in 60% acetonitrile containing 1% TFA using a Polytron PT 1200 CL homogenizer (Kinematica Inc., Cincinatti, OH) and shaken in the same solvent overnight at 4°C. Debris was removed by centrifugation and the supernatants were frozen, lyophilized, and dissolved in 0.1% TFA. The material was further treated as described above for peptide/protein extraction of spleen, including concentration on OASIS cartridges and subsequent lyophilization.
Inhibition zone assay and depletion of antibacterial activity in brain extract.
Thin agarose plates (1 mm) in standard Luria-Bertani (LB) medium containing 6 × 104 test microbes/ml were made, and 3-mm wells were punched in the plates. The test microbe used was Escherichia coli strain D21. From each sample, 3 μl was applied to the wells. After an overnight incubation at 30°C, the diameters of the inhibition zones were measured. Experiments were performed with Medium E, a salt medium worked out for E. coli (40).
Whole mouse brain extract (10 μg/μl) was mixed with an excess of anti-CRAMP serum (5 mg/ml) and incubated at room temperature for 4 h. In parallel, control experiments were performed where buffer replaced the antiserum. After the incubation period, the mixtures were analyzed in the inhibition zone assay against E. coli D21 in agarose plates supplemented with salt (medium E).
Statistical analysis.
The nonparametric Mann-Whitney U test (Microsoft Excel plugin XLStat) was used to analyze differences between groups for statistical significance. Two-tailed tests were used at the alpha-level 0.05. Data are presented as means ± standard deviations (SD).
RESULTS
Mouse CRAMP in the BBB and meninges after meningococcal infection.
The brain is rarely infected by pathogens, and the mechanism(s) behind this protection remains to be elucidated. Thus, we hypothesized that antimicrobial peptides could play a role in the innate defenses of the brain. Consequently, we investigated the expression of the antimicrobial peptide mouse CRAMP in the brain during meningococcal infection. Mice (C57BL/6) were infected i.p. with Neisseria meningitidis (6 × 108 CFU/mouse). Thirty hours postinfection, the mice were euthanized and brains were processed for immunohistochemistry. Strong immunoreactivity (IR) for CRAMP was detected in the blood vessel walls in the olfactory bulb (Fig. 1A) and in the cortex (Fig. 2D to L). In addition, CRAMP-IR was found in cells of the leptomeninges, which are associated with the surface of the brain (Fig. 1C). In the noninfected brain, no staining for CRAMP could be observed (Fig. 1B and D). The kinetics of CRAMP induction in blood vessel walls was investigated in a separate experiment using a slightly lower dose (1 × 108 to 2 × 108 CFU/mouse). CRAMP-IR was detected as early as 6 h postinfection (Fig. 1E and F); however, the staining was more prominent at 12 and 24 h postinfection (Fig. 1G and H). The strongest induction was observed after 30 h of infection and with the high dose of bacterial inoculum (6 × 108 CFU/mouse) (Fig. 1A). These data demonstrate that CRAMP expression in the blood vessel walls is both dose and time dependent. The specificity of the CRAMP-IR was confirmed by preincubation of the CRAMP antiserum with its antigen, which abolished all specific signals (data not shown), except for the staining in the border of some tissue sections, which was concluded as unspecific (Fig. 1E and H). Infected CRAMP-KO mice exhibited no CRAMP-IR in blood vessel walls (Fig. 1I).
FIG. 1.

Detection of CRAMP in cells of the BBB and meninges. Following i.p. injection with 6 × 108 CFU of N. meningitidis, C57BL/6 mice were perfused with fixative 30 h postinfection. Brain sections were stained for CRAMP as described in Materials and Methods. CRAMP-IR was found in blood vessel walls (arrowheads) of the olfactory bulb (A) and in cells (arrowheads) of the leptomeninges (C). No CRAMP staining was found in the corresponding regions of noninfected brains (B and D). The kinetics of the CRAMP induction was investigated at 3, 6, 12, and 24 h postinfection in C57BL/6 mice infected with 1 × 108 to 2 × 108 CFU (E to H). Upregulation of CRAMP in blood vessel walls was first observed at 6 h postinfection (F; arrowheads). Unspecific fluorescence was noted at the border of the tissue (E and H; arrows). Staining of brain sections of infected CRAMP-KO mice resulted in lack of specific staining (I; asterisk), while infected wt 129/SVJ control mice exhibited staining in the blood vessel walls (I; arrowheads) 24 h postinfection. In the liver, CRAMP was only detected in infiltrating immune cells 24 h postinfection (J). Scale bars: panels A to E, I, and J, 100 μm; panels F to H, 50 μm.
FIG. 2.

Detection of CRAMP in endothelial cells of the brain. C57BL/6 mice were infected i.p. with 6 × 108 CFU of N. meningitidis and perfused with fixative 30 h postinfection. Brain sections were stained for CRAMP as described in Materials and Methods. CRAMP-IR was detected in blood vessel walls of the olfactory bulb and was colocalized with CD31, a marker for endothelial cells (A to C). In cortex, CRAMP-IR was also detected in cells of the leptomeninges covering the surface of the brain (D and G; arrowheads). Panels G to I show higher magnification of the box in panel D. Neutrophil staining is shown in panel H (arrowhead), which exhibits CRAMP-IR (I; arrowhead). In the merged figures, meningothelial cells and the neutrophil exhibited complementary staining patterns for CRAMP (F and I). In addition, CRAMP-IR was detected in a large vessel wall in the cortex (J, K, and L). One neutrophil containing CRAMP is indicated in panels K and L (arrowheads). In the olfactory bulb, CRAMP-IR was found mainly in endothelial cells (M and P) but was also in neutrophils (P and Q). In the merged figure, neutrophils and CRAMP-IR endothelial cells are separated (O and R). Panels P to R show higher magnification of the box in panel M, and the arrowheads indicate neutrophils (Neutroph.). Scale bars: panels A to C, 20 μm; panels D to F and M to O, 100 μm; panels G to L, 50 μm; panels P to R, 30 μm.
CRAMP immunoreactivity in brain endothelial cells.
To confirm the identity of the CRAMP-positive cells in the vessel walls of the brain, double staining with an endothelial marker (CD31) was performed. CRAMP-IR was associated with endothelial cells, as depicted in Fig. 2A to C. In addition, cells of the leptomeninges could be identified as a cellular source of CRAMP in the infected brain (Fig. 2D to I). Since neutrophils are known to contain CRAMP, it was important to examine the relationship between neutrophils and the presence of CRAMP-IR in the infected brains. Indeed, the observed CRAMP staining in endothelial and meningothelial cells was clearly separated from neutrophils, as shown both for cortex (Fig. 2D to L) and olfactory bulb (Fig. 2M to R). However, we could confirm the presence of CRAMP-IR in neutrophils in the infected brains (Fig. 2D to L and M to R).
To examine whether expression of CRAMP after meningococcal infection is exclusively associated with the BBB endothelia, sections of the liver, kidney, and adrenal gland were stained with the CRAMP-specific antiserum. In the liver, CRAMP immunoreactivity was only detected in infiltrating immune cells after infection (Fig. 1J). Double staining, using a neutrophil specific marker, confirmed that the CRAMP-positive cells in the liver were neutrophils (data not shown). Also in kidney and adrenal gland, CRAMP staining could only be observed in invading immune cells after infection and not in endothelial cells (data not shown). This suggests that expression of CRAMP in endothelial cells in response to meningococcal infection is restricted to the brain.
The results from the meningococcal challenge experiments in C57BL/6 mice could also be reproduced in the mouse strain 129/SVJ (Fig. 1), demonstrating that CRAMP staining in brain endothelial cells and in the leptomeninges after meningococcal infection was not a strain-specific effect. Thus, CRAMP-KO mice and the corresponding wt 129/SVJ mice could be used to evaluate the biological significance of CRAMP in a meningococcal challenge model.
Meningococcal meningitis in the mouse.
Since CRAMP peptide immunostaining was found in the BBB and meninges in response to meningococcal challenge, we postulated that mice deficient in this antimicrobial peptide would be more susceptible to meningitis. CRAMP-KO and control mice (129/SVJ and C57BL/6) were infected i.p. with 1 × 108 CFU/mouse, and CSF was collected 24 h postinfection. Meningococci were found in CSF from both CRAMP-KO mice and wt 129/SVJ mice but not in CSF from wt C57BL/6 mice (Table 1). Thus, bacterial translocation across the BBB appeared to be independent of CRAMP but dependent on the mouse strain used.
TABLE 1.
Detection of meningococci in wt and CRAMP-KO mice
| Mouse strain | % of mice positive for bacteria in CSF (no. positive/total no.) | % of mice surviving (no. survived/total no.) |
|---|---|---|
| C57BL/6 | 0 (0/8) | 100 (8/8) |
| 129/SVJ | 45 (5/11) | 100 (28/28) |
| CRAMP-KO | 55 (5/9) | 88 (22/25) |
Meningococcal sepsis in the mouse.
When infecting CRAMP-KO mice and wt 129/SVJ mice with 108 N. meningitidis, 3/25 KO mice died, while all wt mice survived the challenge (Table 1). To determine the reason for the death of 12% of the CRAMP-KO mice during the experiments, the effect of meningococcal sepsis was investigated by counting bacteria in blood, liver, and spleen. Blood was collected at 6, 24, and 48 h postinfection, and liver and spleen were removed 6 h postinfection. After 6 h of infection, there was significantly more bacteria in the blood of CRAMP-KO mice (mean, 4.2 × 105 CFU/ml) compared to wt 129/SVJ mice (mean, 2.4 × 105 CFU/ml), as depicted in Fig. 3A (P < 0.001; Mann-Whitney U test). However, after 24 and 48 h of infection, the difference of bacterial counts in the blood between CRAMP-KO mice and wt 129/SVJ mice was not significant (data not shown). The higher bacterial numbers found in blood of CRAMP-KO mice compared to wt controls were also reflected in the liver 6 h postinfection (15.8 × 105 CFU/ml versus 4.66 × 105 CFU/ml; P < 0.05; Fig. 3B). In the spleen there was also a tendency of increased bacterial growth in CRAMP-KO mice, but the difference did not reach statistical significance (17.3 × 105 CFU/ml versus 10.66 × 105 CFU/ml; P > 0.05; Fig. 3B).
FIG. 3.

Bacterial counts in blood, liver, and spleen of CRAMP-KO mice and wt 129/SVJ mice 6 h postinfection. Mice were inoculated i.p. with 1 × 108 to 2 × 108 CFU of N. meningitidis. (A) Blood samples were taken from the tail vein, diluted, and spread on GCB agar plates. The number of CFU was counted the following day. The line indicates the mean of the values. Three separate experiments were carried out, using 28 129/SVJ mice and 25 CRAMP-KO mice. (B) Livers and spleens from six mice were removed 6 h postinfection. The tissues were homogenized, diluted, and spread on GCB agar plates. The number of CFU was counted after an overnight incubation. The data are presented as means ± SD.
Meningococcal growth in blood and serum ex vivo.
To further investigate the observed difference in vivo of meningococcal growth, the antibacterial properties of blood and serum from CRAMP-KO mice and 129/SVJ mice were analyzed in an ex vivo assay. Blood and serum were mixed with bacteria and incubated for 3 or 6 h, respectively. In line with the in vivo data, there was a significant increase in growth of N. meningitidis in blood and serum from CRAMP-KO mice compared with wt 129/SVJ mice (blood, P < 0.001; serum, P < 0.001; Fig. 4).
FIG. 4.

Growth of N. meningitidis in blood and serum. Blood and serum from CRAMP-KO and wt 129/SVJ mice were incubated with 102 CFU of N. meningitidis for 3 h and 6 h, respectively. The incubation mixtures were plated on GCB agar plates, and the number of colonies was counted the following day. Growth index was defined as a ratio between the bacterial counts after the incubation divided by the starting counts. The data are presented as means ± SD of a representative experiment done in triplicate.
Induction of CRAMP in serum after infection.
Serum was further analyzed for the presence of the CRAMP peptide at 6 and 24 h postinfection. A dot blot analysis revealed immunoreactivity for CRAMP at 6 h postinfection in wt 129/SVJ mice but not at 24 h postinfection (Fig. 5A). There was no immunoreactivity for CRAMP in noninfected 129/SVJ mice or in infected CRAMP-KO mice (Fig. 5A). Western blot analysis of the fractions with immunoreactivity showed that CRAMP was predominantly present as its 17-kDa cathelin proform in all serum samples analyzed (Fig. 5B).
FIG. 5.

Detection of CRAMP immunoreactivity in serum. (A) Serum (20 μl) was lyophilized and redissolved in 10 μl 0.1% TFA. One-microliter serum samples from each mouse were applied to a membrane for dot blot analysis using an antiserum against CRAMP(1-39). Lanes 1 to 6 represent six different mice from each group, as indicated to the right. As positive controls, dilution series of CRAMP and CRAMP(1-39) were used (twofold dilutions from 1.7 to 50 ng). (B) Western blot analysis of 2 μl of the immunoreactive serum samples from panel A (second top row), using anti-CRAMP(1-39), revealed that CRAMP mainly was present as its 17-kDa proform (lanes 1 to 6). Serum from a noninfected 129/SVJ mouse (lane 7) or an infected CRAMP-KO mouse (lane 8) exhibited no immunoreactivity for CRAMP. The additional bands of large sizes (lanes 1 to 6) possibly represent complexes of plasma proteins and CRAMP. As positive controls, 25 ng of CRAMP and CRAMP(1-39) were used. inf., infected.
Bactericidal activity of CRAMP against N. meningitidis.
A more detailed analysis of the bactericidal properties of cathelicidins against N. meningitidis was performed in vitro. The synthetic peptide CRAMP, the extended form CRAMP(1-39), and the human cathelicidin LL-37 were evaluated for their killing efficiency against different meningococcal serogroups (A, B, C, and W-135 strains). CRAMP was moderately active against N. meningitidis FAM20 (serogroup C), with 20% killing at the highest concentration of 32 μM (Fig. 6A). Notably, both CRAMP(1-39) and LL-37 exhibited potent bactericidal activity against meningococci, with EC50 values of less than 2 μM (Fig. 6A). This pattern was evident for all meningococcal serogroups investigated (data not shown). In addition, a more detailed in vitro study was performed using carbonate buffers. Carbonate was chosen because this compound accumulates in the circulation during metabolic acidosis, which occurs during septic conditions (10). Therefore, carbonate may be more physiologically relevant as a buffer in antimicrobial assays than the normally utilized phosphate buffers. Moreover, a recent study suggests that carbonate buffers increase the susceptibility of bacteria to antimicrobial peptides (12). Interestingly, our results demonstrate that if the bacteria were grown in carbonate-based GC broth, CRAMP exhibited potent antimicrobial effects with an EC50 of 2 μM (Fig. 6B).
FIG. 6.

Bactericidal activity of cathelicidins against meningococci. (A) The synthetic peptides CRAMP, CRAMP(1-39), and LL-37 were mixed with 105 CFU/ml of N. meningitidis strain FAM20 and incubated for 1 h. The incubation mixtures were plated on GCB agar plates, and the number of surviving colonies was enumerated the following day. CRAMP exhibited moderate bactericidal activity against meningococci, with 20% killing at the highest peptide concentration of 32 μM. CRAMP(1-39) and LL-37 were potent killers of this bacterium, with EC50s of <2 μM peptide. The data are presented as means ± SD of two independent experiments done in triplicate. (B) The effect of carbonate on the bactericidal effect of CRAMP against N. meningitidis strain FAM20. Bacteria were suspended to a concentration of 105 CFU/ml in either phosphate (square)- or carbonate (circle)-based GC liquid. Of this suspension, 180 μl was added to each well of a sterile 96-well polypropylene microtiter plate containing 20 μl of antimicrobial peptide solution at different concentrations. The different concentrations were achieved by twofold serial dilutions in 0.1% trifluoroacetic acid. The final concentration of peptide in each well ranged from 2 μM to 32 μM. The samples were incubated at 37°C, 5% CO2 for 3 h. The percentage of surviving bacteria was expressed as (CFU after peptide incubation)/(CFU after control incubation). The data are presented as means ± SD of two independent experiments done in triplicate.
Evidence for an extended form of CRAMP in vivo.
The finding that an N-terminal extension of CRAMP by five amino acid residues enhanced the bactericidal capacity prompted us to investigate if there exists an additional and extended form(s) of CRAMP in vivo. Western blot analysis of a peptide/protein extract made from C57BL/6 spleen revealed that the mature form of CRAMP is longer than the 34-residue peptide previously isolated from murine bone marrow extract (Fig. 7). In addition, mouse brain extract was analyzed for its antibacterial properties against E. coli. Notably, extract from infected mice (24 h postinoculation) exhibited significantly more antimicrobial activity than brain extract derived from wt noninfected mice (data not shown). Importantly, this activity could be reduced if the extract was coincubated with CRAMP-specific antiserum prior to the antibacterial assay (data not shown). These findings demonstrate that CRAMP is present in its active form in mouse brain.
FIG. 7.
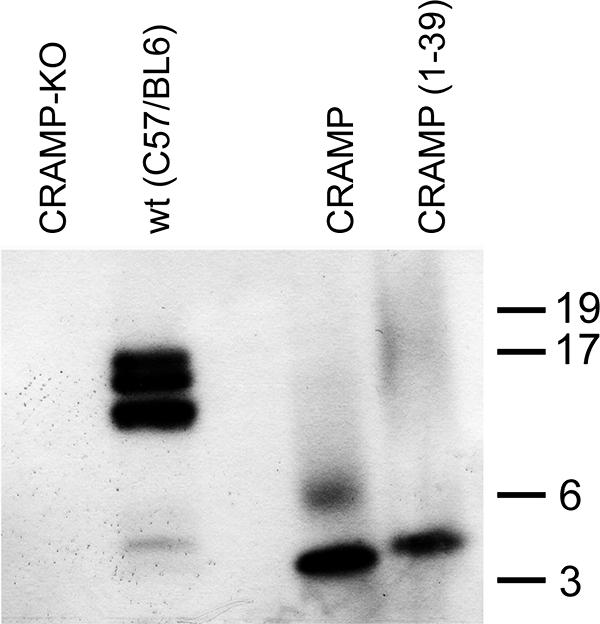
Detection of CRAMP in a spleen extract. Peptide/protein extracts (20 μg) of approximately 5 × 107 spleen cells from noninfected CRAMP-KO and C57BL/6 mice were analyzed by Western blot using CRAMP(1-39) antiserum. A faint band of around 5 kDa is detected in the second left lane, indicating that it may exist as an additional and extended form of CRAMP in vivo. No band was detected in the CRAMP-KO extract (left). As positive controls, 25 ng of CRAMP (molecular mass, 3.878 kDa) and CRAMP(1-39) (molecular mass, 4.419 kDa) were used.
DISCUSSION
In the present study, we demonstrated that the antimicrobial peptide CRAMP is induced in endothelial cells of the blood brain barrier and meninges after meningococcal infection. We also established a protective role for CRAMP against meningococcal sepsis, especially early in the infectious process. Finally, we proposed that carbonate, a compound that accumulates in the circulation during septic conditions, constitutes a novel innate immune factor by making meningococci more susceptible to CRAMP.
Our finding that CRAMP is present in endothelial cells of the BBB is consistent with the established concept that the BBB constitutes the first line of defense against potential invaders of the brain. Several studies have demonstrated that these cells synthesize and secrete an array of cytokines and chemokines in response to bacterial challenge (36, 39). Brain microvascular endothelial cells, an in vitro model of the BBB, have been shown to exhibit bactericidal properties which could be abrogated with a protein synthesis inhibitor (23). Thus, it was of interest to investigate the role of antimicrobial peptides in mice infected with Neisseria meningitidis, a potential neuroinvasive pathogen.
The finding that the BBB itself contains potent antimicrobial molecules may shed light on an old clinical observation that has not yet been resolved. The hallmark of meningitis is an exclusive involvement of the meninges, leaving the brain tissue intact. This indicates that the fight against the microbial invader takes place at the BBB interface and not within the brain tissue itself. Subsequently, the release of proinflammatory, and potentially toxic, substances within the brain parenchyma is prevented. Thus, our present finding that cells of the BBB contain an antimicrobial peptide may explain why bacterial brain infections strictly cause inflammation in the vessel walls and meninges. In contrast, viral brain infections may cause both meningitis and encephalitis, indicating a more complex mode of pathogenesis.
Interestingly, endothelial expression of CRAMP appears to be a brain-specific effect, since a number of peripheral tissues, such as liver, kidney, and adrenal gland, of infected mice did not stain for endothelial expression of CRAMP. This may reflect the specialized immunological niche of the brain, where inflammation must be strictly controlled. Since the cranium limits the brain volume, it is of vital importance to avoid excess swelling in the brain parenchyma, which could lead to detrimental effects, such as brain edema and rapid death of the host. It would be beneficial to the host to arm the perimeter of the brain (the BBB and meninges) with antibacterial peptides which efficiently could keep microbial invaders at a safe distance from vital brain constituents, such as neurons and their projections.
We also demonstrated that CRAMP is expressed in cells of the leptomeninges after meningococcal challenge. These epithelial-like cells have been shown to respond vigorously to meningococcal infection and to synthesize a number of cytokines, chemokines, and adhesion molecules (15). Secretion of the CRAMP peptide from meningothelial cells could have important functions in the subarachnoid space during infection.
Since neutrophils contain high concentrations of the CRAMP peptide, we wanted to exclude the possibility that CRAMP immunoreactivity in endothelial and meningeal cells was originating from circulating neutrophils. Antimicrobial peptides can be released from neutrophils in vivo, and uptake or deposition may occur into different cell types (28, 45). However, double-staining experiments using a neutrophil-specific antibody demonstrated that neutrophils and CRAMP-positive endothelial and meningeal cells do not colocalize (Fig. 2F, I, L, O, and R). If deposition on BBB endothelia was the result of release from distant neutrophils, other endothelia should also exhibit CRAMP immunoreactivity. However, this was not observed in endothelia of liver, kidney, or adrenal gland from infected mice. Therefore, our results indicate that the presence of CRAMP in endothelial and meningeal cells in the brain after meningococcal infection represent de novo synthesis of CRAMP by these cell types.
Knowing that CRAMP is present in the BBB and in cells of the leptomeninges, we wanted to test whether this peptide could protect the brain against meningococcal meningitis by utilizing mice deficient in the CRAMP gene (32). Consequently, CRAMP-KO mice were infected i.p. with N. meningitidis and CSF was collected 24 h postinfection. CSF of both the CRAMP-KO mice and the wt 129/SVJ mice contained bacteria, while in the CSF of C57BL/6 mice no bacteria were present (Table 1). This indicates that 129/SVJ mice are more susceptible to meningococcal meningitis than C57BL/6 mice. The fact that inbred mouse strains differ significantly in their susceptibility to bacterial infection has previously been recognized (27). However, the exact reason for the strain difference regarding meningococcal meningitis is not known, but it may involve an as yet unidentified antibacterial factor(s) or regulatory circuits in innate immunity. Thus, the results from the CSF experiments suggest that CRAMP is not the exclusive determinant for meningococcal translocation across the BBB. Nevertheless, since prominent localization of CRAMP can be observed in the BBB after infection, we propose that CRAMP has a significant, but as yet undefined, role in the innate immune system of the brain.
During the experiments, 12% of the CRAMP-KO mice died from meningococcal infection within the first 24 h of infection, while no wt 129/SVJ mice died (Table 1). To investigate the mechanism behind these deaths, a detailed analysis of bacterial counts in the blood, liver, and spleen of CRAMP-KO mice and wt 129/SVJ mice was performed. This analysis revealed an increased growth of bacteria in CRAMP-KO mice 6 h postinfection. At later time points, there was no significant difference in bacterial counts in the blood between the two mouse strains, suggesting an early, and nonredundant, role of CRAMP in meningococcal sepsis.
Increased growth of N. meningitidis in blood of CRAMP-KO mice in vivo was also observed ex vivo. Blood and serum from CRAMP-KO mice showed significantly decreased capacity to control the growth of N. meningitidis compared to blood and serum from wt mice.
In line with an early role for CRAMP in meningococcal infection, we observed large amounts of the proform in the circulation 6 h after infection. Notably, we did not detect the mature form, which is known to be antimicrobial. How can this observation explain our results from the in vivo experiments? One alternative is that the proform has intrinsic effects. It is known that the holoprotein of cathelicidins do not exhibit antibacterial effects per se, but immunomodulatory effects cannot be ruled out (42). Thus, it is possible that the large amount of CRAMP proform that we detected control meningococcal growth by other means than being directly antibacterial. Another explanation is that the proform serves as a reservoir for mature CRAMP, which may be released locally followed by rapid proteolytic degradation.
Considering that mature CRAMP may kill meningococci in vivo, it was important to further investigate and quantify this bactericidal capacity. In vitro experiments revealed that CRAMP in fact killed meningococci but at concentrations that were too high to be relevant in a physiological setting. In contrast, the extended-version CRAMP(1-39) and the human counterpart LL-37 exhibited potent bactericidal effects against meningococci. The finding that synthetic CRAMP peptide only exhibited moderate bactericidal activity against N. meningitidis while CRAMP(1-39) was significantly more potent in this respect prompted us to investigate the length of the mature peptide present in vivo. Interestingly, our current data indicate the presence of an additional form of mouse CRAMP in a spleen extract of mice. Notably, this unexpected finding is consistent with data from the rat where rCRAMP(1-43) recently was found to be more active against N. meningitidis than the shorter variant rCRAMP(1-34) (4).
To investigate the bactericidal properties of CRAMP against meningococci under more in vivo-like conditions, the in vitro assay was modified to involve different culture conditions. Interestingly, when the phosphate-based GC broth was replaced by a carbonate-based GC broth, the bactericidal capacity of the CRAMP peptide against meningococci was significantly improved. This finding may have physiological implications, since carbonate is formed in the brain and in the circulation during the development of metabolic acidosis associated with anoxic conditions, such as septic shock (10). In such situations of anoxia, carbon dioxide accumulates and reacts with water to form carbonic acid, which leads to the release of large amounts of carbonate ions and protons. Recently, carbonate has been demonstrated to make bacteria more susceptible to CRAMP (12). Hence, our current data lend support to the notion that carbonate may serve as a novel innate effector molecule during meningococcal sepsis.
In conclusion, we have for the first time demonstrated that an antimicrobial peptide is expressed in the blood brain barrier and in meningeal cells after meningococcal infection. This finding is consistent with the protective function of the BBB and the meninges and might explain previous observations of bactericidal properties of the BBB. However, our experiments indicate that CRAMP is not the exclusive determinant in the protection against meningococcal meningitis, i.e., translocation of N. meningitidis across the BBB. It is possible that CRAMP has other functions in the brain, such as being chemotactic or serving as a mediator of the inflammatory response associated with meningococcal meningitis. During meningococcal sepsis, however, we observe a massive release of CRAMP proform in serum 6 h after infection. This finding, together with the death of 12% of CRAMP-KO mice, suggests an early and nonredundant role for CRAMP in meningococcal sepsis. Finally, we propose that carbonate constitutes a possible innate immune mediator by making meningococci more susceptible to CRAMP.
Acknowledgments
Monica Lindh and the late Katarina Åman are gratefully acknowledged for their valuable technical assistance.
Financial support was received from the Swedish Research Council (O4X-2887, O6X-11217, O6X-10846, and O6B1-15302), Marianne and Marcus Wallenberg's Foundation, Knut and Alice Wallenberg's foundation, Swedish Cancer Society, Swedish Foundation for International Cooperation in Research and Higher Education (STINT), The Swedish Foundation for Strategic Research, and Karolinska Institutet.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 9 October 2006.
REFERENCES
- 1.Adams, J. C. 1992. Biotin amplification of biotin and horseradish peroxidase signals in histochemical stains. J. Histochem. Cytochem. 40:1457-1463. [DOI] [PubMed] [Google Scholar]
- 2.Agerberth, B., H. Gunne, J. Odeberg, P. Kogner, H. G. Boman, and G. H. Gudmundsson. 1995. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc. Natl. Acad. Sci. USA 92:195-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bals, R., X. Wang, M. Zasloff, and J. M. Wilson. 1998. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. USA 95:9541-9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergman, P., S. Termen, L. Johansson, L. Nystrom, E. Arenas, A. B. Jonsson, T. Hokfelt, G. H. Gudmundsson, and B. Agerberth. 2005. The antimicrobial peptide rCRAMP is present in the central nervous system of the rat. J. Neurochem. 93:1132-1140. [DOI] [PubMed] [Google Scholar]
- 5.Biragyn, A., P. A. Ruffini, C. A. Leifer, E. Klyushnenkova, A. Shakhov, O. Chertov, A. K. Shirakawa, J. M. Farber, D. M. Segal, J. J. Oppenheim, and L. W. Kwak. 2002. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298:1025-1029. [DOI] [PubMed] [Google Scholar]
- 6.Bowdish, D. M., D. J. Davidson, and R. E. Hancock. 2005. A re-evaluation of the role of host defence peptides in mammalian immunity. Curr. Protein Pept. Sci. 6:35-51. [DOI] [PubMed] [Google Scholar]
- 7.Brumovsky, P. R., T. J. Shi, H. Matsuda, J. Kopp, M. J. Villar, and T. Hokfelt. 2002. NPY Y1 receptors are present in axonal processes of DRG neurons. Exp. Neurol. 174:1-10. [DOI] [PubMed] [Google Scholar]
- 8.Carp, R. I., A. L. Davidson, and P. A. Merz. 1971. A method for obtaining cerebrospinal fluid from mice. Res. Vet. Sci. 12:499. [PubMed] [Google Scholar]
- 9.Coons, A. H. 1958. Fluorescent antibody methods, p. 399-422. In J. F. Danielli (ed.), General cytochemical methods. Academic Press, New York, N.Y. [PubMed]
- 10.Desai, V. S., M. H. Weil, W. Tang, R. Gazmuri, and J. Bisera. 1995. Hepatic, renal, and cerebral tissue hypercarbia during sepsis and shock in rats. J. Lab. Clin. Med. 125:456-461. [PubMed] [Google Scholar]
- 11.De Yang, Q. Chen, A. P. Schmidt, G. M. Anderson, J. M. Wang, J. Wooters, J. J. Oppenheim, and O. Chertov. 2000. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 192:1069-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dorschner, R. A., B. Lopez-Garcia, A. Peschel, D. Kraus, K. Morikawa, V. Nizet, and R. L. Gallo. 2006. The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J. 20:35-42. [DOI] [PubMed] [Google Scholar]
- 13.Dorschner, R. A., V. K. Pestonjamasp, S. Tamakuwala, T. Ohtake, J. Rudisill, V. Nizet, B. Agerberth, G. H. Gudmundsson, and R. L. Gallo. 2001. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A streptococcus. J. Investig. Dermatol. 117:91-97. [DOI] [PubMed] [Google Scholar]
- 14.Dyer, D. W., W. McKenna, J. P. Woods, and P. F. Sparling. 1987. Isolation by streptonigrin enrichment and characterization of a transferrin-specific iron uptake mutant of Neisseria meningitidis. Microb. Pathog. 3:351-363. [DOI] [PubMed] [Google Scholar]
- 15.Fowler, M. I., R. O. Weller, J. E. Heckels, and M. Christodoulides. 2004. Different meningitis-causing bacteria induce distinct inflammatory responses on interaction with cells of the human meninges. Cell Microbiol. 6:555-567. [DOI] [PubMed] [Google Scholar]
- 16.Gallo, R. L., K. J. Kim, M. Bernfield, C. A. Kozak, M. Zanetti, L. Merluzzi, and R. Gennaro. 1997. Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J. Biol. Chem. 272:13088-13093. [DOI] [PubMed] [Google Scholar]
- 17.Ganz, T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3:710-720. [DOI] [PubMed] [Google Scholar]
- 18.Gudmundsson, G. H., and B. Agerberth. 2004. Biology and expression of the human cathelicidin LL-37, p. 139-160. In D. A. Devine (ed.), Mammalian host defense peptides. Cambridge University Press, Cambridge, United Kingdom.
- 19.Gudmundsson, G. H., B. Agerberth, J. Odeberg, T. Bergman, B. Olsson, and R. Salcedo. 1996. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur. J. Biochem. 238:325-332. [DOI] [PubMed] [Google Scholar]
- 20.Hao, H. N., J. Zhao, G. Lotoczky, W. E. Grever, and W. D. Lyman. 2001. Induction of human beta-defensin-2 expression in human astrocytes by lipopolysaccharide and cytokines. J. Neurochem. 77:1027-1035. [DOI] [PubMed] [Google Scholar]
- 21.Heilborn, J. D., M. F. Nilsson, G. Kratz, G. Weber, O. Sorensen, N. Borregaard, and M. Stahle-Backdahl. 2003. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J. Investig. Dermatol. 120:379-389. [DOI] [PubMed] [Google Scholar]
- 22.Hiratsuka, T., M. Nakazato, Y. Date, H. Mukae, and S. Matsukura. 2001. Nucleotide sequence and expression of rat beta-defensin-1: its significance in diabetic rodent models. Nephron 88:65-70. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman, J. A., C. Wass, M. F. Stins, and K. S. Kim. 1999. The capsule supports survival but not traversal of Escherichia coli K1 across the blood-brain barrier. Infect. Immun. 67:3566-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huttner, K. M., C. A. Kozak, and C. L. Bevins. 1997. The mouse genome encodes a single homolog of the antimicrobial peptide human beta-defensin 1. FEBS Lett. 413:45-49. [DOI] [PubMed] [Google Scholar]
- 25.Kellogg, D. S., Jr., I. R. Cohen, L. C. Norins, A. L. Schroeter, and G. Reising. 1968. Neisseria gonorrhoeae. II. Colonial variation and pathogenicity during 35 months in vitro. J. Bacteriol. 96:596-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koczulla, R., G. von Degenfeld, C. Kupatt, F. Krotz, S. Zahler, T. Gloe, K. Issbrucker, P. Unterberger, M. Zaiou, C. Lebherz, A. Karl, P. Raake, A. Pfosser, P. Boekstegers, U. Welsch, P. S. Hiemstra, C. Vogelmeier, R. L. Gallo, M. Clauss, and R. Bals. 2003. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J. Clin. Investig. 111:1665-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam-Yuk-Tseung, S., and P. Gros. 2003. Genetic control of susceptibility to bacterial infections in mouse models. Cell Microbiol. 5:299-313. [DOI] [PubMed] [Google Scholar]
- 28.Lau, Y. E., A. Rozek, M. G. Scott, D. L. Goosney, D. J. Davidson, and R. E. Hancock. 2005. Interaction and cellular localization of the human host defense peptide LL-37 with lung epithelial cells. Infect. Immun. 73:583-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison, G., F. Kilanowski, D. Davidson, and J. Dorin. 2002. Characterization of the mouse beta defensin 1, Defb1, mutant mouse model. Infect. Immun. 70:3053-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moser, C., D. J. Weiner, E. Lysenko, R. Bals, J. N. Weiser, and J. M. Wilson. 2002. β-Defensin 1 contributes to pulmonary innate immunity in mice. Infect. Immun. 70:3068-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nassif, X., S. Bourdoulous, E. Eugene, and P. O. Couraud. 2002. How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol. 10:227-232. [DOI] [PubMed] [Google Scholar]
- 32.Nizet, V., T. Ohtake, X. Lauth, J. Trowbridge, J. Rudisill, R. A. Dorschner, V. Pestonjamasp, J. Piraino, K. Huttner, and R. L. Gallo. 2001. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414:454-457. [DOI] [PubMed] [Google Scholar]
- 33.Rosenstein, N. E., B. A. Perkins, D. S. Stephens, T. Popovic, and J. M. Hughes. 2001. Meningococcal disease. N. Engl. J. Med. 344:1378-1388. [DOI] [PubMed] [Google Scholar]
- 34.Rubin, L. L., and J. M. Staddon. 1999. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 22:11-28. [DOI] [PubMed] [Google Scholar]
- 35.Salzman, N. H., D. Ghosh, K. M. Huttner, Y. Paterson, and C. L. Bevins. 2003. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422:522-526. [DOI] [PubMed] [Google Scholar]
- 36.Sokolova, O., N. Heppel, R. Jagerhuber, K. S. Kim, M. Frosch, M. Eigenthaler, and A. Schubert-Unkmeir. 2004. Interaction of Neisseria meningitidis with human brain microvascular endothelial cells: role of MAP- and tyrosine kinases in invasion and inflammatory cytokine release. Cell Microbiol. 6:1153-1166. [DOI] [PubMed] [Google Scholar]
- 37.Stolzenberg, E. D., G. M. Anderson, M. R. Ackermann, R. H. Whitlock, and M. Zasloff. 1997. Epithelial antibiotic induced in states of disease. Proc. Natl. Acad. Sci. USA 94:8686-8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Termen, S., M. Tollin, B. Olsson, T. Svenberg, B. Agerberth, and G. H. Gudmundsson. 2003. Phylogeny, processing and expression of the rat cathelicidin rCRAMP: a model for innate antimicrobial peptides. Cell Mol. Life Sci. 60:536-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wells, D. B., P. J. Tighe, K. G. Wooldridge, K. Robinson, and D. A. Ala' Aldeen. 2001. Differential gene expression during meningeal-meningococcal interaction: evidence for self-defense and early release of cytokines and chemokines. Infect. Immun. 69:2718-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel, H. J., and D. M. Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97-106. [PubMed] [Google Scholar]
- 41.Yang, D., O. Chertov, S. N. Bykovskaia, Q. Chen, M. J. Buffo, J. Shogan, M. Anderson, J. M. Schroder, J. M. Wang, O. M. Howard, and J. J. Oppenheim. 1999. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286:525-528. [DOI] [PubMed] [Google Scholar]
- 42.Zaiou, M., V. Nizet, and R. L. Gallo. 2003. Antimicrobial and protease inhibitory functions of the human cathelicidin (hCAP18/LL-37) prosequence. J. Investig. Dermatol. 120:810-816. [DOI] [PubMed] [Google Scholar]
- 43.Zanetti, M. 2004. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 75:39-48. [DOI] [PubMed] [Google Scholar]
- 44.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389-395. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, L., W. Yu, T. He, J. Yu, R. E. Caffrey, E. A. Dalmasso, S. Fu, T. Pham, J. Mei, J. J. Ho, W. Zhang, P. Lopez, and D. D. Ho. 2002. Contribution of human alpha-defensin 1, 2, and 3 to the anti-HIV-1 activity of CD8 antiviral factor. Science 298:995-1000. [DOI] [PubMed] [Google Scholar]