

Abstract
The two-component alkanesulfonate monooxygenase system utilizes reduced flavin as a substrate to catalyze a unique desulfonation reaction during times of sulfur starvation. The importance of protein-protein interactions in the mechanism of flavin transfer was analyzed in these studies. The results from affinity chromatography and cross-linking experiments support the formation of a stable complex between the flavin mononucleotide (FMN) reductase (SsuE) and monooxygenase (SsuD). Interactions between the two proteins do not lead to overall conformational changes in protein structure, as indicated by the results from circular dichroism spectroscopy in the far-UV region. However, subtle changes in the flavin environment of FMN-bound SsuE that occur in the presence of SsuD were identified by circular dichroism spectroscopy in the visible region. These data are supported by the results from fluorescent spectroscopy experiments, where a dissociation constant of 0.0022 ± 0.0010 μM was obtained for the binding of SsuE to SsuD. Based on these studies, the stoichiometry for protein-protein interactions is proposed to involve a 1:1 monomeric association of SsuE with SsuD.
All living organisms need sulfur for various metabolic processes. Sulfur is a primary component of cysteine and methionine and plays an essential role as a substituent in certain enzyme cofactors. Bacterial organisms typically utilize inorganic sulfate or cysteine as their primary sulfur source (15). In conditions where sulfur in the environment is limiting, specific proteins which enable the bacteria to utilize sulfur from alternate sources are synthesized (16). The ssuEADCB operon is one of two gene clusters in Escherichia coli that are induced by sulfate starvation. Expression of this operon leads to the synthesis of an NAD(P)H-dependent flavin mononucleotide (FMN) reductase (SsuE) and monooxygenase (SsuD) that are involved in the acquisition of sulfite from alkanesulfonates (29). SsuE provides reduced flavin to SsuD for the desulfonation of alkanesulfonates in the presence of molecular oxygen (Fig. 1) (5). In this enzyme system the flavin does not function as a prosthetic group but serves as a substrate for both SsuE and SsuD (28). The alkanesulfonate system has been identified in a wide range of bacterial organisms since the initial characterization of this system from E. coli (5).
FIG. 1.

Mechanism of desulfonation by the two-component alkanesulfonate monooxygenase system from E. coli.
There are two important mechanisms associated with the alkanesulfonate monooxygenase system that have not been fully addressed. The first is the mechanism of desulfonation by SsuD to form an aldehyde and sulfite product. The E. coli alkanesulfonate monooxygenase system is the first enzymatic system reported which is capable of oxygenolytic cleavage of the C-S bond of 1-alkanesulfonates by an oxygenase mechanism (5). The second interesting feature is the mechanism of reduced flavin transfer from SsuE to SsuD. In general, the mechanism of reduced flavin transfer could occur either through free diffusion or by direct channeling from SsuE to SsuD, requiring the interaction of the two proteins. Because reduced flavin can be rapidly oxidized, generating oxygen radicals, the mechanism involving direct transfer of the reduced flavin would likely be more favorable in a cellular system (3, 11, 30). Therefore, in the alkanesulfonate monooxygenase system the transfer of FMNH2 may be tightly controlled through protein interactions and specific channeling between SsuE and SsuD.
Alternative results describing the transfer of reduced flavin have been reported depending on the system being studied. An altered kinetic mechanism for SsuE has been observed in the presence and absence of SsuD and an alkanesulfonate substrate (7). In single-enzyme kinetic assays, SsuE follows an ordered sequential mechanism, with NADPH as the first substrate to bind and NADP+ as the last product to dissociate. However, in the presence of SsuD and octanesulfonate, the kinetic mechanism of SsuE is altered to a-rapid equilibrium ordered mechanism. Direct transfer of reduced flavin has been demonstrated through kinetic analyses with NADPH-dependent flavin reductase (FRP) and bacterial luciferase (12, 13, 18). The mechanism of FRP is altered from a ping-pong to a sequential kinetic mechanism in the luciferase-coupled assay, providing strong support for complex formation. These experimental results are further maintained by the observed decrease in the Km values for both FMN and NAD(P)H in the luciferase-coupled assay compared to the single FRP assay. Conversely, using in vitro kinetic and fluorescence analyses, there was no substantial change in the apparent Km value for FAD in the NAD(P)H-flavin oxidoreductase (HpaC) and 4-hydroxyphenylacetate 3-monooxygenase (HpaB) coupled assay in E. coli (19). These results suggest that protein-protein interactions between HpaC and HpaB do not play a role in the coordinated production and utilization of FADH2 in E. coli. Another study on the mechanism of reduced flavin transfer had been performed based on numerical simulations of the steady-state mechanism of styrene monooxygenase (SMO) from Pseudomonas putida S12 (14). The observed coupling of NADH to styrene oxidation can be best explained by a model that includes both the direct transfer and passive diffusion of reduced FAD from the NADH-specific flavin reductase (SMOB) to the FAD-specific styrene epoxidase (SMOA). The mechanism involves the formation of a transient protein complex with both the reductase and monooxygenase enzymes associated with reduced FAD.
Based on prior studies of two-component flavoenzymes, the mechanism of reduced flavin transfer in these systems appears to be diverse and is dependent on the specific system (7, 13, 14, 19). Although steady-state kinetic studies support a model for direct flavin transfer from SsuE to SsuD (7), the actual mechanism has not been directly addressed in this system. The detailed mechanism of desulfonation is highly important given the critical role of this system in the acquisition of sulfite from alkanesulfonates in a broad range of bacterial organisms. The studies described here were performed to determine if static protein-protein interactions between SsuE and SsuD could be identified. Stable interactions between the two proteins would support a mechanism for direct flavin transfer. Interactions between the alkanesulfonate monooxygenase proteins in these studies were identified through affinity chromatography, chemical cross-linking, and spectroscopic experiments. The reported results will provide the basis for future studies to determine the kinetics of flavin transfer in the desulfonation reaction by the alkanesulfonate monooxygenase system.
MATERIALS AND METHODS
Materials.
All reagents were purchased from Sigma-Aldrich, Bio-Rad, or Fisher. The chemical cross-linking reagent ProFound Label Transfer Sulfo-SBED Protein:Protein Interaction Agent was from Pierce (Rockford, IL). The buffer solution contained 25 mM potassium phosphate (pH 7.5) and 10% glycerol unless otherwise noted.
Construction, expression, and purification of recombinant proteins.
The T7 RNA polymerase-dependent expression vector pET21a carrying either the ssuE or ssuD gene was utilized for protein expression and purification as previously described (7). The concentrations of SsuE and SsuD proteins in the solution with different unit concentrations were calculated by considering either the molar extinction coefficients of the two proteins (20.3 mM−1 cm−1 and 46.9 mM−1 cm−1 at 280 nm, respectively) or the previously determined molecular masses of SsuE and SsuD proteins (21.3 kDa and 41.6 kDa, respectively) (5, 7).
The pET21a (Novagen, Madison, WI) vector containing the C-terminally His-tagged ssuD gene was constructed by mutation of the native ssuD stop codon. The native construct was PCR amplified using the primers 5′ CGT AAA GTC GCG CAA AGC GCG CTC GAG CAC CAC CAC 3′ and 5′ GTG GTG GTG CTC GAG CGC GCT TTG CGC GAC TTT ACG 3′. The mutations were generated utilizing the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), and the generated mutation was verified by sequence analysis at Davis Sequencing (University of California, Davis).
Affinity chromatography binding assay.
The native SsuE and His-tagged SsuD proteins were expressed as previously described with the following exceptions (7). The native SsuE and His-tagged SsuD cell cultures were grown separately in LB medium containing 100 μg/ml ampicillin (LB-Amp). A single colony of E. coli BL21(DE3) containing the appropriate expression plasmid was used to inoculate 5 ml LB-Amp medium, with incubation overnight at 37°C. A 1% inoculum of the 5-ml cell culture was used to inoculate 100 ml LB-Amp, and the culture was incubated at 37°C until the A600 reached 0.4. The flask was then transferred from 37°C to 18°C, and the protein was expressed by the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 400 μM. The incubation was continued for 6 h, and cells were harvested by centrifugation at 5,000 rpm for 15 min at 4°C and stored at −80°C. Each protein was purified according to a previously published protocol (7).
A 50-ml cell lysate containing the His-tagged SsuD protein was loaded onto a column containing Ni-nitrilotriacetic acid (NTA) Superflow resin (QIAGEN, Valencia, CA), and the column was washed with 100 ml buffer to remove unbound protein prior to loading the cell lysate containing native SsuE. The column was then washed again with 100 ml buffer after loading the native SsuE protein. The unbound protein was eluted from the column with 100 ml of buffer containing 125 mM imidazole, followed by elution of the His-tagged SsuD protein with 100 ml of buffer containing 300 mM imidazole. The eluted proteins from both buffer concentrations were collected and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Two sets of control experiments were performed to ensure that the complex formation between SsuE and His-tagged SsuD protein in the Ni-NTA column was not due to nonspecific binding or protein aggregations. The first control experiment was performed by loading the cell lysate containing native SsuE protein onto a Ni-NTA column in the absence of the His-tagged SsuD protein. The second control experiment was performed by loading a protein available in our laboratory, the rat cysteine dioxygenase protein, onto a Ni-NTA column in the presence of His-tagged SsuD protein. For each control, the column was washed with 100 ml of buffer, followed by a second wash with 100 ml of buffer containing 125 mM of imidazole. A final wash with 100 ml of buffer containing 300 mM of imidazole was performed and the protein fractions collected and analyzed by SDS-PAGE.
Far-UV circular dichroism (CD).
The spectrum of SsuE was obtained with 2.4 μM SsuE in 25 mM potassium phosphate buffer (pH 7.5), 10% glycerol, and 100 mM NaCl at 25°C. The SsuD spectrum was obtained with 1.2 μM SsuD in 25 mM phosphate buffer (pH 7.5) at 25°C. The SsuE/SsuD spectrum was obtained with 2.4 μM SsuE and 1.2 μM SsuD. Spectra were recorded on a Jasco J-810 Spectropolarimeter (Easton, MD). Measurements were taken in 0.1-nm increments from 300 to 185 nm in a 0.1-cm-path-length cuvette with a bandwidth of 1 nm and a scanning speed of 50 nm/min. Each spectrum is the average of four scans; smoothing of the data was performed using the default parameters within the Jasco J-720 software.
Visible CD.
The spectrum of FMN was obtained with 20 μM FMN in 25 mM potassium phosphate buffer (2.0-ml total volume) (pH 7.5), 10% glycerol, and 100 mM NaCl at 25°C. The FMN/SsuE spectrum was obtained with 20 μM of SsuE and FMN in 25 mM potassium phosphate buffer (2.0-ml total volume) (pH 7.5), 10% glycerol, and 100 mM NaCl at 25°C. The FMN/SsuE/SsuD spectrum was obtained with a 1:1 stoichiometric addition of 20 μM SsuD to 20 μM FMN-bound SsuE (2.0-ml total volume). Spectra were recorded on a Jasco J-810 Spectropolarimeter (Easton, MD). Measurements were taken in 0.2-nm increments from 550 to 300 nm in a 1-cm-path-length cuvette with a bandwidth of 1 nm and a scanning speed of 50 nm/min. Each spectrum is the average of eight scans; smoothing of the data was performed using the default parameters within the Jasco J-720 software. The concentration of FMN was calculated using a molar extinction coefficient of 12.2 mM−1 cm−1 at 450 nm.
Fluorescence spectroscopy.
Binding of FMN to SsuE was determined by spectrofluorometric titrations, monitoring the decrease in the relative intensity of the FMN spectra due to fluorescence quenching of the flavin upon binding SsuE. SsuD interactions with FMN-bound SsuE were monitored by spectrofluorometric titration, monitoring the increase in the relative intensity of the FMN spectra due to interactions between the two proteins. Spectra were recorded on a Perkin-Elmer LS 55 luminescence spectrometer (Palo Alto, CA) with both excitation and emission slit widths set at 10 nm.
Fluorometric titration of FMN with SsuE.
A 0.04 μM FMN solution in 25 mM potassium phosphate buffer (pH 7.5) (1.0-ml total volume) was titrated with 1-μl aliquots of SsuE for a total of 10 to 15 titrations (0.04 to 0.40 μM). Emission intensity measurements from 470 to 650 nm were made using an excitation wavelength of 450 nm.
Fluorometric titration of FMN-bound SsuE with SsuD.
A 0.40 μM FMN-bound SsuE sample in 25 mM potassium phosphate buffer (pH 7.5) (1.0-ml total volume) was titrated with 1-μl aliquots of SsuD for a total of 25 to 30 titrations (0.02 to 0.95 μM). Emission intensities from 470 to 650 nm were measured using an excitation wavelength of 450 nm. The concentration of SsuD bound to SsuE was determined by applying the following equation (19): [SsuD]bound = [SsuE][(Ic − I0)/(If − I0)], where [SsuE] represents the initial concentration of enzyme, I0 is the initial fluorescence intensity of FMN prior to addition of SsuD, Ic is the fluorescence intensity of FMN following each addition of SsuD, and If is the final fluorescence intensity. The concentration of SsuD bound ([SsuD]bound, y) was plotted against the total SsuD ([SsuD]total, x) to obtain the dissociation constant (KD) for the binding between SsuE and SsuD according to equation 1 below, where n is the binding capacity of SsuE (19):
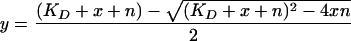 |
(1) |
Filtration of FMN-bound SsuE.
The flavin spectra were obtained at 450 nm before and after filtration with 20 μM FMN in 25 mM potassium phosphate (pH 7.5) 10% glycerol, and 100 mM NaCl at 25°C in the presence of 20 μM of SsuE and SsuD proteins in a 2.0-ml total volume. The filtration was performed by centrifugation at 3,000 rpm for 15 min, utilizing a 10,000-molecular-weight-cutoff Amicon Ultra-4 centrifugation filter from Millipore (Bedford, MA). Control experiments with only FMN were performed by monitoring the spectra at 450 nm before and after filtration.
Chemical cross-linking.
Chemical cross-linking experiments were performed using the ProFound Label Transfer Sulfo-SBED {sulfosuccinimidyl[2,6-(biotinamido)-2-(p-azidobenzamido)-hexanoamido]-ethyl-1,3′-dithiopropionate} Protein:Protein Interaction Agent (Pierce, Rockford, IL) (see Fig. 5A). A 10 μM sample of the SsuE protein was incubated in the dark at room temperature for 30 min with 1 mM of cross-linking reagent in a total volume of 500 μl. The unreacted reagent was removed by washing the sample twice by centrifugation at 3,000 rpm for 15 min, utilizing a 10,000-molecular-weight-cutoff Amicon Ultra-4 centrifugation filter from Millipore (Bedford, MA). The labeled SsuE protein was mixed with 10 μM of SsuD in a total volume of 1 ml and incubated in the dark for 15 min. The reaction mixture was exposed to UV light at a distance of 5 cm for 15 min, followed by the addition of 20 mM dithiothreitol (DTT) (final concentration). A sample (10 μl) was collected following each step for further analysis by nondenaturing and nonreducing polyacrylamide gel electrophoresis (in the absence of SDS and β-mercaptoethanol). Control experiments similar to those described above were performed using either SsuE or SsuD to determine which bands represent intrasubunit cross-linking.
FIG. 5.

Chemical cross-linking of SsuE and SsuD. (A) structure of the trifunctional cross-linker reagent (ProFound Label Transfer Sulfo-SBED). The reagent contains an amine reactive site, a photoreactive arylazide, a biotin label, and a cleavable disulfide bond. (B) Silver-stained SDS-PAGE (10% acrylamide) from cross-linking reactions. Lanes 1 and 6, protein markers; lane 2, SsuE and SsuD proteins; lane 3, treated SsuE; lane 4, cross-linked SsuE and SsuD; lane 5, DTT-treated cross-linked SsuE and SsuD; lane 7, native SsuE and SsuD. The samples were mixed with nondenaturing and nonreducing sample buffer except for protein markers (lanes 1 and 6) and SsuE and SsuD proteins (lane 2).
RESULTS
Affinity chromatography.
The transfer of reduced flavin between the two-component monooxygenase enzymes could occur either by a diffusion mechanism or by direct flavin transfer due to protein-protein interactions. Affinity chromatography experiments with His-tagged SsuD and native SsuE were performed to identify static protein interactions. His-tagged SsuD from a cell lysate was loaded onto a Ni-NTA column. Following a phosphate buffer wash to remove unbound protein, native SsuE from a cell lysate was loaded onto the Ni-NTA column containing bound His-tagged SsuD. The column was first washed with 125 mM imidazole buffer to remove any unbound protein, followed by a second wash with 300 mM imidazole buffer to remove bound His-tagged SsuD. The fractions collected from the column after applying the 125 mM and 300 mM imidazole buffers were collected and analyzed by SDS-PAGE. The results showed that His-tagged SsuD and native SsuE coeluted from the column with 300 mM imidazole buffer (Fig. 2, lane 4). The data suggest that the native SsuE protein remains bound to the column after the 125 mM imidazole wash due to protein-protein interactions with His-tagged SsuD. Control experiments showed that native SsuE from a cell lysate was unable to bind to the Ni-NTA column, and the protein was eluted off the column with 125 mM imidazole buffer. These interactions were specific and were not caused by random aggregation, based on similar experiments between an unrelated protein and His-tagged SsuD as a control that did not result in the coelution of the two proteins with 300 mM imidazole buffer.
FIG. 2.

SDS-PAGE (10% acrylamide) from affinity chromatography experiments with SsuE and His-tagged SsuD. Lane 1, protein standard marker; lane 2, native SsuD; lane 3, native SsuE; lane 4, protein fractions collected after washing with 300 mM imidazole buffer containing SsuE and His-tagged SsuD.
Structural changes due to protein-protein interactions.
The CD spectroscopy experiments in the far-UV (190- to 250-nm) and visible (300- to 500-nm) regions were performed to monitor any changes in the gross secondary structure or flavin environment caused by interactions between FMN-bound SsuE and SsuD. The individual CD spectra of SsuE and SsuD in the far UV region were initially obtained to determine if there were any observable alterations in the secondary structure of each protein compared to the spectrum obtained with both proteins (Fig. 3). The results showed that there were no significant changes in the secondary structure, and the additive individual spectra of SsuE and SsuD closely resemble the spectrum obtained with both proteins present (Fig. 3). When spectra in the visible absorbance region were obtained, oxidized flavin (FMN) exhibited a characteristic spectrum with a weak signal typical for free FMN (Fig. 4). A significant change of the flavin spectrum was observed upon binding of FMN to SsuE, providing an intense signal with both positive and negative ellipticity (Fig. 4). When SsuD was included in the solution, the molar ellipticity between 300 to 375 nm decreased (Fig. 4). This suggested that the flavin environment was affected by the presence of SsuD protein. However, the CD spectrum of FMN did not change in the presence of SsuD alone (data not shown). Therefore, the protein-protein interactions cause a slight change in the flavin environment but do not cause significant changes in the overall gross secondary structure.
FIG. 3.

Far-UV CD spectroscopy of SsuE and SsuD. The spectrum of SsuE (solid line) was obtained with 2.4 μM SsuE in 25 mM phosphate buffer (pH 7.5) at 25°C. The SsuD spectrum (dotted line) was obtained with 1.2 μM SsuD in 25 mM phosphate buffer (pH 7.5) at 25°C. The SsuE/SsuD spectrum (dashed line) was obtained with 2.4 μM of SsuE and 1.2 μM SsuD. The additive spectrum was from individual spectra of SsuE and SsuD (dashed line). Measurements were taken in 0.1-nm increments from 300 to 185 nm in a 0.1-cm-path-length cuvette. Each spectrum is the average of four scans; smoothing of the data was performed using the default parameters within the Jasco J-720 software.
FIG. 4.

Visible CD spectra for FMN in the absence and presence of SsuE and SsuD. The spectrum of FMN (solid line) was obtained with 20 μM FMN in 25 mM potassium phosphate buffer (2.0-ml total volume) (pH 7.5), 10% glycerol, and 100 mM NaCl at 25°C. The FMN/SsuE spectrum (dashed line) was obtained with 40 nmol of FMN and 20 μM of SsuE in 25 mM potassium phosphate buffer (2.0-ml total volume) (pH 7.5), 10% glycerol, and 100 mM NaCl at 25°C. The FMN/SsuE/SsuD spectrum (dotted line) was obtained with a 1:1 stoichiometric addition of 20 μM SsuD to 40 μM FMN-bound SsuE (2.0-ml total volume). Measurements were taken in 0.2-nm increments from 550 to 300 nm in a 1-cm-path-length cuvette. Each spectrum is the average of eight scans; smoothing of the data was performed using the default parameters within the Jasco J-720 software.
Chemical cross-linking.
Cross-linking experiments were performed to investigate any physical interactions between SsuE and SsuD proteins, utilizing a trifunctional cross-linker reagent. The trifunctional cross-linker (sulfo-SBED) contains an amine reactive site, a photoreactive arylazide, a biotin label, and a cleavable disulfide bond (Fig. 5A). The trifunctional cross-linker was first reacted with amine groups on SsuE before the addition of SsuD. While SsuD contains no cysteine residues for cross-linking, analysis of the distribution of amino acids in the three-dimensional structure shows several surface-exposed amine-containing residues that are randomly distributed (4). Following SsuE labeling, the SsuD protein was added and the sample exposed to UV light to cross-link SsuD with the photoreactive group. The addition of DTT following photoactivation cleaves the disulfide bond in the amine reactive arm, resulting in biotin-labeled SsuD. Samples were taken after each step for analysis by native gel electrophoresis. The sample after the activation of the photoreactive group contained a protein band with an apparent molecular mass of approximately 63 kDa, corresponding to the monomeric cross-linking of SsuE (21.3 kDa) and SsuD (41.6 kDa) (Fig. 5B, lane 4). The two bands observed in both lane 4 and lane 7 at approximately 43 and 80 kDa were due to dimerization of SsuE and SsuD, respectively, as similar results were observed in control experiments. The sample following DTT treatment showed a decrease in the intensity of the 63-kDa band representing the putative cross-linked SsuE-SsuD species, with a concomitant increase in the intensity of the monomeric SsuE and SsuD bands (Fig. 5B, lane 5). Experiments involving the cross-linking of only SsuE were performed to determine if a 63-kDa band was observed. A cross-linked trimer of SsuE would have a molecular mass similar to that of a cross-linked monomer of SsuE and SsuD. There was no band observed at 63 kDa with SsuE only, which indicated that the 63-kDa band represented the cross-linked monomers of SsuE and SsuD. In addition, cross-linking experiments with SsuE and bovine serum albumin were performed to verify that the higher-molecular-mass band was not caused by protein aggregation. The higher-molecular-mass band at 63 kDa was not observed with any of the controls tested (data not shown), further confirming the specificity of protein-protein interactions between SsuE and SsuD.
Binding of SsuD to FMN-bound SsuE.
Fluorescence spectroscopy experiments were performed to determine the KD for protein-protein interactions and binding stoichiometry. When FMN binds SsuE, there is a decrease in the flavin fluorescence at an excitation wavelength of 450 nm. The KD of SsuD binding to SsuE was determined by titrating the FMN-bound SsuE solution with SsuD. The increase in flavin fluorescence was monitored at an excitation wavelength of 450 nm following each addition of SsuD (Fig. 6A). The concentration of SsuD bound was plotted against the total SsuD to obtain a KD value of 0.0022 ± 0.0010 μM (Fig. 6B) with a 1:1 stoichiometric ratio between SsuE and SsuD. The increase in fluorescence associated with SsuD binding to FMN-bound SsuE could be caused by release of the bound flavin. Filtration experiments utilizing a 10,000-molecular-weight-cutoff Amicon Ultra-4 centrifugation filter from Millipore (Bedford, MA) were performed to confirm that the flavin is still bound to SsuE following the addition of SsuD. If the flavin is still bound, then it should be retained with the protein in the retentate, while free flavin would be located in the filtrate. A UV-visible spectrum following filtration showed that the flavin was retained in the retentate, indicating that the FMN remains bound to SsuE in the presence of SsuD (Fig. 7). The flavin was located in the filtrate in filtration experiments with free flavin only. These results confirmed that the increase in the intrinsic fluorescence associated with SsuD binding is due to protein-protein interactions. These data are consistent with the results from CD spectroscopy experiments in which the FMN-bound SsuE environment is altered in the presence of SsuD.
FIG. 6.

Fluorometric titration of SsuD to FMN-bound SsuE. (A) Emission intensity of fluorometric titration of FMN-bound SsuE with SsuD. A 0.40 μM concentration of FMN-bound SsuE was titrated with between 0.02 and 0.95 μM SsuD. Emission intensity measurements from 470 to 650 nm were made using excitation at 450 nm. (B) The concentration of SsuD bound was plotted against the total SsuD and was fit to equation 1 (see text) in experimental procedures using the Kaleidagraph software (Abelbeck Software, Reading, PA).
FIG. 7.

UV-visible spectra for filtration of FMN in the presence of SsuE and SsuD proteins. The flavin spectra were obtained with 20 μM FMN in 25 mM phosphate buffer (pH 7.5), 10% glycerol, and 100 mM NaCl at 25°C in the presence of 20 μM of SsuE and SsuD proteins in a 2.0-ml total volume. The spectra were taken before filtration (solid line) and after filtration (dashed line) on a UV-visible spectrophotometer at a wavelength of 450 nm.
DISCUSSION
Reduced flavins are utilized by various organisms for a wide range of biological processes. For most flavoproteins the reductive and oxidative half-reactions of the flavin occur within the same protein. Interestingly, the production and utilization of this intermediate species in certain microbial systems involve both a reductase and an oxygenase enzyme that independently catalyze each half-reaction. Although the role of reduced flavin in redox reactions has been known for some time, the mechanism of reduced flavin transfer in two-component enzyme systems is still poorly understood. Therefore, understanding the mechanism of reduced flavin transfer is extremely important to explore the role of reduced flavin in cellular systems. Free reduced flavin is typically considered unstable under aerobic conditions and is easily oxidized, generating hydrogen peroxide (3, 11, 30). The hydrogen peroxide generated can be further oxidized leading to the production of oxygen radicals that can damage cellular macromolecules. Due to its unique properties, it is important to stabilize the reduced flavin from unproductive oxidation. In most flavoproteins the flavin is a bound prosthetic group, and the active site of the protein protects the reduced flavin from further oxidation. In systems where the reductive and oxidative half-reactions occur in separate proteins, a possible mechanism of reduced flavin transfer is through protein-protein interactions between the reductase and oxygenase components. A channeling mechanism would protect the reduced flavin from futile oxidation before catalysis can occur.
The focus of these studies was to investigate the role of protein-protein interactions between the alkanesulfonate monooxygenase proteins through several biophysical approaches. Results from the affinity chromatography experiments showed that the two proteins interact with a relatively strong binding affinity, as the proteins were shown to coelute from the column (Fig. 2, lane 4). This result is highly convincing, since a large volume of buffer (10 to 20 times the column volume) was used to wash the column prior to elution. The possibility that SsuE and SsuD coelute due to nonspecific protein interactions and binding was demonstrated in control experiments. If SsuD was not preloaded on the Ni-NTA column, the SsuE protein eluted from the column with a lower concentration of imidazole buffer. In addition, a catalytically unrelated protein was unable to interact with His-tagged SsuD. This supports the argument that the coelution of SsuE and SsuD was due to protein interactions and was not caused by protein aggregation or nonspecific interactions of SsuE with the column. While the amount of SsuE was not equimolar with SsuD, this would be expected if the protein binding sites on SsuD were inaccessible due to alternative arrangements of SsuD on the column.
The identification of SsuE and SsuD interactions was further probed with a trifunctional cross-linker. The silver-stained gel showed a protein band at approximately 63 kDa, which correlates with monomers of SsuE and SsuD covalently bound with the cross-linker reagent. In control experiments with SsuE or SsuD only, there was no higher-molecular-mass band observed at 63 kDa. Therefore, the band observed likely corresponds to the SsuE and SsuD proteins. The results from these studies further support the presence of protein interactions between SsuE and SsuD and provide an approach to determine the locations and identities of specific amino acids involved in protein interactions.
Although the two proteins are shown to interact in the absence of substrates, this interaction was not caused by any major conformational changes in the secondary structure of each protein as indicated by far-UV CD spectroscopy. Any changes that occur due to protein-protein interactions may lead to only minor perturbations in the protein structure that would not be translated to overall secondary structural changes. Several groups have utilized visible CD spectroscopy to analyze perturbations in the flavin environment within the active site (2, 8). The 100-fold-greater affinity for flavin binding to SsuE over SsuD provided an ideal means to analyze changes in the FMN-bound SsuE active site upon SsuD binding (7). While there were no major changes in the visible absorbance from 375 to 550 nm, a slight decrease in the negative molar ellipticity was observed between 300 and 375 nm with SsuD present. The change was not due to protein aggregation, because bovine serum albumin did not elicit the same decrease. These results suggest that the change in the flavin environment was due to specific protein interactions between SsuE and SsuD and not to random aggregations. This argument was supported by the data that the flavin environment did not change in the presence of SsuD alone (K. Abdurachim and H. R. Ellis, unpublished data). An alteration in the flavin environment would be expected if SsuE exists in a more open conformation in the presence of SsuD.
The decreased molar ellipticity observed by visible CD spectroscopy suggested that there was a change in the flavin environment that could be further analyzed by fluorescence spectroscopy. There is an observed decrease in fluorescence upon flavin binding to SsuE, which was previously used to determine the KD for binding of the oxidized flavin to SsuE. Titration of FMN-bound SsuE with SsuD resulted in an increase in fluorescence that reached a saturation point. Independent filtration experiments confirmed that the flavin was still bound to SsuE in the presence of SsuD, and therefore the increase in absorbance was not caused by the release of the bound flavin. The KD value determined from these studies was 0.0022 ± 0.0010 μM, which indicates a relatively strong interaction between these proteins. Analytical ultracentrifugation studies have shown that SsuE is a dimer and SsuD is a tetramer at the concentration used in these studies (7). Therefore, the 1:1 stoichiometric ratio for monomeric binding between SsuE and SsuD supports a structural model involving four dimers of SsuE bound to a tetramer of SsuD.
The results obtained suggest that static interactions occur between SsuE and SsuD and that these interactions may play a role in the transfer of reduced flavin. If these static interactions occur during catalysis, the results are consistent with a channeling mechanism for reduced flavin transfer between SsuE and SsuD. This observation supports previous findings in our laboratory that the catalytic mechanism of SsuE is altered in the presence of SsuD, and it provides physical evidence for these interactions (7). A change in the catalytic mechanism between a single enzyme and an enzyme-coupled reaction is often an indicator of protein channeling (13). It was previously reported that SsuD expressed in crude cell lysate is able to accept reduced flavins from alternate flavin reductases in addition to SsuE. These results suggest that there is no specificity for flavin transfer between SsuE and SsuD. However, it is not known if other flavin reductases are expressed to any significant level under sulfur starvation conditions. The ability of monooxygenase enzymes from two-component systems to accept reduced flavin from oxidoreductases associated with other systems could suggest a common binding site unique to this family of enzymes. Steady-state kinetic studies and fluorescence spectroscopy have also provided evidence for protein interactions between FMN reductase and bacterial luciferase from Vibrio harveyi (27). The flavin reductases specific for bacterial luciferase show little sequence identity with SsuE; however, bacterial luciferase and SsuD show very similar overall structures even though they possess only 15% amino acid identity. Further analysis has shown that many of the conserved residues located in the active site of bacterial luciferase are also found in the putative active site of SsuD (6). The conserved structure of these proteins may suggest that they share a common mechanism for flavin transfer between the reductase and oxygenase components. An alternative to a direct channeling mechanism has been proposed for the styrene monooxygenase two-component system through kinetic studies, which provide evidence for the formation of transient protein interactions during flavin transfer (14). In this mechanism the isoalloxazine ring of the flavin is reduced by the reductase component while still bound to the monooxygenase.
Substrate channeling has become a recognized concept to explain the transfer of a metabolite between proteins (9, 22-27). The formation of a molecular channel has some physiological advantages for cellular systems, including the protection of unstable intermediates from the solvent or breakdown due to side reactions during transfer (23). One classic example in support of a substrate channeling mechanism is found in tryptophan synthase from Salmonella enterica serovar Typhimurium, where the metabolic intermediate (indole) is thought to channel from the active site of the α subunit to the active site of the β subunit (1, 10, 20, 22). Substrate channeling has also been well characterized in carbamoyl phosphate synthase, a protein with three distinct active sites (17, 21, 26). It has been fully established that reduced flavin is a very reactive intermediate and is easily oxidized in the presence of molecular oxygen, generating species toxic to the cells. There are advantages for substrate channeling that are relevant to the properties of reduced flavin, including the protection of a reactive intermediate, the sequestering of toxic molecules, and the improvement of catalytic efficiency (1, 20, 23-25). The existence of protein-protein interactions between SsuE and SsuD supports a mechanism where the flavin is channeled or shared by the two proteins but eliminates the possibility of a diffusion mechanism. In the case of the alkanesulfonate monooxygenase system, the latter can be investigated through pre-steady-state kinetic analysis to determine if the active sites of SsuE and SsuD proteins are involved in allosteric communication.
Acknowledgments
We thank Douglas Goodwin and Benlian Gao for helpful discussions on the manuscript.
This research was supported in part by the National Science Foundation (grant MCB-0545048 to H.R.E.).
Footnotes
Published ahead of print on 22 September 2006.
REFERENCES
- 1.Anderson, K. S., E. W. Miles, and K. A. Johnson. 1991. Serine modulates substrate channeling in tryptophan synthase. A novel intersubunit triggering mechanism. J. Biol. Chem. 266:8020-8033. [PubMed] [Google Scholar]
- 2.Auer, H. E., and F. E. Frerman. 1980. Circular dichroism studies of acyl-CoA dehydrogenase and electron transfer flavoprotein. J. Biol. Chem. 255:8157-8163. [PubMed] [Google Scholar]
- 3.Bruice, T. C. 1982. A progress report on studies of the activation of molecular oxygen by dihydroflavins, p. 265-277. In V. Massey and C. H. Williams (ed.), Flavins and flavoproteins. Elsevier North-Holland, Inc., New York, N.Y.
- 4.Eichhorn, E., C. A. Davey, D. F. Sargent, T. Leisinger, and T. J. Richmond. 2002. Crystal structure of Escherichia coli alkanesulfonate monooxygenase SsuD. J. Mol. Biol. 324:457-468. [DOI] [PubMed] [Google Scholar]
- 5.Eichhorn, E., J. R. van der Ploeg, and T. Leisinger. 1999. Characterization of a two-component alkanesulfonate monooxygenase from Escherichia coli. J. Biol. Chem. 247:26639-26646. [DOI] [PubMed] [Google Scholar]
- 6.Fisher, A. J., T. B. Thompson, J. B. Thoden, T. O. Baldwin, and I. Rayment. 1996. The 1.5-Å resolution crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem. 271:21956-21968. [DOI] [PubMed] [Google Scholar]
- 7.Gao, B., and H. R. Ellis. 2005. Altered mechanism of the alkanesulfonate FMN reductase with the monooxygenase enzyme. Biochem. Biophys. Res. Commun. 331:1137-1145. [DOI] [PubMed] [Google Scholar]
- 8.Goetzman, E. S., M. He, T. V. Nguyen, and J. Vockley. 2006. Functional analysis of acyl-CoA dehydrogenase catalytic residue mutants using surface plasmon resonance and circular dichroism. Mol. Gen. Metabol. 87:233-242. [DOI] [PubMed] [Google Scholar]
- 9.Huang, X., H. M. Holden, and F. M. Raushel. 2001. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 70:149-180. [DOI] [PubMed] [Google Scholar]
- 10.Hyde, C. C., S. A. Ahmed, E. A. Padlan, E. W. Miles, and D. R. Davies. 1988. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 263:17857-17871. [PubMed] [Google Scholar]
- 11.Imlay, J. A. 2003. Pathways of oxidative damage. Annu. Rev. Microbiol. 57:395-418. [DOI] [PubMed] [Google Scholar]
- 12.Jeffers, C. E., and S.-C. Tu. 2001. Differential transfers of reduced flavin cofactor and product by bacterial flavin reductase to luciferase. Biochemistry 40:1749-1754. [DOI] [PubMed] [Google Scholar]
- 13.Jeffers, C. E., J. C. Nichols, and S.-C. Tu. 2003. Complex formation between Vibrio harveyi luciferase and monomeric NADPH:FMN oxidoreductase. Biochemistry 42:529-534. [DOI] [PubMed] [Google Scholar]
- 14.Kantz, A., F. Chin, N. Nallamothu, T. Nguyen, and G. T. Gassner. 2005. Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch. Biochem. Biophys. 442:102-116. [DOI] [PubMed] [Google Scholar]
- 15.Kertesz, M. A. 2000. Riding the sulfur cycle—metabolism of sulfonates and sulfate esters in gram-negative bacteria. FEMS Microbiol. Rev. 24:135-175. [DOI] [PubMed] [Google Scholar]
- 16.Kertesz, M. A., T. Leisinger, and A. M. Cook. 1993. Proteins induced by sulfate limitation in Escherichia coli, Pseudomonas putida, or Staphylococcus aureus. J. Bacteriol. 175:1187-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim, J., and F. M. Raushel. 2004. Perforation of the tunnel wall in carbamoyl phosphate synthetase derails the passage of ammonia between sequential active sites. Biochemistry 43:5334-5340. [DOI] [PubMed] [Google Scholar]
- 18.Lei, B., and S.-C. Tu. 1998. Mechanism of reduced flavin transfer from Vibrio harveyi NADPH:FMN oxidoreductase to luciferase. Biochemistry 37:14623-14629. [DOI] [PubMed] [Google Scholar]
- 19.Louie, T. M., X. S. Xie, and L. Xun. 2003. Coordinate production and utilization of FADH2 by NAD(P)H-flavin oxidoreductase and 4-hydroxyphenylacetate 3-monooxygenase. Biochemistry 42:7509-7517. [DOI] [PubMed] [Google Scholar]
- 20.Miles, E. W. 2001. Tryptophan synthase: a multienzyme complex with an intramolecular tunnel. Chem. Rec. 1:140-151. [DOI] [PubMed] [Google Scholar]
- 21.Miles, B. W., and F. M. Raushel. 2000. Synchronization of the three reaction centers within carbamoyl phosphate synthetase. Biochemistry 39:5051-5056. [DOI] [PubMed] [Google Scholar]
- 22.Miles, E. W., S. Rhee, and D. R. Davies. 1999. The molecular basis of substrate channeling. J. Biol. Chem. 274:12193-12196. [DOI] [PubMed] [Google Scholar]
- 23.Ovadi, J. 1991. Physiological significance of metabolic channelling. J. Theor. Biol. 152:1-22. [PubMed] [Google Scholar]
- 24.Raushel, F. M., J. B. Thoden, and H. M. Holden. 2003. Enzymes with molecular tunnels. Acc. Chem. Res. 36:539-548. [DOI] [PubMed] [Google Scholar]
- 25.Srere, P. A. 1987. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56:89-124. [DOI] [PubMed] [Google Scholar]
- 26.Thoden, J. B., X. Huang, F. M. Raushel, and H. M. Holden. 2002. Carbamoyl-phosphate synthetase. Creation of an escape route for ammonia. J. Biol. Chem. 277:39722-39727. [DOI] [PubMed] [Google Scholar]
- 27.Tu, S.-C. 2001. Reduced flavin: donor and acceptor enzymes and mechanisms of channeling. Antioxid. Redox Signal. 3:881-897. [DOI] [PubMed] [Google Scholar]
- 28.van der Ploeg, J. R., E. Eichhorn, and T. Leisinger. 2001. Sulfonate-sulfur metabolism and its regulation in Escherichia coli. Arch. Microbiol. 176:1-8. [DOI] [PubMed] [Google Scholar]
- 29.van Der Ploeg, J. R., R. Iwanicka-Nowicka, T. Bykowski, M. M. Hryniewicz, and T. Leisinger. 1999. The Escherichia coli ssuEADCB gene cluster is required for the utilization of sulfur from aliphatic sulfonates and is regulated by the transcriptional activator Cbl. J. Biol. Chem. 274:29358-29365. [DOI] [PubMed] [Google Scholar]
- 30.Walsh, C. 1978. Chemical approaches to the study of enzymes catalyzing redox transformations. Annu. Rev. Biochem. 47:881-931. [DOI] [PubMed] [Google Scholar]