

Abstract
Cisplatin is one of the most effective chemotherapeutics, but its usefulness is limited by its toxicity to normal tissues, including cells of the kidney proximal tubule. The purpose of these studies was to determine the mechanism of cisplatin cytotoxicity. It was shown in vivo that cisplatin administration induces upregulation of the gene for the p21 cyclin-dependent kinase (cdk) inhibitor in kidney cells. This protein is a positive effector on the fate of cisplatin-exposed renal tubule cells in vivo and in vitro; adenoviral transduction of p21 completely protected proximal tubule cells from cisplatin toxicity. Herein is reported that cdk2 inhibitory drugs protect kidney cells in vivo and in vitro, that transduction of kidney cells in vitro with dominant-negative cdk2 also protected, and that cdk2 knockout cells were resistant to cisplatin. The cdk2 knockout cells regained cisplatin sensitivity after transduction with wild-type cdk2. It is concluded that cisplatin cytotoxicity depends on cdk2 activation and that the mechanism of p21 protection is by direct inhibition of cdk2. This demonstrated the involvement of a protein that previously was associated with cell-cycle progression with pathways of apoptosis. It also was demonstrated that this pathway of cisplatin-induced cell death can be interceded in vivo to prevent nephrotoxicity.
Cisplatin is one of the most effective chemotherapeutic agents against testicular and bladder tumors, head and neck, ovarian, breast, and lung cancers, and refractory non-Hodgkin’s lymphomas (1,2). The major adverse effect of cisplatin use is nephrotoxicity, in which kidney proximal tubule cells are especially sensitive (3). It is likely that its anticancer activity depends on formation of DNA intrastrand cross-links (4). Several distinct mechanisms have been proposed for cisplatin cytotoxicity in renal tubule cells, including direct DNA damage (5), caspase activation (6), mitochondrial dysfunction (7), formation of reactive oxygen species (8), effects on the endoplasmic reticulum (9), and activation of TNF-α apoptotic pathways (10). However, it is unclear whether cisplatin nephrotoxicity depends on any of these pathways or these apoptotic cascades merely amplify more proximal initiated cell death signals.
We have shown in vivo that kidney cells entered the cell cycle after cisplatin administration and that the gene for the p21Cip1/WAF1 cell-cycle inhibitor was induced simultaneously (11). The p21 protein interacts with several members of the cell cycle to regulate cell-cycle progression (12–14), and its induction is a positive effector on the fate of renal tubule cells both in vivo and in vitro (15,16). In addition, we recently reported that cells cultured from mouse proximal tubules were completely protected from cisplatin cytotoxicity by adenoviral transduction of human p21 (17). The activity of cyclin-dependent kinase 2 (cdk2), a serine/threonine kinase whose main function is the phosphorylation of substrates necessary for cell-cycle progression (reviewed in reference [18]), is inhibited by binding with p21. We now report that cisplatin cytotoxicity in vitro depends on cdk2 activity and that both functional and morphologic nephrotoxicity in vivo can be prevented using cdk2-inhibitory drugs. The dependence of the apoptotic pathway on cdk2 was demonstrated by (1) using separate interactive moieties of p21, in which only the cdk2 binding region effectively protected (19); (2) using different cdk2 inhibitory drugs (17); (3) using a transducing cdk2 dominant negative adenovirus; and (4) using cdk2 knockout cells. These results demonstrated that induction of cisplatin cytotoxicity in kidney cells depends on cdk2 activity and can be prevented both in vitro and in vivo by cdk2 inhibition.
Materials and Methods
Animals and Administration of Drugs
Experiments were performed on 10- to 12-wk-old wild-type 129Sv mice that weighed 22 to 28 g. The mice were maintained on a standard diet, and water was available freely. Cisplatin was administered by a single intraperitoneal injection of 20 mg/kg, a dosage that produces severe acute renal failure in mice (11). Some mice also received daily intravenous injections of purvalanol (Tocris Cookson, Inc., Ellisville, MO) at 30 mg/kg dissolved at 4 mg/ml in a 5:7:13 (vol/vol/vol) mixture of DMSO:PEG400:10% Kollidon 17PF. Animals were killed painlessly with methods of euthanasia approved by the Panel on Euthanasia of the American Veterinary Medical Association. The induction of acute renal failure was monitored by following creatinine concentration in serum that was obtained by retro-orbital bleeding using a commercial kit (Sigma Diagnostics No. 555, St. Louis, MO).
Morphologic Assessment
At various times after cisplatin treatment, kidneys were removed, immersed in 4% neutral-buffered formaldehyde, and fixed for 48 to 72 h. The tissues were paraffin embedded and processed for light microscopy. Sections of 5 μm were stained with hematoxylin-eosin and periodic acid-Schiff, and histologic criteria were determined (20). The following parameters were chosen as indicative of morphologic damage to the kidney after cisplatin injection: Brush border loss, red blood cell extravasation, tubule dilation, tubule degeneration, tubule necrosis, and tubular cast formation. These parameters were evaluated as described previously (20) on a scale from 0 to 4: Not present (0), mild (1), moderate (2), severe (3), and very severe (4). Each parameter was determined on at least five different animals by a histologist who was blinded to the source of the sections. Statistical significance was assessed by the two-sided t test for independent samples.
Cell Culture
Cells were grown at 37°C in 5% CO2. Mouse kidney proximal tubule cells (TKPTS) (21) were cultured in DMEM + Ham’s F-12 medium supplemented with 50 μU/ml insulin and 7% FBS. Mouse embryonic fibroblasts (MEF) that were derived from cdk2 knockout mice (22) were grown in DMEM with 10% FBS. These cells were obtained from cdk2 (−/−) mouse embryos (13.5 d postcoitus) and were spontaneously immortalized in a 3T3 protocol after approximately 10 passages (22). Cisplatin was added to cultures, when indicated, to a final concentration of 25 μM (TKPTS) or 35 μM (MEF) when cells were approximately 75% confluent, and the cells were grown for an additional 24 h. TKPTS and MEF that expressed wild-type cdk2 had similar sensitivity to cisplatin; apoptotic cell death occurred to the same extent 24 h after exposure to 25 μM cisplatin. Adenoviruses were added to a final multiplicity of infection of 100 18 h before cisplatin. Purvalanol and roscovitine were dissolved in DMSO and added 4 h before cisplatin to a final concentration of 9 or 45 μM, respectively. As reported previously (17), roscovitine extended the cell cycle, rather than causing cell-cycle arrest. Purvalanol had a similar effect on the TKPTS. Except as noted, culture conditions followed the same schedule: Cultured cells were maintained for 30 h after splitting before adenovirus was added, cell-cycle inhibitors were added at 44 h, cisplatin was added at 48 h, and treatments were as indicated. The cells were harvested 72 h after splitting.
Cell Death Determination
FACS Analysis
Cells were harvested by trypsinization, pooled with the culture medium that contained floating cells, and collected by centrifugation (10 min, 500 × g). The cell pellets were resuspended in 0.3 ml of PBS that contained 5 mM EDTA, and 0.7 ml of ethanol was added. Cells were incubated at 4°C for 16 h, collected by centrifugation (10 min, 2000 rpm), and resuspended in 0.5 ml of PBS-EDTA. RNase A was added (50 μl, 10 mg/ml) and the suspension was incubated at 25°C for 30 min. Propidium iodide was added (450 μl, 100 μg/ml), and the samples were analyzed using FACSCalibur (Becton Dickinson, Rockville, MD). At least 10,000 cells were analyzed for each culture condition (Figures 2 and 3), and values (in Figure 2) were based on three separate cultures. The percentage of cells in sub-G1/G0 (apoptotic fraction) (17,23), G1/G0, S, and G2/M phases was determined using a cell-cycle analysis program (WinMDI 2.8; Scripps Institute, San Diego, CA). The cells in the subdiploid (sub-G1/G0) region of the histogram are classified as apoptotic, probably as a result of the generation and loss of low molecular weight DNA fragments during apoptosis. These analyses were performed on at least four separate cultures.
Figure 2.
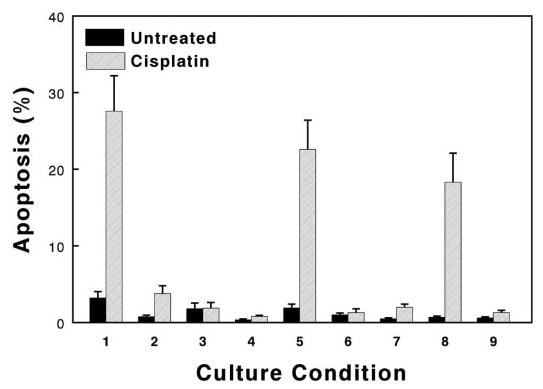
Bar graph of FACS analyses for apoptosis (percentage of cells in sub-G1/G0 fraction). Values represent means (± SE) of cells that were treated with and without 24 h of cisplatin (as indicated), using either TKPTS (lanes 1 through 6) or mouse embryonic fibroblasts (MEF) that were derived from cdk2 knockout mice (lanes 7 through 9). In addition to possible cisplatin exposure, cells were untreated (lanes 1 and 7); treated with 100 multiplicity of infection adenovirus that expressed p21 (lane 2), wild-type cdk2 (lanes 5, 8, and 9), or DN-cdk2 (lane 6); treated with 45 μM roscovitine (Lane 3); or treated with 9 μM purvalanol (lanes 4 and 9), as described in Materials and Methods.
Figure 3.

Representative analyses for apoptosis. Cells (TKPTS) were untreated (A, C, and E) or treated with 25 μg/ml cisplatin for 24 h (B, D, and F). (A and B) Light microscopy of cells before harvesting (scale indicated). (C and D) Hoechst 33258 staining of formaldehyde-fixed cells that were grown on coverslips (scale indicated; typical apoptotic nuclei indicated by arrows); (E and F) FACS analysis of propidium iodide–stained ethanol-fixed cells. Cell-cycle analysis (cells in G0 + G1, S, and G2 + M phases) shown in first diagram.
Hoechst Staining
Cells were grown on coverslips as described above. After times as indicated, cells were rinsed with PBS, fixed for 5 min with neutral-buffered formaldehyde, and stained with Hoechst 33258 (10 μg/ml in PBS) for 10 min. Under these conditions, Hoechst 33258 stains the nuclei of all cells blue when examined by fluorescence microscopy using a DAPI filter. The nuclei of apoptotic cells display chromatin condensation and/or nuclear fragmentation, whereas normal cell nuclei are stained homogeneously. Apoptosis was assessed in at least 300 cells using a fluorescence microscope with a DAPI filter and a ×40 objective.
Light Microscopy
Cells were photographed with an inverted microscope (Nikon Eclipse TE200, Melville, NY) using Hoffman optics before harvesting.
Adenoviruses
The p21-expression adenovirus was obtained from Dr. Wafik El-Deiry (University of Pennsylvania, Philadelphia, PA [13]). Green fluorescence protein (GFP)-expression adenoviruses that expressed p21, p21 fragments, and cdk2 were constructed in our laboratory according to protocols and materials supplied by Dr. Bert Vogelstein (Johns Hopkins, Baltimore, MD [24]). Human p21 cDNA was obtained from Dr. Bert Vogelstein (13); human cdk2 wild-type and cdk2 dominant negative (DN-cdk2) cDNA were obtained from Dr. Sander van den Heuvel (Massachusetts General Hospital, Boston, MA [25]). The wild-type and DN-cdk2 adenoviruses were constructed by insertion of a BamHI fragment that contained the cDNA for the protein into the Bgl II site of the pAdTrack-CMV plasmid. After selection for clones with the proper orientation of cDNA insertion by restriction analysis, adenoviruses were constructed as described above.
Western Blot Analysis
Proteins were extracted from TKPTS and MEF using a lysis buffer that contained 50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 0.5% NP-40 with phosphatase inhibitor I and II, and proteinase inhibitor (Sigma, St. Louis, MO). Western blot analyses were as described previously (17,19). In brief, protein concentration was determined using a Bio-Rad protein assay (Hercules, CA). Protein (100 μg/lane) was electrophoresed using 12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membrane. After blocking with 5% nonfat dry milk in TBST, the membrane was incubated at 4°C overnight with primary antibody. After washing, horseradish peroxidase–conjugated secondary antibody was applied. Proteins that bound to the secondary antibody were visualized using ECL (Amersham Biosciences, Piscataway, NJ).
Kinase Assay for cdk2
For assay of in vivo activity, kidneys were homogenized with a Teflon glass homogenizer in buffer that contained 10 mM HEPES (pH 7.6), 25 mM KCl, 1 mM EDTA, 10% glycerol, 1.8 M sucrose, 0.15 mM spermine, 0.5 mM spermidine, 0.5 mM dithiothreitol, and phosphatase and proteinase inhibitors as described above. The homogenate was layered over 2.2 ml of this buffer, and nuclei were pelleted by centrifugation at 24,000 rpm in an SW60 rotor. Nuclei from two mouse kidneys were resuspended in 1 ml of cold lysis buffer (as discussed in Western Blot Analysis), sonicated, and centrifuged for 10 min in an Eppendorf centrifuge. The supernatant was used for histone H1 kinase activity (see next). For assay of in vitro activity, TKPTS were washed twice with PBS and lysed in cold lysis buffer (as discussed in Western Blot Analysis). Samples were kept on ice for 30 min and centrifuged for 30 min at 10,000 × g. The supernatant was used for histone H1 kinase activity (see next).
Kinase activity of cdk2 was assayed by a modified histone H1 kinase assay (19,26,27). Briefly, protein extracts (200 μg) were immunoprecipitated by agarose-immobilized anti-cdk2 antibody for 4 h at 4°C with constant rocking and washed three times with lysis buffer and once with kinase buffer that contained 20 mM Tris-Cl (pH 7.4), 10 mM MgCl2, and 1 mM dithiothreitol. Agarose beads were resuspended in 20 μl of kinase buffer that contained 2 μg of histone H1, 20 μM ATP, and 10 μCi γ-32P-ATP and incubated for 30 min at 30°C. Samples were boiled and electrophoresed by PAGE as described above and autoradiographed.
Results
Correlation of cdk2 Activity with Cell Death after Cisplatin Exposure In Vitro and In Vivo
Cdk2 activity was determined in TKPTS and in mouse kidney cell nuclei before and after cisplatin treatment as a function of histone H1 kinase activity associated with an anti-cdk2 immunoprecipitate (Figure 1). The background cdk2 activity in TKPTS (Figure 1A, lane 1) was unaffected by transduction with a GFP-expression adenovirus (lane 2) but was increased by treatment with cisplatin (lane 3). Transduction of cells with adenoviruses that expressed full-length p21 (lane 4), the cdk2-inhibitory region of p21 (lane 5), or DN-cdk2 (lane 7) before cisplatin exposure lowered cdk2 activity to below background levels. Similarly, treatment with purvalanol and cisplatin virtually eliminated cdk2 activity (lane 9). However, transduction with adenovirus that expressed a p21 protein that was missing the cdk2-inhibitory region (lane 6) or wild-type cdk2 (lane 8) had no effect on the rise of cisplatin-induced cdk2 activity. A similar effect on cdk2 activity by cisplatin and purvalanol also was found in vivo (Figure 1B). Without cisplatin treatment, a background level of cdk2 activity was measured (Figure 1B, lanes 1, 2, and 7). Treatment with cisplatin for 24 or 48 h increased the cdk2 activity (lanes 3 and 5, respectively). Treatment with purvalanol without cisplatin (lane 2), with 24 h of cisplatin (lane 4), or with 48 h of cisplatin (lane 6) lowered cdk2 activity to control levels, although not to the extreme extent that was found in vitro. A control in which immunoprecipitation was performed without anti-cdk2 antibody did not contain any measurable kinase activity (lane 8).
Figure 1.
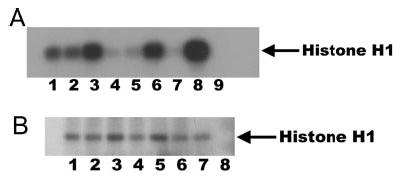
(A) Cyclin-dependent kinase (Cdk2) activity in mouse kidney proximal tubule cells (TKPTS) correlates with cell death after cisplatin treatment. Background activity in untreated cells (lane 1), cells after transduction with a green fluorescence protein (GFP)-expression adenovirus (lane 2), or cells that were treated with 9 μM purvalanol (lane 9). Cells were treated for 24 h with cisplatin (lanes 3 through 9). In addition to cisplatin, cells were transduced with adenoviruses that expressed full-length p21 (lane 4), the cdk2-inhibitory region of p21 (lane 5), the C-terminal region of p21 missing the cdk2-inhibitory region (lane 6), dominant negative cdk2 (DN-cdk2; lane 7), or wild-type cdk2 (lane 8). (B) Cdk2 activity in mouse kidney nuclei after cisplatin treatment. Mice either were not treated with cisplatin (lanes 1, 2, and 7) or were treated with 20 mg/kg cisplatin (lanes 3 through 6) for 24 h (lanes 3 and 4) or 48 h (lanes 5 and 6). In addition, some mice received daily intravenous injections of purvalanol at 30 mg/kg (lanes 2, 4, and 6) as described in Materials and Methods. A control was included in which agarose beads that did not contain anti-cdk2 antibody was used for immunoprecipitation of extract (lane 8).
Protection of Cultured TKPTS by cdk2 Inhibition
Apoptosis was determined by FACS analysis (Figures 2 and 3) of propidium iodide–stained TKPTS, as described in Materials and Methods. Morphologic changes were confirmed by light microscopy, and apoptotic changes in nuclear structure were confirmed by fluorescence microscopy of Hoechst 33258–stained cells (Figure 3).
TKPTS that were cultured for 72 h in the absence of cisplatin had a low level of apoptosis (3.2 ± 0.85%) as determined by FACS analysis (Figure 2, lane 1). Transduction with p21-adenovirus lowered the percentage of apoptotic cells to 0.76 ± 0.20% (Figure 2, lane 2). The addition of cdk inhibitors had little effect on background apoptosis; roscovitine slightly lowered the level of apoptosis to 1.82 ± 0.74% (Figure 2, lane 3), and purvalanol decreased the percentage of apoptotic cells to 0.36 ± 0.10% (Figure 2, lane 4).
Administration of cisplatin in the absence of cdk inhibition increased apoptosis to 27.6 ± 4.6% of the cells (Figure 2, lane 1). However, addition of p21-adenovirus 18 h before cisplatin treatment or treatment with roscovitine or purvalanol 4 h before cisplatin reduced the level of apoptosis to background levels: 3.8 ± 1.0% (Figure 2, lane 2), 1.89 ± 0.74 (Figure 2, lane 3), and 0.8 ± 0.15% (Figure 2, lane 4), respectively. Addition of adenovirus-expressing GFP had no effect on the percentage of cells in apoptosis (data not shown).
Because the p21 protein had been reported to bind and repress several proteins with proapoptotic functions and chemical cdk2 inhibitors may inhibit other kinases with potential proapoptotic functions, we transduced TKPTS with wild-type and DN-cdk2 to determine whether specifically inhibiting cellular cdk2 activity would protect from cisplatin cytotoxicity. Transduction using adenovirus that expressed these cdk2 had no effect on the low levels of background apoptosis that was detected without cisplatin treatment (Figure 2, lanes 5 and 6). Similarly, wild-type cdk2 had little effect on cytotoxicity in the presence of cisplatin, in which 22.6 ± 3.8% of the cells were apoptotic (Figure 2, lane 5). However, transduction with DN-cdk2 reduced the cisplatin-induced apoptosis to 1.3 ± 0.5% (Figure 2, lane 6).
The recent demonstration of the viability of mice that are homozygotic for a knockout of the cdk2 gene showed that this kinase is not required for cell-cycle progression (22,28). Embryonic fibroblasts that were derived from cdk2 knockout mice were resistant to cisplatin, in which apoptosis occurred in only 2.0 ± 0.4% of cells (Figure 2, lane 7). To determine whether this relative insensitivity to cisplatin cytotoxicity depended on the lack of cdk2, the cells were transduced with an adenovirus that expressed wild-type cdk2. This converted the cells to cisplatin sensitive, in which cisplatin induced apoptosis in 18.3 ± 3.8% of the cells (Figure 2, lane 8). Because exposure of cells to adenovirus per se also can induce apoptosis, we confirmed that the increased sensitivity to cisplatin was induced by cdk2 and not by adenovirus by exposing these cells to cdk2 inhibitor (purvalanol), which lowered the cisplatin-induced apoptosis to 1.3 ± 0.3% in the presence of transduced wild-type cdk2 (Figure 2, lane 9). We also confirmed that the MEF knockout cells reacted to cisplatin by inducing p21 (Figure 4) to the same extent that TKPTS are stimulated, even though these knockout cells largely are protected from cisplatin cytotoxicity.
Figure 4.
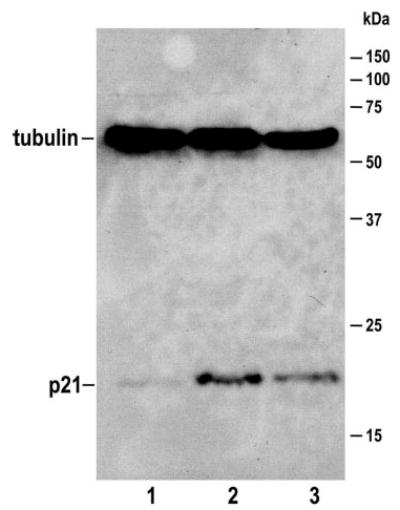
Induction of p21 in cultured cells. Cdk2 knockout MEF were untreated (lane 1), or treated with 25 μM cisplatin for 24 h (lane 2). TKPTS were treated with 25 μM cisplatin for 24 h (lane 3) as a positive control for p21 induction. Proteins were electrophoresed using 12% SDS-PAGE and transferred to polyvinylidene difluoride membrane. After blocking with 5% nonfat dry milk in TBST, membranes were incubated at 4°C overnight with primary antibodies to p21 and tubulin. After washing, horseradish peroxidase–conjugated second antibody was applied. Proteins that bound to the second antibody were visualized using ECL. Tubulin (55 kD) was used as a loading control. The 21-kD p21 and 55-kD tubulin are indicated.
Cdk2 Inhibition Ameliorates Cisplatin Nephrotoxicity
We next determined whether a similar mechanism of cdk2 dependence was responsible for cisplatin-induced nephrotoxicity in vivo. Purvalanol (0.8 mg) was injected into the tail vein of mice (129/Sv, male, 22 to 28 g) followed by cisplatin injection (intraperitoneally, 20 mg/kg) 24 h later. Mice were given purvalanol daily and killed 1, 2, or 3 d after cisplatin injection. Creatinine (Figure 5) and blood urea nitrogen (BUN) levels were determined using serum that was collected from the retro-orbital vein. Mice that received cisplatin but were not treated with purvalanol had elevated creatinine levels at day 2 (1.33 ± 0.11) and day 3 (2.17 ± 0.19). Mice that were treated with purvalanol cdk2 inhibitor had significantly lower creatinine levels at day 2 (0.55 ± 0.10; P = 0.0005) and day 3 (0.66 ± 0.22; P = 0.0008). In fact, creatinine levels in the mice that were treated with cisplatin plus cdk2 inhibitor were not significantly different from levels in untreated control mice (P = 0.12 and P = 0.19 at days 2 and 3, respectively). The rise in BUN also was significantly reduced by purvalanol (control 28.3 ± 1.7, day 2 88.6 ± 4.1 versus 67.5 ± 6.1 [P = 0.01]; day 3 149.8 ± 3.5 versus 75.9 ± 13.6 [P = 0.0007] comparing mice without and with purvalanol, respectively). After 5 d of cisplatin, creatinine levels in purvalanol-treated mice still were within the normal range, whereas creatinine levels in mice without purvalanol treatment were starting to decline. Similarly, BUN levels in both populations were declining (data not shown).
Figure 5.
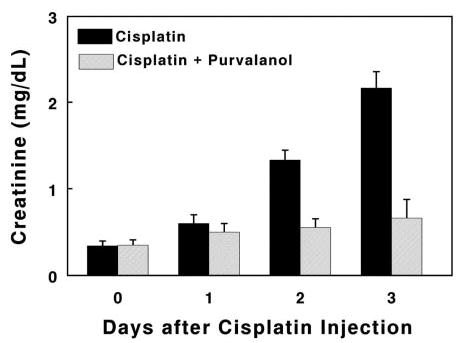
Serum creatinine levels after cisplatin administration. Values, in milligrams per deciliter, at each time point represent means (± SE) of at least six mice. Comparing statistical differences between non–purvalanol-treated mice and untreated control, P = 0.00005 at day 2 and P = 0.000015 at day 3; between non–purvalanol- and purvalanol-treated mice, P = 0.0005 at day 2 and 0.0008 at day 3; between purvalanol-treated mice and untreated control, P = 0.12 at day 2 and 0.19 at day 3.
Necrosis also was markedly reduced in purvalanol-treated mice, showing that both kidney function and morphology (Figure 6) were protected. An assessment of the morphologic differences (Figure 7) demonstrated that all measured parameters of nephrotoxicity were lowered by purvalanol, most significantly necrosis, red blood cell extravasation, and tubular dilation. The quantitative morphology shown in Figure 7 seems to depict more severe injury than illustrated by the morphology displayed in Figure 6 or by the functional injury (creatinine) in Figure 5. In the cisplatin + purvalanol group, one mouse had areas of the kidney that were injured much greater than we observed in other mice from this group. Although the morphologic injury to the kidney was not severe enough to affect physiologic function (BUN and creatinine levels), inclusion of these data affected quantitative morphology values, although there still were statistical differences between treated and untreated groups. Because this damage was not typical in the cisplatin + purvalanol group, it was not illustrated in Figure 6.
Figure 6.
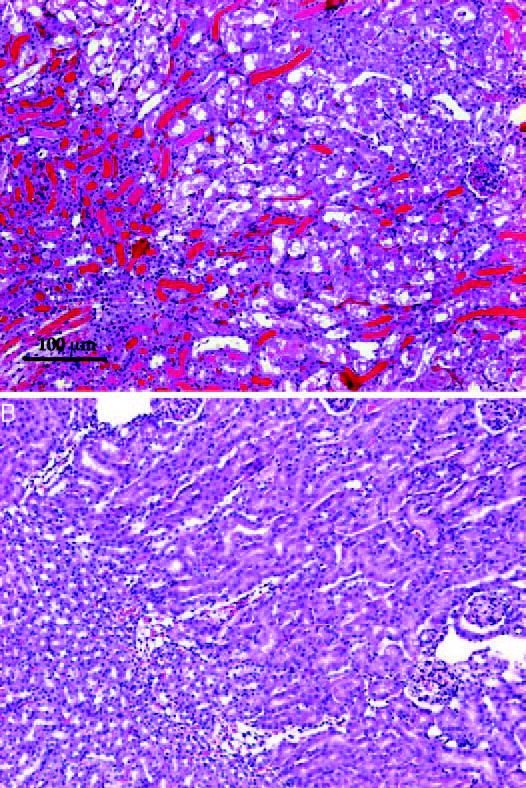
Morphology of kidney 72 h after cisplatin injection. Representative sections of cisplatin treated (top) or purvalanol + cisplatin treated (bottom). Scale is indicated.
Figure 7.
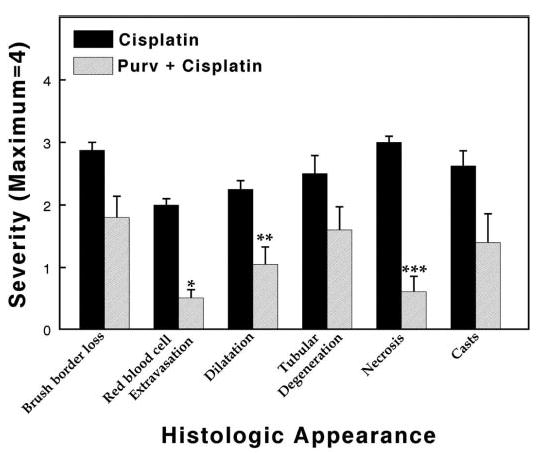
Morphologic evaluation of mouse kidneys 72 h after cisplatin injection expressed on a scale from 0 to 4. Values represent means (± SE) of kidney sections from at least five mice from each treatment. Statistically significant differences are indicated: *P = 0.00001; **P = 0.02; ***P = 0.00009.
We have reported that p21 is highly induced in kidney after cisplatin administration and that p21 induction by itself is protective (20). Therefore, we investigated whether purvalanol induced p21 and whether purvalanol protection depended on p21 by using p21 knockout mice. Purvalanol did not induce p21 higher than background in wild-type mice that were not treated with cisplatin (data not shown), and the cdk2 inhibitor protected p21 knockout mice from cisplatin nephrotoxicity (BUN after 3 d of cisplatin: 154.5 ± 8.1 without purvalanol; 106.6 ± 16.5 with purvalanol; P = 0.010), showing that this inhibitor is not dependent on p21 induction to protect kidney cells in vivo. The BUN values after cisplatin treatment were higher in p21 knockout than those that we found in p21-competent mice, probably because (1) p21 knockout mice are more sensitive to cisplatin-induced nephrotoxicity (20) and (2) purvalanol is not as effective in eliminating cdk2 activity in vivo as it is in vitro (compare Figure 1B, lanes 1 and 2, with Figure 1A, lanes 1 and 9), although it lowers cdk2 activity (compare Figure 1B, lanes 3 and 4 or lanes 5 and 6) so that p21 induction, combined with purvalanol treatment, more efficiently lowers cdk2 activity.
Discussion
We previously reported that induction of the p21 cdk inhibitor in kidney after acute renal failure ameliorates the nephrotoxic effects of both cisplatin administration and ischemia-reperfusion (15,20). Using cultured cells that were derived from mouse kidney proximal tubules, we also demonstrated that p21 transduction and cdk2 inhibitors before cisplatin exposure could protect from cytotoxicity (17). We now demonstrate that cisplatin exposure caused an increase in cdk2 activity, both in vitro and in vivo (Figure 1), that the mechanism of cisplatin cytotoxicity depends on cdk2 activity, and that protection from cisplatin toxicity by p21 is directly dependent on cdk2 inhibition (Figure 2). This mechanism was confirmed by the following evidence: (1) Several different cdk2-inhibitory drugs and proteins are protective, (2) transduction using DN-cdk2 is cytoprotective, and (3) cdk2 knockout cells are not sensitive to doses of cisplatin that cause apoptosis in wild-type cells.
The expression of p21 has been associated with both protection from and induction of apoptosis (17,29, and references within). Similarly, cell-cycle inhibitory drugs such as roscovitine, olomoucine, and purvalanol, which are potent and selective inhibitors of cell-cycle kinases cdk2 and cdc2, prevent apoptosis in a variety of cell types but induce apoptosis in others (17,30 and references within). The spectrum of kinases inhibited by these agents include not only cdk2 but also cdk1 (cdc2) and cdk5 (27,31–35). Because of the similarity of the proteins, they also are likely to inhibit cdk3, although this was not investigated. In addition, the p21 protein interacts with several other proteins through other domains (36). The cdk2 interactive domain was localized between amino acids 49 and 71 (37) by deletion analysis and 49 and 79 (38) by crystallography. We have reported (19) that a 54–amino acid fragment of p21 (amino acids 38 to 91) binds and inhibits cdk2 and also protects from cisplatin cytotoxicity. It therefore was likely that cdk2 inhibition was a primary mechanism for protection by p21 and cdk2 inhibitory drugs, but solely on the basis of these results, other kinase inhibition could not be ruled out.
The cDNA and the gene for the cdk2 protein have been isolated (39), and have led to the development of a DN-cdk2 (40) that consists of a single amino acid change that inactivates the phospho-transfer reaction. These DN-cdk2 mutants have provided protection from growth factor deprivation–induced apoptosis in human endothelial cells (41), staurosporine- or TNF-α–induced apoptosis in HeLa cells (42,43), and from hypoxia-induced apoptosis in cardiomyocytes (44). At the same time, these results do not provide definite proof of the involvement of cdk2 in the apoptotic pathway. First, DN-cdk1 and -cdk3 have provided similar protection from apoptosis as DN-cdk2 (42,43), opening the possibility that these dominant negative mutants could inhibit other kinases. It also was possible that these other mutants bound and thereby lowered the availability of cdk2-interacting cyclins, which are required for cdk2 activity. Second, neither genetically removing the cdk2 gene (22,28) nor inhibiting cdk2 with siRNA or antisense oligonucleotides (40) arrests the cell cycle, but DN-cdk2 prevents growth of several cell types in culture (25,45). Growth inhibition by DN-cdk2 could arise by inhibition of activities other than cdk2. Because of these uncertainties, we have complemented our results in which DN-cdk2 protected from cisplatin apoptosis by examining cdk2 knockout cells for cisplatin sensitivity. These cells were protected from apoptosis in the presence of cisplatin that causes death in wild-type cells (Figures 2 and 3). The dependence of the apoptotic pathway on cdk2 was established further by adenoviral transduction of the knockout cells with wild-type cdk2, which converted them to cisplatin sensitive. Furthermore, we demonstrated that wild-type cdk2 and not adenoviral infection was stimulating the cisplatin-induced cell death by treating the cells with cdk2 inhibitor, which protected them from cisplatin cytotoxicity (Figures 2 and 3).
These experiments provided definitive proof of the dependence of the cisplatin-induced apoptosis pathway on cdk2 in vitro. However, most of the cell death in vivo in kidney after cisplatin administration is morphologically characterized as necrosis. Recent hypotheses have proposed that necrosis could proceed by pathways similar to those described for apoptosis (46–48) and that the type of cell death that is induced by DNA alkylating agents is a “necrotic form of programmed cell death” (49). We therefore extended our investigations to determine whether cdk2 inhibition would protect from cisplatin nephrotoxicity in vivo. As shown in Figure 3, the loss of kidney function that is associated with cisplatin administration and results in an increase in serum creatinine was almost completely abolished; creatinine levels in mice that were treated with purvalanol and cisplatin were not significantly elevated from levels in untreated control mice. Also, the cdk2 inhibitor protected from the morphologic damage that was caused by cisplatin (Figures 6 and 7) in which there was a readily apparent decrease in cellular necrosis in kidneys of treated mice. It therefore is possible that cdk2 activity is necessary for many different pathways of apoptosis in vitro and also for similar pathways of necrosis in vivo.
We report that treatment with purvalanol prevented elevation of serum creatinine after cisplatin treatment, so these levels were statistically indistinguishable from untreated control values. At the same time, BUN levels were elevated, although not to the extent in mice that did not receive purvalanol. There are several possible explanations for this discrepancy. First, cisplatin causes systemic toxicity in animals that is not limited to nephrotoxicity (50), which may affect blood urea levels more than GFR. Organs and/or cells types other than kidney could be less protected by purvalanol from cisplatin toxicity. Second, creatinine is more of a reliable estimate of GFR than is BUN (51), which can be affected by hydration, diet, and other types of nonrenal tissue trauma.
Many different cancers are susceptible to cisplatin chemotherapy, and cdk inhibitors currently are undergoing clinical trials as anticancer agents (52,53). Because of the beneficial effects of cdk inhibitors on the nephrotoxic adverse effects of cisplatin use, combined with their intrinsic chemotherapeutic potential, cisplatin and cdk2 inhibitors should naturally complement each other clinically and possibly augment each other’s beneficial effects as chemotherapeutics. In addition, the use of a cdk2 inhibitor to ameliorate the effects of acute renal failure in vivo mimics the activity of the p21 protein, which is naturally induced in kidney cells as a protective response to renal injury and stress. The sensitivity of cdk2 to inactivation in vitro by purvalanol likely is much greater than cdk2 inactivation in vivo. This is evident in comparing Figure 1A, lane 9 (in vitro inactivation), with Figure 1B, lane 2, 4, or 6 (in vivo inactivation), even though background in vitro activity (Figure 1A, lane 1) is greater than background in vivo activity (Figure 1B, lane 1 or 7). This probably is a result of the efficiency of delivery of the drug to cells in the kidney and/or partial inactivation/binding of the drug before it can be effective. The cytotoxicity of cisplatin in vitro can be controlled effectively by cdk2 inhibition, by drugs, proteins, or genetic manipulation. Development of more efficient means of cdk2 inhibition in vivo is likely to provide effective therapy from cisplatin nephrotoxicity.
Acknowledgments
This work was supported in part by research grants from the National Institutes of Health (R01 DK54471 and P01 DK058324) and the Biomedical Research Foundation and with resources and the use of facilities at the John L. McClennan Memorial Veterans’ Hospital (Little Rock, AR).
We thank Dr. Wafik El-Deiry (University of Pennsylvania Medical School) for initially providing the p21-adenovirus, Dr. Bert Vogelstein (Johns Hopkins University School of Medicine) for providing the clone of human p21 cDNA and adenoviral construction materials and protocols, Drs. Sander van den Heuvel and Ed Harlow (Massachusetts General Hospital) for providing clones of human wild-type and DN-cdk2 cDNA, Dr. Elsa Bello-Reuss (University of Texas Medical Branch) for providing the TKPTS, and Cyril Berthet for discussion and support. We also thank Kimberly Henning and Wasson Snow for valuable assistance during the in vivo drug injections.
References
- 1.Giaccone G. Clinical perspectives on platinum resistance. Drugs. 2000;59:9–17. doi: 10.2165/00003495-200059004-00002. [DOI] [PubMed] [Google Scholar]
- 2.Loehrer PJ, Einhorn LH. Cisplatin: Diagnosis and treatment—Drugs five years later. Ann Intern Med. 1984;100:704–713. doi: 10.7326/0003-4819-100-5-704. [DOI] [PubMed] [Google Scholar]
- 3.Safirstein RL. Renal diseases induced by anti-neoplastic agents. In: Schrier RW, editor. Diseases of the Kidney and Urinary Tract. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 1175–1188. [Google Scholar]
- 4.Lippard SJ. 40 Years of the DNA Double Helix. Houston: Robert A. Welch Foundation; 1993. Structural and biological consequences of platinum anticancer drug binding to DNA; pp. 48–60. [Google Scholar]
- 5.Leibbrandt ME, Wolfgang GH, Metz AL, Ozobia AA, Haskins JR. Critical subcellular targets of cisplatin and related platinum analogs in rat renal proximal tubule cells. Kidney Int. 1995;48:761–770. doi: 10.1038/ki.1995.348. [DOI] [PubMed] [Google Scholar]
- 6.Kaushal GP, Kaushal V, Hong X, Shah SV. Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 2001;60:1726–1736. doi: 10.1046/j.1523-1755.2001.00026.x. [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama S, Hayakawa M, Kato T, Hanaki Y, Shimizu K, Ozawa T. Adverse effects of anti-tumor drug, cisplatin, on rat kidney mitochondria: Disturbances in glutathione peroxidase activity. Biochem Biophys Res Commun. 1989;159:1121–1127. doi: 10.1016/0006-291x(89)92225-0. [DOI] [PubMed] [Google Scholar]
- 8.Matsushima H, Yonemura K, Ohishi K, Hishida A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J Lab Clin Med. 1998;131:518–526. doi: 10.1016/s0022-2143(98)90060-9. [DOI] [PubMed] [Google Scholar]
- 9.Baliga R, Liu H. Activation of caspase 12 by cisplatin (CP) induced endoplasmic reticulum (ER) stress mediates apoptosis in LLC-PK1 cells [Abstract] J Am Soc Nephrol. 2004;15:39. doi: 10.1681/ASN.2004090768. [DOI] [PubMed] [Google Scholar]
- 10.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Megyesi J, Udvarhelyi N, Safirstein RL, Price PM. p53-independent activation of transcription of p21WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol. 1996;271:F1211–F1216. doi: 10.1152/ajprenal.1996.271.6.F1211. [DOI] [PubMed] [Google Scholar]
- 12.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF-1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 14.Harper JW, Adami GP, Wei N, Keyomarsi K, Elledge SJ. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 15.Megyesi J, Andrade L, Vieira J, Safirstein RL, Price PM. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001;60:2164–2172. doi: 10.1046/j.1523-1755.2001.00044.x. [DOI] [PubMed] [Google Scholar]
- 16.Megyesi J, Andrade L, Vieira JM, Safirstein RL, Price PM. Coordination of the cell cycle is an important determinant of the syndrome of acute renal failure. Am J Physiol Renal Physiol. 2002;283:F810–F816. doi: 10.1152/ajprenal.00078.2002. [DOI] [PubMed] [Google Scholar]
- 17.Price PM, Safirstein RL, Megyesi J. Protection of renal cells from cisplatin toxicity by cell cycle inhibitors. Am J Physiol Renal Physiol. 2004;286:F378–F384. doi: 10.1152/ajprenal.00192.2003. [DOI] [PubMed] [Google Scholar]
- 18.Morgan DO. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 19.Yu F, Megyesi J, Safirstein RL, Price PM. Isolation of the functional domain of p21 that protects from cisplatin cytotoxicity. Am J Physiol Renal Physiol. 2005;289:F514–F520. doi: 10.1152/ajprenal.00101.2005. [DOI] [PubMed] [Google Scholar]
- 20.Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest. 1998;101:777–782. doi: 10.1172/JCI1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernest S, Bello-Reuss E. Expression and function of P-glycoprotein in a mouse kidney cell line. Am J Physiol Cell Physiol. 1995;269:C323–C333. doi: 10.1152/ajpcell.1995.269.2.C323. [DOI] [PubMed] [Google Scholar]
- 22.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 23.Darzynkiewicz Z, Li X, Gong J. Assays of cell viability: Discrimination of cells dying by apoptosis. Methods Cell Biol. 1994;41:15–38. doi: 10.1016/s0091-679x(08)61707-0. [DOI] [PubMed] [Google Scholar]
- 24.He TC, Zhou S, da Costa LY, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 26.Kranenburg O, de Groot RP, Van der Eb AJ, Zantema A. Differentiation of P19 EC cells leads to differential modulation of cyclin-dependent kinase activities and to changes in the cell cycle profile. Oncogene. 1995;10:87–95. [PubMed] [Google Scholar]
- 27.Gray NS, Wodicka L, Thunnissen AMWH, Norman TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L, Kim SH, Lockhart DJ, Schultz PG. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281:533–538. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- 28.Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- 29.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 30.Villerbu N, Gaben AM, Redeuilh G, Mester J. Cellular effects of purvalanol A: A specific inhibitor of cyclin-dependent kinase activities. Int J Cancer. 2002;97:761–769. doi: 10.1002/ijc.10125. [DOI] [PubMed] [Google Scholar]
- 31.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: An update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, Fergusson D, Mottram J, Soete M, Dubremetz JF, Le Roch K, Doerig C, Schultz PG, Meijer L. Intracellular targets of cyclin-dependent kinase inhibitors: Identification by affinity chromatography using immobilised inhibitors. Chem Biol. 2000;7:411–422. doi: 10.1016/s1074-5521(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 33.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki M, Delcros JG, Molinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 & cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 34.Sherr CJ, Roberts LM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 35.Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S. Inhibition of cyclin-dependent kinases by purine derivatives. Eur J Biochem. 1994;224:771–786. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- 36.Dotto GP. p21WAF1/Cip1: More than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–M56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 37.Nakanishi M, Robetorye RS, Adami GR, Pereira-Smith OM, Smith JR. Identification of the active region of the DNA synthesis inhibitory gene p21Sdi1/CIP1/WAF1. EMBO J. 1995;14:555–563. doi: 10.1002/j.1460-2075.1995.tb07031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russo AA, Jeffrey PD, Patter AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 39.Tsai LH, Harlow E, Meyerson M. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature. 1991;353:174–177. doi: 10.1038/353174a0. [DOI] [PubMed] [Google Scholar]
- 40.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3:233–245. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 41.Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM, Ross R. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol Cell. 1998;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- 42.Harvey KJ, Lukovic D, Ucker DS. Caspase-dependent cdk activity is a requisite effector of apoptotic death events. J Cell Biol. 2000;148:59–72. doi: 10.1083/jcb.148.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meikrantz W, Schlegel R. Suppression of apoptosis by dominant negative mutants of cyclin-dependent protein kinases. J Biol Chem. 1996;271:10205–10209. doi: 10.1074/jbc.271.17.10205. [DOI] [PubMed] [Google Scholar]
- 44.Adachi S, Ito H, Tamamori-Adachi M, Ono Y, Nozato T, Abe S, Ikeda MA, Marumo F, Hiroe M. Cyclin A/cdk2 activation is involved in hypoxia-induced apoptosis in cardiomyocytes. Circ Res. 2001;88:408–414. doi: 10.1161/01.res.88.4.408. [DOI] [PubMed] [Google Scholar]
- 45.Hu B, Mitra J, van den Heuvel S, Enders G. S and G2 phase roles for Cdk2 revealed by inducible expression of a dominant-negative mutant in human cells. Mol Cell Biol. 2001;21:2755–2766. doi: 10.1128/MCB.21.8.2755-2766.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edinger AL, Thompson CB. Death by design: Apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 47.Padanilam BJ. Cell death induced by acute renal injury: A perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol. 2003;284:F608–F627. doi: 10.1152/ajprenal.00284.2002. [DOI] [PubMed] [Google Scholar]
- 48.Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: A specific form of programmed cell death? Exp Cell Res. 2003;283:1–16. doi: 10.1016/s0014-4827(02)00027-7. [DOI] [PubMed] [Google Scholar]
- 49.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hacker MP. Platinum induced nephrotoxicity, overview. In: Hacker MP, Lazo JS, Tritton TR, editors. Organ Directed Toxicities of Anticancer Drugs. Boston: Martinus Nijhoff; 1988. pp. 155–158. [Google Scholar]
- 51.Brenner BM, Deen WM, Robertson CR. Glomerular filtration. In: Brenner BM, Rector FC Jr, editors. The Kidney. Philadelphia: Saunders; 1976. pp. 251–271. [Google Scholar]
- 52.Cohen P. Protein kinases: The major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 53.Dai Y, Grant S. Small molecule inhibitors targeting cyclin-dependent kinases as anticancer agents. Curr Oncol Rep. 2004;6:123–130. doi: 10.1007/s11912-004-0024-3. [DOI] [PubMed] [Google Scholar]