

Abstract
Bacterial growth with short-chain aliphatic alkenes requires coenzyme M (CoM) (2-mercaptoethanesulfonic acid), which serves as the nucleophile for activation and conversion of epoxide products formed from alkene oxidation to central metabolites. In the present work the CoM analog 2-bromoethanesulfonate (BES) was shown to be a specific inhibitor of propylene-dependent growth of and epoxypropane metabolism by Xanthobacter autotrophicus strain Py2. BES (at low [millimolar] concentrations) completely prevented growth with propylene but had no effect on growth with acetone or n-propanol. Propylene consumption by cells was largely unaffected by the presence of BES, but epoxypropane accumulated in the medium in a time-dependent fashion with BES present. The addition of BES to cells resulted in time-dependent loss of epoxypropane degradation activity that was restored upon removal of BES and addition of CoM. Exposure of cells to BES resulted in a loss of epoxypropane-dependent CO2 fixation activity that was restored only upon synthesis of new protein. Addition of BES to cell extracts resulted in an irreversible loss of epoxide carboxylase activity that was restored by addition of purified 2-ketopropyl-CoM carboxylase/oxidoreductase (2-KPCC), the terminal enzyme of epoxide carboxylation, but not by addition of epoxyalkane:CoM transferase or 2-hydroxypropyl-CoM dehydrogenase, the enzymes which catalyze the first two reactions of epoxide carboxylation. Comparative studies of the propylene-oxidizing actinomycete Rhodococcus rhodochrous strain B276 showed that BES is an inhibitor of propylene-dependent growth in this organism as well but is not an inhibitor of CoM-independent growth with propane. These results suggest that BES inhibits propylene-dependent growth and epoxide metabolism via irreversible inactivation of the key CO2-fixing enzyme 2-KPCC.
Large quantities of gaseous short-chain alkenes, such as ethylene, propylene, and butylenes, are produced by both biogenic and anthropogenic processes (32-34). A variety of microorganisms are capable of growth on short-chain alkenes and are thought to play an important role in the remineralization of this carbon source (19, 25, 34).
The gram-negative proteobacterium Xanthobacter autotrophicus strain Py2 and the gram-positive actinomycete Rhodococcus rhodochrous strain B276 are two nutritionally versatile bacteria that have been characterized extensively with regard to their abilities to grow using propylene as a carbon source (19). The first step in propylene metabolism in these organisms is insertion of a single oxygen atom into the olefin bond of propylene, forming (R)- and (S)-epoxypropane enantiomers (22, 30, 36, 46). The epoxypropane enantiomers are subsequently metabolized by a three-step linear pathway that uses four enzymes and the atypical cofactor coenzyme M (CoM) (2-mercaptoethanesulfonic acid) to catalyze the net carboxylation of epoxypropane to form the central metabolite acetoacetate, as shown in Fig. 1 (1, 2, 4, 5, 20, 27). Recent studies of ethylene- and vinyl chloride-utilizing bacteria have shown that CoM serves as the cofactor for the utilization of these short-chain alkenes as well (14-16, 29). For these strains, epoxyalkane:CoM transferase (EaCoMT) forms hydroxyethylthioether conjugates which are believed to undergo subsequent dehydrogenation and conversion to acetyl-CoA rather than carboxylation (15). The recently discovered roles for CoM in propylene, ethylene, and vinyl chloride metabolism are the only known roles for this cofactor outside the methanogenic archaea, where CoM serves as the methyl group carrier in the terminal reactions of methanogenesis (41, 42, 44).
FIG. 1.

Pathway of propylene metabolism in X. autotrophicus strain Py2 and R. rhodochrous strain B276.
Traditional molecular biology probes of community structure allow researchers to discover which organisms are present, but they do not allow determination of how the removal of a specific organism affects the community. 2-Bromoethanesulfonate (BES), a structural analogue of CoM in which the thiol group of CoM is replaced by a bromine atom, is a potent inhibitor of methane production by methanogens and has been used extensively as a probe for environmental sampling and investigating community structure (7, 8, 24, 39, 47). Investigations of the inhibition of methanogenesis have focused on methyl coenzyme M reductase, the terminal enzyme involved in methane production (24). Because CoM utilization was previously attributed only to methanogens, BES was thought to be a methanogen-specific inhibitor. Recently, however, BES has been shown to affect the dechlorination of polychlorinated biphenyls and chlorinated ethylenes by nonmethanogens in mixed-culture bioreactors (9, 28, 45). These studies reinforced the importance of determining the specificity of bacterial BES inhibition, as well as the scope of CoM utilization, for processes other than methanogenesis. Thus, it is possible that BES may be used to probe environmental communities to determine how inhibition of bacterial CoM utilization pathways affects community structure and carbon flow.
In this paper, we present evidence that BES is capable of inhibiting bacterial growth on, and metabolism of, propylene. Moreover, the effects of BES on propylene and epoxypropane degradation by whole-cell suspensions and clarified cellular extracts were examined in this study. Our data provide evidence that inhibition of propylene metabolism is largely the result of inactivation of the terminal epoxide carboxylation enzyme 2-ketopropyl-CoM carboxylase/oxidoreductase (2-KPCC).
MATERIALS AND METHODS
Materials.
2-Bromoethanesulfonate (sodium salt, 98%) and 2-mercaptoethanesulfonic acid were purchased from Sigma-Aldrich. Racemic epoxypropane (a mixture of R and S enantiomers) and (R)-epoxypropane were also obtained from Sigma-Aldrich. Ascarite II was purchased from Thomas Scientific, Swedesboro, NJ. All other chemicals used were the highest purity available. Unless otherwise noted, all organic substrates used for assays were prepared as 500 mM stock solutions in H2O.
Small-scale growth of X. autotrophicus and R. rhodochrous and measurement of cell growth.
X. autotrophicus strain Py2 and R. rhodochrous strain B276 were cultured at 30°C using a mineral salts medium (13) and growth conditions described previously (13, 35, 37). Cultures were grown in 25 ml of medium in sealed 250-ml shake flasks that had been modified by addition of a Klett-compatible side arm for measuring optical densities and by replacement of the flask openings with crimp-sealable (20-mm) tops and side arms for introduction of volatile growth substrates and oxygen and for gas sampling. Cultures were inoculated using 0.5 to 3 ml of cells grown to the mid-logarithmic phase on the carbon source of interest. The following carbon sources were used for growth: 15% (vol/vol) propylene (gas phase), 25% (vol/vol) propane (gas phase), 40 mM acetone, and 32 mM n-propanol. For addition of BES to cultures, a 500 mM stock solution was prepared and filter sterilized using a 0.2-μm acrodisc filter. BES was then added from the stock solution so that the final concentration in the medium was between 1 and 5 mM. The levels of volatile organic compounds and O2 were monitored by flame ionization detector (FID) and thermal conductivity detector (TCD) gas chromatography as described below. The O2 concentration in the gas phase of cultures was maintained between 10 and 20% (vol/vol) by addition of sterile O2 through the side arms of culture flasks. The optical densities of the cultures were determined at desired times by placing the side arms of the culture flasks in a Klett-Summerson photoelectric colorimeter with a no. 66 filter. The Klett-Summerson colorimeter was standardized using a Shimadzu model UV-2101 spectrophotometer for conversion of Klett readings to absorbance values (optical density at 600 nm).
Large-scale growth of X. autotrophicus.
X. autotrophicus cells to be used for whole-cell propylene and epoxypropane degradation assays, for preparation of cell extracts, and for enzyme purification were grown with propylene (10%, vol/vol) as the carbon source in a 15-liter Microferm fermentor (New Brunswick Scientific) as described previously (2). Cells were harvested when the optical density was between 2.5 and 4 using an A/G Technologies polysulfone membrane cartridge filtration system. Concentrated cell suspensions were pelleted by centrifugation, washed once with 50 mM potassium phosphate buffer (pH 7.2), and either stored on ice for use within 24 h or flash frozen in liquid nitrogen and stored at −80°C.
Growth of Escherichia coli expressing recombinant proteins.
E. coli strains were grown at 37°C on Luria-Bertani (LB) medium unless otherwise noted. All cells used for enzyme expression were grown in 12 liters of medium in a 15-liter Microferm fermentor (New Brunswick Scientific). Bacteria containing expressed recombinant proteins were concentrated using a tangential flow filtration system (Millipore Corp.), washed once with 50 mM potassium phosphate buffer (pH 7.2), flash frozen in liquid nitrogen, and stored at −80°C.
For expression of EaCoMT, cells were transformed with pJK1, the expression vector for EaCoMT, as described previously (27). Freshly transformed E. coli BL21(DE3) was grown at 37°C in LB broth until the optical density (A600) was 0.8 to 1.0. Then 1 mM ZnCl2 was added to the medium, the fermentor was cooled to 30°C, and isopropyl-β-d-thiogalactopyranoside (IPTG) was added for expression of the recombinant enzyme as described previously (26).
For expression of 2-(R)-hydroxypropyl CoM dehydrogenase (R-HPCDH), E. coli BL21(DE3) CodonPlus (Stratagene) was transformed with plasmid pXD28 (12). Frozen cell stocks of transformed E. coli were resuspended in 25 ml of LB broth and grown at 37°C to an optical density (A600) of 0.6. The sample was transferred to a fermentor containing LB broth and grown at 30°C until the optical density (A600) was 1.0. Then 2-mercaptoethanol was added to a concentration of 1 mM, and the fermentor was rapidly cooled to 15°C. The cells were induced by addition of 0.25 mM IPTG and harvested 4 to 5 h after induction of protein synthesis.
Purification of proteins.
Recombinant EaCoMT and R-HPCDH and native 2-KPCC were purified to homogeneity (>95% pure, as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis) as described previously (3, 12, 26).
Gas chromatography.
Propylene, epoxypropane, acetone, and n-propanol were quantified by FID gas chromatography as described previously (2, 35, 37). Oxygen was quantified using a Shimadzu GC-8A gas chromatograph equipped with a molecular sieve 5A column (0.3 by 100 cm) equipped with a TCD. The injector, column, and detector temperatures were 100, 60, and 60°C, respectively. For TCD gas chromatography, 0.1-ml samples from the headspaces of culture vessels were analyzed and compared to standards for quantification of oxygen.
Whole-cell propylene and epoxypropane degradation assays.
All assays were conducted at 30°C in a shaking water bath (200 cycles min−1) using 9-ml sealed serum vials with a total liquid volume of 1 ml. The assay buffer was 50 mM potassium phosphate (pH 7.0). The assay mixtures contained whole cells (0.12 to 0.15 mg total cellular protein), 40 mM KHCO3, 10 mM CO2 gas (50 mM total HCO3− plus CO2), and 0 to 10 mM BES. The assays were initiated by addition of 2 to 4 μmol of propylene gas or epoxypropane. Unless otherwise noted, the source of epoxypropane for assays was the commercially available racemic mixture of R and S enantiomers. At the desired times, samples (50 μl) were removed from the headspace, and the amounts of propylene and epoxypropane were determined by FID gas chromatography.
To remove BES and residual epoxypropane from cell suspensions at the conclusion of epoxypropane degradation assays, the assay contents were transferred to 1.5-ml microcentrifuge tubes and pelleted by microcentrifugation. The cells were washed once with 1 ml of potassium phosphate buffer, resuspended to obtain a volume of 1 ml, and transferred to a fresh 9-ml serum vial for subsequent assays. The vials were sealed, and this was followed by addition of CO2 and KHCO3 as described above. Assays were reinitiated by addition of epoxypropane as described above.
Incorporation of 14C from 14CO2 into BES-treated cell suspensions.
X. autotrophicus cells (optical density, 0.9; 0.15 mg total cellular protein in 1 ml) were incubated for 260 min using the assay conditions described above in the presence of 4 μmol epoxypropane, in the presence of 50 mM CO2 plus KHCO3, and in the presence or absence of 5 mM BES. Epoxypropane degradation was monitored during this 260-min incubation. At the conclusion of incubation, the cells were pelleted by microcentrifugation, washed once with phosphate buffer, resuspended to obtain a volume of 1 ml using mineral salts medium containing 10 mM succinate (included in the assay vials as a carbon and energy source to promote new protein synthesis) and 5 mM CoM, and transferred to a new 9-ml assay vial. Chloramphenicol (0.4 mg) was added to some assay vials to prevent new protein synthesis. CO2 gas and NaH14CO3 from a 500 mM stock solution were transferred to the sealed serum vials at the ratio described above to obtain a final concentration of CO2 plus NaHCO3 of 50 mM. The specific activity of 14C in the assay mixtures was 55 μCi/(mmol CO2 plus NaHCO3). After 1 min of preincubation, assays were initiated by addition of 3 μmol of racemic epoxypropane. At the desired times, 25 μl of the assay mixture was removed and added to 200 μl of an ethanol-acetic acid mixture (95:5, vol/vol) in 1.5-ml microcentrifuge tubes. The quenched reaction mixtures were vortexed, dried by overnight incubation (≥8 h) at 50°C using a heating block, and placed in scintillation vials containing 10 ml of liquid scintillation fluid (ScintiSafe Econo 1; Fisher Scientific). The radioactivity of the samples was measured using a Beckman LS 600 scintillation counter. Portions of the radiolabeled bicarbonate stock solution were diluted, added to 10 ml of liquid scintillation fluid, and used to construct standard curves for converting disintegrations per minute to nanomoles of acid-stable carbon fixed in the samples.
BES treatment of, and epoxypropane degradation by, clarified cell extracts.
Cell extracts of propylene-grown X. autotrophicus were prepared as described previously (2). All treatments and assays involving cell extracts were performed under anaerobic and CO2-free conditions using 50 mM Tris buffer (pH 7.4). Solutions and vials were made anoxic and CO2 was depleted by sparging the solutions and vials with nitrogen gas or by repeated evacuation and flushing using a manifold. The nitrogen gas was passed through a heated copper-based catalyst (for removal of residual O2) and through a column of ascarite II (for removal of residual CO2). Portions (5 ml) of cell extract (16.2 mg/ml protein) were incubated at room temperature for 4 h in Tris buffer containing 10 mM dithiothreitol (DTT) in the absence or presence of 5 mM BES. The cell extracts were then dialyzed for 16 h in individual dialysis bags (SpectraPor; molecular weight cutoff, 6,000 to 8,000) in 5 liters of buffer at 4°C. The cell extracts treated in this way were then assayed for epoxide isomerase activity (conversion of epoxypropane to acetone, which occurs in the absence of CO2) as described below. The assays were conducted in 9-ml serum vials with 1-ml (total volume) mixtures containing cell extract (2 mg protein), 5 mM CoM, 10 mM DTT, 4 mM pyruvate, 5 U (1 U = 1 μmol/min) of lactate dehydrogenase, 2 mM NAD+, and 3 μmol (R)-epoxypropane. As indicated below, EaCoMT (0.5 mg of purified enzyme, 6 U of activity), R-HPCDH (0.5 mg of purified enzyme, 25 U of activity), or 2-KPCC (0.5 mg of purified protein, 0.2 U of activity), purified as described above, was included in some assays. Epoxide isomerase activity was monitored by determining the disappearance of epoxypropane and the formation of acetone using gas chromatography as described above.
Protein determination.
Protein concentrations of cell extracts and whole-cell suspensions were determined by a modified biuret assay (10) using bovine serum albumin as the standard. For whole-cell suspensions, the cells were solubilized in 3 M NaOH for 30 min at 65°C prior to protein determination.
RESULTS AND DISCUSSION
Effect of BES on growth of X. autotrophicus.
BES is a structural analog of CoM and a potent inhibitor of methanogenic growth and methanogenesis (7, 8, 24, 39, 47). Since aerobic alkene-oxidizing bacteria are the only organisms other than methanogens known to use CoM as a cofactor, it was of interest to determine whether BES is a selective inhibitor of bacterial alkene metabolism. To investigate this, the growth of X. autotrophicus on propylene was examined in the absence and presence of several concentrations of BES. As shown in Fig. 2A, the presence of 3 or 5 mM CoM completely prevented growth of the bacteria with propylene as the carbon source. When the concentration of BES was 1 mM, little or no effect on the growth rate was observed (Fig. 2A). Thus, a threshold concentration between 1 and 3 mM completely inhibited growth. The concentration of BES required for inhibition of growth was significantly higher than the concentration reported for methanogens, where inhibition was observed at concentrations of 0.44 to 10 μM (24).
FIG. 2.

Effects of BES on growth of X. autotrophicus with propylene, acetone, and n-propanol as carbon sources. (A) Growth with propylene. Symbols: ▴, no BES; ○, 1 mM BES; □, 3 mM BES; •, 5 mM BES. (B) Growth with n-propanol (circles) and acetone (triangles). Solid symbols, no BES present; open symbols, 5 mM BES present. The data are the averages of measurements for two cultures.
In order to investigate the specificity of BES inhibition, X. autotrophicus was grown in the absence or presence of BES with acetone and n-propanol, two C3 compounds that do not require CoM for metabolism, growth conditions under which detectable levels of CoM are not present (27). As shown in Fig. 2B, the presence of 5 mM BES had no effect on growth with acetone or n-propanol as a carbon source. Thus, inhibition of X. autotrophicus growth by BES correlates directly with growth conditions requiring CoM, suggesting that BES inhibits growth by acting as a CoM analog. These results agree with the results of the studies of Sparling and Daniels, conducted prior to the discovery of CoM in alkene metabolism, in which the growth of various eubacteria and nonmethanogenic archaea, as well as the yeast Saccharomyces cerevisiae, was shown to be not inhibited by BES at a concentration of 25 mM (39).
Effect of BES on propylene and epoxypropane degradation by whole-cell suspensions.
As shown in Fig. 3, addition of 5 mM BES had no effect on the rate of propylene degradation by cell suspensions during the first 120 min of the experiment. After this, there was a slight decrease in the rate of propylene degradation. At the same time, significant levels of epoxypropane, the initial product of propylene metabolism (Fig. 1), began to accumulate in the cell suspensions containing BES, but detectable levels did not accumulate in the cell suspensions lacking BES (Fig. 3). Upon complete degradation of 2 mM propylene (after 530 min [Fig. 3]), 0.75 mM epoxypropane was formed in the presence of BES. Thus, BES appears to have an inhibitory effect on epoxypropane degradation but not on propylene conversion to epoxypropane, consistent with the idea that BES acts as a CoM analog.
FIG. 3.

Effect of BES on propylene degradation and epoxypropane accumulation in cell suspensions of X. autotrophicus. Assays were performed with propylene-grown cells (0.15 mg protein). The solid symbols indicate the propylene remaining, and the open symbols indicate the epoxypropane formed. Symbols: ▪, buffer-only control; • and ○, no BES present; ▴ and ▵, 5 mM BES present.
The inhibition of epoxypropane degradation by BES was investigated in more detail by examining the time courses for epoxypropane degradation by cell suspensions in the presence of various concentrations of BES. As shown in Fig. 4, a time-dependent loss of epoxypropane degradation activity was observed, the magnitude and rate of which were proportional to the concentration of BES included in the assay mixtures. Thus, under the conditions of the assays whose results are shown in Fig. 3 and 4, BES appears to have a time-dependent inhibitory effect on epoxypropane catabolism.
FIG. 4.

Concentration dependence of BES inactivation of epoxypropane degradation in cell suspensions of X. autotrophicus. Assays were performed with propylene-grown cells (0.12 mg protein). Symbols: □, buffer-only control; •, no BES present; ○, 1 mM BES present; ▾, 2.5 mM BES present; ▵, 5 mM BES present; ▪, 10 mM BES present.
Coenzyme M restores epoxypropane degradation activity in BES-treated cell suspensions.
As shown in Fig. 1, epoxypropane metabolism is a three-step pathway catalyzed by four distinct enzymes that effect the net carboxylation of epoxypropane enantiomers to acetoacetate. The assays described above relied on measurement of epoxypropane consumption, a reaction requiring only active EaCoMT and a source of free CoM. Conceivably, any one or more of the enzymes could be inhibited or inactivated by BES, since each enzyme uses CoM or a CoM conjugate as a substrate (Fig. 1). Inactivation of the enzyme(s) of any one of the three steps of the pathway would result in the observed time-dependent loss of epoxypropane degradation activity, since CoM must be recycled in order to sustain the initial conjugation of epoxypropane and CoM catalyzed by EaCoMT. In order to determine whether the observed loss of epoxypropane degradation activity resulted from depletion of the free CoM pool due to inactivation of an enzyme downstream from EaCoMT, the following experiment was performed.
Cells that had been incubated with epoxypropane and with or without 5 mM BES for 320 min, at which point all epoxypropane degradation activity had been lost in the BES-treated cells (Fig. 4), were pelleted, washed to remove residual BES, and resuspended in fresh buffer. The ability of the cells to degrade epoxypropane was then determined in the absence and presence of exogenous CoM. As shown in Fig. 5, addition of CoM to the control cells (preincubated without BES) resulted in a slight increase in the epoxypropane degradation rate, suggesting that CoM availability was at least partially rate limiting in the washed cell suspensions. More importantly, epoxypropane degradation activity was largely restored in the BES-treated cells when exogenous CoM was added, but no activity was observed when CoM was omitted from the assay mixture (Fig. 5). The specific rates of epoxypropane degradation with CoM present were 0.193 U/mg and 0.129 U/mg for the control and BES-treated cells, respectively, which translates to 33% lower activity in the BES-treated cells.
FIG. 5.

Coenzyme M restores epoxypropane degradation activity in BES-treated X. autotrophicus cell suspensions. Cells (0.12 mg protein) were preincubated with 2 μmol epoxypropane in the absence or presence of BES for 300 min, as shown in Fig. 4. The cells were then washed and resuspended in fresh medium, and epoxypropane degradation activity was assayed as described in Materials and Methods. The solid symbols show the results of assays performed with cells preincubated without BES, while the open symbols show the results of assays performed with cells preincubated with 5 mM BES. Triangles, assays conducted without supplemental CoM; squares, assays conducted in the presence of 5 mM CoM.
Effect of BES treatment and chloramphenicol on epoxypropane-dependent 14CO2 fixation activity.
The data shown in Fig. 5 suggest that the observed time-dependent loss of epoxide-degrading activity in cell suspensions can be largely attributed to depletion of the free CoM required to sustain the reaction catalyzed by the first enzyme of the epoxide consumption pathway, i.e., EaCoMT. This in turn suggests that the primary target of BES inactivation is one or more of the enzymes downstream of the reaction catalyzed by EaCoMT (Fig. 1). To investigate this in more detail, the fixation of 14CO2 into acid-stable cell products, indicative of the complete epoxide carboxylase reaction catalyzed by all four enzyme components (37), was measured after pretreatment of cells with or without BES as described above. CoM (5 mM) was included in the assay mixtures to ensure that any differences in 14CO2 fixation rates could be attributed to loss of enzymatic activity rather than to depletion of the CoM pool, and succinate was included as a conventional carbon and energy source to promote new protein synthesis. Additionally, some assays were performed in the presence of chloramphenicol, which prevents new protein synthesis in X. autotrophicus under these conditions (18), in order to determine whether new protein synthesis was necessary to restore epoxide carboxylase activity.
For the non-BES-treated cells lacking chloramphenicol, the yield of 14CO2 fixed to epoxypropane degraded was 82% after 150 min of incubation when 3 μmol epoxypropane was used as the substrate (data not shown). This result is virtually identical to the value obtained in our previous studies of epoxypropane-dependent 14CO2 fixation (the additional 18% 14CO2 fixed is believed to be rereleased as 14CO2 due to cellular respiration) (37). The presence of chloramphenicol led to a slight decrease in the rate of 14CO2 fixation in the non-BES-treated cells. However, there was an approximately threefold decrease in the initial rate of epoxypropane-dependent 14CO2 fixation in BES-treated cells compared with the rate in non-BES-treated cells (data not shown). For the BES-treated cells, a time-dependent increase in 14CO2 fixation activity was observed for the cells lacking chloramphenicol, in which new protein synthesis could occur, and by 150 min the BES-treated cells were fixing 14CO2 at a rate comparable to the rate of the non-BES-treated cells (data not shown). These results demonstrate that new protein synthesis was required for recovery of epoxide carboxylase activity after BES pretreatment.
Identification of the target of BES inactivation in cell extracts of X. autotrophicus.
The whole-cell studies described above provided evidence that BES irreversibly inactivates one or more enzymes downstream of EaCoMT in the epoxide carboxylation pathway (Fig. 1). To shed more light on the specific enzyme(s) that is inactivated, studies were conducted using cell extracts and purified enzymes. For this analysis, we used an in vitro assay that measures the isomerization of epoxypropane to acetone rather than carboxylation of epoxypropane to acetoacetate (2, 43). Epoxide isomerization requires the same enzymes and assay conditions that are required for epoxide carboxylation, with the important distinction that CO2 and bicarbonate are not included in the assay mixture (2, 11). Under these conditions, 2-KPCC, the terminal enzyme of the epoxide carboxylation pathway, catalyzes the reductive cleavage and protonation (rather than carboxylation) of 2-ketopropyl-CoM to form acetone as a product, as shown in equation 1 (compare the terminal reaction in Fig. 1) (2, 11):
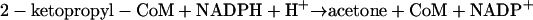 |
(1) |
The advantage of the epoxide isomerization assay is that acetone is much easier to detect and quantify (by gas chromatography analysis) than acetoacetate formed by substrate carboxylation. To further simplify the in vitro assay, (R)-epoxypropane was used as the assay substrate, so that 2-(R)-hydroxypropyl-CoM was the only product of the EaCoMT-catalyzed reaction (Fig. 1). Thus, with (R)-epoxypropane as the substrate, the in vitro assay required only EaCoMT, R-HPCDH, and 2-KPCC and did not require 2-(S)-hydroxypropyl CoM dehydrogenase. Since NADPH, the substrate for 2-KPCC, is rapidly oxidized in cell extracts by various oxidoreductases, DTT, which can serve as an alternative reductant for 2-KPCC (2, 3, 5, 40), was substituted for NADPH. Finally, lactate dehydrogenase and pyruvate were included in the assay mixtures to regenerate oxidized NAD+ from reduced NADH. In summary, the reaction measured in cell extracts, dependent on active EaCoMT, R-HPCDH, 2-KPCC, and a source of CoM, was as follows:
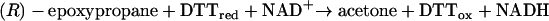 |
(2) |
The initial rate (i.e., the rate in the first few minutes of the assay) of acetone formation from (R)-epoxypropane in dialyzed cell extracts was 60% lower in assays containing 5 mM BES than in assays conducted in the absence of BES (data not shown). Thus, when product formation from all three enzymes was measured, a significant rapid-equilibrium inhibition was observed that was not apparent in whole-cell assays of epoxypropane degradation (Fig. 4). To further investigate the inactivation of the enzyme system, cell extracts were incubated for 4 h with and without BES and then dialyzed to remove residual BES. As shown in Fig. 6A, a significant amount (>75%) of epoxide isomerase activity was lost in the cells preincubated with BES, indicating that one or more of the three required enzymes (EaCoMT, R-HPCDH, and 2-KPCC) is irreversibly inactivated by BES treatment.
FIG. 6.

BES inactivation of epoxide isomerase activity in cell extracts of X. autotrophicus and restoration of epoxide isomerase activity in BES-treated cell extracts by addition of purified 2-KPCC. Cell extracts were prepared and pretreated with or without 5 mM BES present, and epoxide isomerase assays were conducted as described in Materials and Methods. The data show the time course for (R)-epoxypropane isomerization to acetone, a reaction requiring active EaCoMT, R-HPCDH, and 2-KPCC. (A) Activity in the absence of purified enzymes. Symbols: •, no BES present in pretreatment; ○, 5 mM BES present in pretreatment. (B) Activity in BES-pretreated extracts in the presence of purified enzymes. Symbols: •, no purified enzyme added; ▾, purified EaCoMT added; ▵, purified R-HPCDH added; ○, purified 2-KPCC added.
As shown in Fig. 6B, addition of purified 2-KPCC, but not addition of purified EaCoMT or R-HPCDH, restored epoxide carboxylase activity in BES-treated extracts. The level of restored activity was the same as the level observed in cell extracts not treated with BES. Thus, 2-KPCC appears to be irreversibly inactivated to a large degree by BES treatment in cell extracts, while EaCoMT and R-HCDH are apparently not affected by prior BES treatment.
BES inhibits propylene-dependent growth but not propane-dependent growth of R. rhodochrous B276.
Another microorganism in which the CoM-dependent pathway of propylene metabolism has been identified and characterized is R. rhodochrous B276, a gram-positive actinomycete (4, 6, 27). R. rhodochrous B276 is a particularly interesting organism because it is one of only two bacteria reported to be capable of growth with both short-chain saturated and unsaturated hydrocarbons (i.e., the bacterium grows using both propylene and propane as carbon sources) (21, 34). While propylene metabolism in R. rhodochrous occurs via the CoM-dependent pathway identified for X. autotrophicus (Fig. 1), propane metabolism is believed to occur via a distinct pathway in which propane is subterminally hydroxylated to isopropanol, followed by oxidation to acetone (13; D. Clark and S. Ensign, unpublished results). Thus, R. rhodochrous provides an excellent model system for evaluating the specificity of BES as an inhibitor of unsaturated hydrocarbon metabolism.
As shown in Fig. 7A, the presence of 5 mM BES completely prevented growth of R. rhodochrous B276 with propylene as the carbon source. In contrast, 5 mM BES had no effect on growth when propane or acetone was used as the carbon source (Fig. 7B). These comparative studies with R. rhodochrous B276 reinforced the idea that BES can be viewed as a specific inhibitor of bacterial alkene metabolism.
FIG. 7.

Effects of BES on growth of R. rhodochrous strain B276 with propylene, acetone, and propane as carbon sources. (A) Growth with propylene. Symbols: •, no BES present; ○, 5 mM BES present. (B) Growth with acetone (triangles) and propane (squares). Solid symbols, no BES present; open symbols, 5 mM BES present. The data are the averages of measurements for two cultures.
Comparison of BES as an inhibitor of methanogenesis and alkene metabolism.
A comparison of our results with the results for BES inhibition of methanogenesis revealed some interesting similarities and differences in the observed effects. For methanogens, the specific target of BES has been shown to be methyl-CoM reductase (MCR), which is inhibited in both a reversible fashion and an irreversible fashion by BES (23, 24, 38). MCR catalyzes the reduction of methyl-CoM to methane using a using a second thiol substrate, CoB, as shown in equation 3 (17, 42, 44):
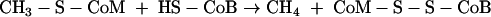 |
(3) |
The inactivation of MCR, the target enzyme of BES in methanogens, has been shown to be due to the interaction between BES and the unique nickel tetrapyrrole cofactor F430 at the active site of the enzyme. Goenrich and coworkers have proposed a mechanism for inactivation of MCR involving oxidation of active Ni(I) F430 to inactive Ni(II) F430 by BES when it is bound in the active site as a CoM analog (23). The tetrapyrrole F430 is unique to methanogens and is not found in the CoM-dependent pathway of epoxide carboxylation. The results of this study suggest instead that 2-KPCC, the terminal enzyme of epoxide carboxylation (Fig. 1), is a target enzyme for inactivation by BES. In this regard, it is tempting to speculate that BES exerts its inhibitory effect as a substrate analog for 2-KPCC and as an alkylating agent in the 2-KPCC active site. Importantly, the active site of 2-KPCC, a member of the DSOR family of enzymes, is defined by the presence of a redox-active cysteine pair that participates in catalysis (11, 31). Alkylation of one or both of these active site cysteine residues would account of the observed irreversible loss of activity. The molecular basis of BES inactivation of epoxypropane carboxylation, including the possibility alluded to above, is the subject of additional studies that are in progress.
As noted above, other alkene-oxidizing bacteria, specifically those that grow using ethylene and vinyl chloride as carbon sources, have been shown to use CoM for epoxide metabolism (14-16, 29). For these bacteria, EaCoMT catalyzes the reaction of epoxyethane or chloroepoxyethane with CoM to form the initial products hydroxyethyl-CoM and chlorohydroxyethyl-CoM. These products are believed to be further metabolized by dehydrogenation (and dehalogenation in the case of chlorohydroxyethyl-CoM) in a step analogous to the reactions catalyzed by R-HPCDH and 2-(S)-hydroxypropyl CoM dehydrogenase, followed by conversion to acetyl-CoA (15). Just recently, BES was tested as a possible inhibitor of EaCoMT activity in cell extracts of the ethylene oxidizer Pseudomonas putida strain AJ (16). BES concentrations as high as 100 mM had no effect on EaCoMT activity. This result agrees with our results showing that EaCoMT does not appear to be a target of BES inhibition or inactivation. In their studies of P. putida, Danko et al. did not test for BES inhibition of growth or for inhibition of the other enzymes believed to be involved in epoxyethane metabolism. Thus, it remains to be determined whether BES is a specific inhibitor of alkene-utilizing bacteria incorporating the carboxylation reaction unique to propylene (and longer-chain) alkenes or whether it is an effective inhibitor of ethylene and epoxyethane catabolism as well.
In summary, in this study we identified and characterized BES as the first selective inhibitor of bacterial short-chain alkene metabolism. The high degree of specificity of BES for inhibition of propylene-dependent growth of X. autotrophicus Py2 and R. rhodochrous B276 suggests that it may be useful as an environmental probe for the identification and characterization of alkene-oxidizing bacteria. Our findings raise the interesting possibility that BES may be useful for identifying new CoM-utilizing bacteria in addition to the methanogens and alkene oxidizers identified thus far.
Acknowledgments
This work was supported by National Institutes of Health grant GM51805.
Footnotes
Published ahead of print on 22 September 2006.
REFERENCES
- 1.Allen, J. R., D. D. Clark, J. G. Krum, and S. A. Ensign. 1999. A role for coenzyme M (2-mercaptoethansulfonic acid) in a bacterial pathway of aliphatic epoxide carboxylation. Proc. Natl. Acad. Sci. USA 96:8432-8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen, J. R., and S. A. Ensign. 1996. Carboxylation of epoxides to β-keto acids in cell extracts of Xanthobacter strain Py2. J. Bacteriol. 178:1469-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, J. R., and S. A. Ensign. 1997. Characterization of three protein components required for functional reconstitution of the epoxide carboxylase multienzyme complex from Xanthobacter strain Py2. J. Bacteriol. 179:3110-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen, J. R., and S. A. Ensign. 1998. Identification and characterization of epoxide carboxylase activity in cell extracts of Nocardia corallina strain B276. J. Bacteriol. 180:2072-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen, J. R., and S. A. Ensign. 1997. Purification to homogeneity and reconstitution of the individual components of the epoxide carboxylase multiprotein enzyme complex from Xanthobacter strain Py2. J. Biol. Chem. 272:32121-32128. [DOI] [PubMed] [Google Scholar]
- 6.Allen, J. R., and S. A. Ensign. 1999. Two short-chain dehydrogenases confer stereoselectivity for enantiomers of epoxypropane in the multiprotein epoxide carboxylating systems of Xanthobacter strain Py2 and Nocardia corallina B276. Biochemistry 38:247-256. [DOI] [PubMed] [Google Scholar]
- 7.Balch, W. E., and R. S. Wolfe. 1979. Specificity and biological distribution of coenzyme M (2-mercaptoethanesulfonic acid). J. Bacteriol. 137:256-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balch, W. E., and R. S. Wolfe. 1979. Transport of coenzyme M (2-mercaptoethanesulfonic acid) in Methanobacterium ruminantium. J. Bacteriol. 137:264-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu, P. C., and M. Lee. 2001. 2-Bromoethanesulfonate affects bacteria in a trichloroethene-dechlorinating culture. Appl. Environ. Microbiol. 67:2371-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chromy, V., J. Fischer, and V. Kulhanek. 1974. Re-evaluation of EDTA-chelated biuret reagent. Clin. Chem. 20:1362-1363. [PubMed] [Google Scholar]
- 11.Clark, D. D., J. R. Allen, and S. A. Ensign. 2000. Characterization of five catalytic activities associated with the NADPH:2-ketopropyl-coenzyme M [2-(2-ketopropylthio)ethanesulfonate] oxidoreductase/carboxylase of the Xanthobacter strain Py2 epoxide carboxylase. Biochemistry 39:1294-1304. [DOI] [PubMed] [Google Scholar]
- 12.Clark, D. D., and S. A. Ensign. 2002. Characterization of the 2-(R)-2-hydroxypropylthio ethane sulfonate dehydrogenase from Xanthobacter strain Py2: product inhibition, pH dependence of kinetic parameters, site-directed mutagenesis, rapid equilibrium inhibition, and chemical modification. Biochemistry 41:2727-2740. [DOI] [PubMed] [Google Scholar]
- 13.Clark, D. D., and S. A. Ensign. 1999. Evidence for an inducible nucleotide-dependent acetone carboxylase in Rhodococcus rhodochrous B276. J. Bacteriol. 181:2752-2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coleman, N. V., and J. C. Spain. 2003. Distribution of the coenzyme M pathway of epoxide metabolism among ethene- and vinyl chloride-degrading Mycobacterium strains. Appl. Environ Microbiol. 69:6041-6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman, N. V., and J. C. Spain. 2003. Epoxyalkane: coenzyme M transferase in the ethene and vinyl chloride biodegradation pathways of Mycobacterium strain JS60. J. Bacteriol. 185:5536-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danko, A. S., C. A. Saski, J. P. Tomkins, and D. L. Freedman. 2006. Involvement of coenzyme M during aerobic biodegradation of vinyl chloride and ethene by Pseudomonas putida strain AJ and Ochrobactrum sp. strain TD. Appl. Environ. Microbiol. 72:3756-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiMarco, A. A., T. A. Bobik, and R. S. Wolfe. 1990. Unusual coenzymes of methanogenesis. Annu. Rev. Biochem. 59:355-394. [DOI] [PubMed] [Google Scholar]
- 18.Ensign, S. A. 1996. Aliphatic and chlorinated alkenes and epoxides as inducers of alkene monooxygenase and epoxidase activities in Xanthobacter strain Py2. Appl. Environ. Microbiol. 62:61-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ensign, S. A. 2001. Microbial metabolism of aliphatic alkenes. Biochemistry 40:5845-5853. [DOI] [PubMed] [Google Scholar]
- 20.Ensign, S. A., and J. R. Allen. 2003. Aliphatic epoxide carboxylation. Annu. Rev. Biochem. 72:55-76. [DOI] [PubMed] [Google Scholar]
- 21.Furuhashi, K., A. Taoka, S. Uchida, I. Karube, and S. Suzuki. 1981. Production of 1,2-epoxyalkanes from 1-alkenes by Nocardia corallina B-276. Eur. J. Appl. Microbiol. Biotechnol. 12:39-45. [Google Scholar]
- 22.Gallagher, S. C., R. Cammack, and H. Dalton. 1997. Alkene monooxygenase from Nocardia corallina B-276 is a member of the class of dinuclear iron proteins capable of stereospecific epoxygenation reactions. Eur. J. Biochem. 247:635-641. [DOI] [PubMed] [Google Scholar]
- 23.Goenrich, M., F. Mahlert, E. C. Duin, C. Bauer, B. Jaun, and R. K. Thauer. 2004. Probing the reactivity of Ni in the active site of methyl-coenzyme M reductase with substrate analogues. J. Biol. Inorg. Chem. 9:691-705. [DOI] [PubMed] [Google Scholar]
- 24.Gunsalus, R. P., J. A. Romesser, and R. S. Wolfe. 1978. Preparation of coenzyme M analogues and their activity in the methyl coenzyme M reductase system of Methanobacterium thermoautotrophicum. J. Bacteriol. 17:2374-2377. [DOI] [PubMed] [Google Scholar]
- 25.Hartmans, S., J. A. M. de Bont, and W. Harder. 1989. Microbial metabolism of short-chain unsaturated hydrocarbons. FEMS Microbiol. Rev. 63:235-264. [DOI] [PubMed] [Google Scholar]
- 26.Krum, J. G., H. Ellsworth, R. R. Sargeant, G. Rich, and S. A. Ensign. 2002. Kinetic and microcalorimetric analysis of substrate and cofactor interactions in epoxyalkane:CoM transferase, a zinc-dependent epoxidase. Biochemistry 41:5005-5014. [DOI] [PubMed] [Google Scholar]
- 27.Krum, J. G., and S. A. Ensign. 2000. Heterologous expression of bacterial epoxyalkane:coenzyme M transferase and inducible coenzyme M biosynthesis in Xanthobacter strain Py2 and Rhodococcus rhodochrous B276. J. Bacteriol. 182:2629-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loffler, F. E., K. M. Ritalahti, and J. M. Tiedje. 1997. Dechlorination of chloroethenes is inhibited by 2-bromoethanesulfonate in the absence of methanogens. Appl. Environ. Microbiol. 63:4982-4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattes, T. E., N. V. Coleman, J. C. Spain, and J. M. Gossett. 2005. Physiological and molecular genetic analyses of vinyl chloride and ethene biodegradation in Nocardioides sp. strain JS614. Arch. Microbiol. 183:95-106. [DOI] [PubMed] [Google Scholar]
- 30.Miura, A., and H. Dalton. 1995. Purification and characterization of the alkene monooxygenase from Nocardia corallina B-276. Biosci. Biotechnol. Biochem. 59:853-859. [Google Scholar]
- 31.Nocek, B., S. B. Jang, M. S. Jeong, D. D. Clark, S. A. Ensign, and J. W. Peters. 2002. Structural basis for CO2 fixation by a novel member of the disulfide oxidoreductase family of enzymes: 2-ketopropyl-coenzyme M oxidoreductase/carboxylase. Biochemistry 41:12907-12913. [DOI] [PubMed] [Google Scholar]
- 32.Sawada, S., and T. Totsuka. 1986. Natural and anthropogenic sources and fate of atmospheric ethylene. Atmos. Environ. 20:821-832. [Google Scholar]
- 33.Sexton, K., and H. Westberg. 1984. Nonmethane hydrocarbon composition of urban and rural atmospheres. Atmos. Environ. 18:1125-1132. [Google Scholar]
- 34.Shennan, J. L. 2006. Utilisation of C-2-C-4 gaseous hydrocarbons and isoprene by microorganisms. J. Chem. Technol. Biotechnol. 81:237-256. [Google Scholar]
- 35.Sluis, M. K., F. J. Small, J. R. Allen, and S. A. Ensign. 1996. Involvement of an ATP-dependent carboxylase in a CO2-dependent pathway of acetone metabolism by Xanthobacter strain Py2. J. Bacteriol. 178:4020-4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Small, F. J., and S. A. Ensign. 1997. Alkene monooxygenase from Xanthobacter strain Py2: purification and characterization of a four-component system central to the bacterial metabolism of aliphatic alkenes. J. Biol. Chem. 272:24913-24920. [DOI] [PubMed] [Google Scholar]
- 37.Small, F. J., and S. A. Ensign. 1995. Carbon dioxide fixation in the metabolism of propylene and propylene oxide by Xanthobacter strain Py2. J. Bacteriol. 177:6170-6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, M. R. 1983. Reversal of 2-bromoethanesulfonate inhibition of methanogenesis in Methanosarcina sp. J. Bacteriol. 156:516-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sparling, R., and L. Daniels. 1987. The specificity of growth inhibition of methanogenic bacteria by bromoethanesulfonate. Can. J. Microbiol. 33:1132-1136. [Google Scholar]
- 40.Swaving, J., J. A. M. Debont, A. Westphal, and A. Dekok. 1996. A novel type of pyridine nucleotide-disulfide oxidoreductase is essential for NAD+- and NADPH-dependent degradation of epoxyalkanes by Xanthobacter strain Py2. J. Bacteriol. 178:6644-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor, C. D., B. C. McBride, R. S. Wolfe, and M. P. Bryant. 1974. Coenzyme M, essential for growth of a rumen strain of Methanobacterium ruminatium. J. Bacteriol. 120:974-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thauer, R. K. 1998. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144:2377-2406. [DOI] [PubMed] [Google Scholar]
- 43.Weijers, C. A. G. M., H. Jongejan, M. C. R. Franssen, A. de Groot, and J. A. M. de Bont. 1995. Dithiol- and NAD-dependent degradation of epoxyalkanes by Xanthobacter Py2. Appl. Microbiol. Biotechnol. 42:775-781. [Google Scholar]
- 44.Wolfe, R. S. 1991. My kind of biology. Annu. Rev. Microbiol. 45:1-35. [DOI] [PubMed] [Google Scholar]
- 45.Ye, D., J. F. R. Quensen, J. M. Tiedje, and S. A. Boyd. 1999. 2-Bromoethanesulfonate, sulfate, molybdate, and ethanesulfonate inhibit anaerobic dechlorination of polychlorobiphenyls by pasteurized microorganisms. Appl. Environ. Microbiol. 65:327-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou, N. Y., C. K. C. K. Chion, and D. J. Leak. 1996. Cloning and expression of the genes encoding the propene monooxygenase from Xanthobacter Py2. Appl. Microbiol. Biotechnol. 44:582-588. [Google Scholar]
- 47.Zinder, S. H., T. Anguish, and S. C. Cardwell. 1984. Selective inhibition by 2-bromoethanesulfonate of methanogenesis from acetate in a thermophilic anaerobic digestor. Appl. Environ. Microbiol. 47:1343-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]