

Abstract
Gut microbiota shows host-specific diversity and temporal stability and significantly contributes to maintenance of a healthy gut. However, in inflammatory bowel disease, this microbiota has been implicated as a contributory factor to the illness. This study compared bacterial dynamics in Crohn's disease patients to those in a control group using a culture-independent method to assess the temporal stability, relative diversity, and similarity of the dominant fecal microbiota, Clostridium spp., Bacteroides spp., Bifidobacterium spp., and lactic acid bacteria spp. (LAB) for all individuals. Fecal samples were collected over several time points from individuals with Crohn's disease who were in remission (n = 11), from Crohn's disease patients who relapsed into an active Crohn's disease state (n = 5), and from a control group (n = 18). Denaturing gradient gel electrophoresis profiles were generated for the different microbial groups by specifically targeting different regions of the 16S rRNA gene and were compared on the basis of similarity and diversity. The temporal stability of dominant species for all Crohn's disease patients was significantly lower (P < 0.005) than that for the control group. Analysis of group-specific profiles for Bifidobacterium spp. found that they were similar in all samples, while the diversity of the LAB varied significantly between the groups, but temporal stability was not significantly altered. We observed significant changes in two functionally important mutualistic groups of bacteria, viz., Clostridium and Bacteroides spp., which may have implications for the host's gut health, since some genera are involved in production of short-chain fatty acid, e.g., butyrate.
The mutualistic arrangement which has evolved between the gut microbiota and the human host has resulted in the former making significant contributions to the ability of the host to resist colonization by certain pathogens, metabolize recalcitrant carbon sources, e.g., cellulose, maintain mucosal immune architecture and function, and obtain essential nutrients, such as vitamins (2). However, the opposite side of this association is that the host, while providing a suitable environment for the microbes, has to tolerate the presence of a large community of potentially opportunistic pathogens. If this mutualism is disturbed, the resulting situation can have severe consequences for the host. An example of such a “disturbance” is Crohn's disease (CD) (18, 32). While susceptibility genes contribute (20, 41), it is evident from concordance rates in monozygotic twin studies (42, 61) and the changing epidemiology over time that environmental factors also contribute. Although a role for a specific pathogen has not been excluded, the weight of evidence implicates the gastrointestinal microbiota or components thereof in the setting of a permissive genetic susceptibility background (19, 26, 48, 55).
A significant factor which may be contributing to the lack of any definitive pathogen in Crohn's disease is the inability to grow the majority of the bacteria which reside in the gastrointestinal tract (5, 10, 49, 68). The current estimate is that approximately 70 to 80% of the bacteria are unculturable (2, 12); thus, alternative culture-independent methods need to be used in order to understand the dynamics of this large, complex, and metabolically active community and its contribution of gut homeostasis. Once such approach, denaturing gradient gel electrophoresis (DGGE) of 16S rRNA genes (36, 37), has provided an insight into this ecosystem. Consequently, increased appreciation of microbial diversity and dynamics in the gastrointestinal tract has been realized (29, 63, 68).
Several key observations have been reported from these studies: that the diversity of the microbiota present in a healthy human gastrointestinal tract is a result of natural selection operating at both the microbial level and the host level (2), that a high level of diversity is present and is considered desirable for ecosystem stability (35), and finally that the intestinal microbiota is resistant to alteration and is stable over time (68). The high level of diversity enables the ecosystem to maintain a level of functional redundancy, which ensures that the community is able to perform key processes (25, 27, 35), e.g., synthesis of short-chain fatty acids, such as butyrate (3). Any significant changes to the bacterial diversity may have direct functional consequences for the ecosystem and hence the host (16, 27, 35). Since the dominant bacterial diversity in Crohn's disease has been shown to be disturbed (53), we wished to determine whether we could identify which functionally important bacterial groups were changing and whether any temporal variability was evident. Previous culture-dependent investigations of Crohn's disease have provided a consensus view that gram-negative anaerobes increase in Crohn's disease compared to control levels (59). However, conflicting reports do exist where levels of Bacteroides and lactobacilli showed no difference between Crohn's disease samples and control samples, while the bifidobacteria were significantly decreased in numbers (13). Furthermore, culture-independent investigations of the dynamics of the intestinal bacterial community in Crohn's disease have either concentrated on the dominant species (24, 44, 45, 52) or quantified the species in specific groups (53, 58), and each analysis has used only a single-time-point sample with no temporal data presented. The consensus of opinion from these investigations is that there is a reduction in the diversity of the dominant bacterial species in Crohn's disease. However, to our knowledge no culture-independent studies of temporal stability and diversity in Crohn's disease have been undertaken. In this exploratory investigation, we report on the temporal stability and diversity of the dominant bacterial species, the Bacteroides fragilis subgroup (which is involved in metabolism of indigestible dietary polysaccharides [2] and may play a role in inflammatory bowel disease [30]), the Clostridium leptum subgroup and the Clostridium coccoides subgroup (these groups contain the majority of butyrate producers [3]), and Bifidobacterium spp. and the lactic acid bacteria spp. (which have been commonly used as probiotics and have immunomodulatory activity [6, 43]), from Crohn's disease patients in remission and relapse and compare their DGGE species profiles to a those for a control group.
MATERIALS AND METHODS
Collection of stool samples for culture-independent analysis.
Sixteen Crohn's disease patients provided fecal samples over several time points (Table 1). All subjects were diagnosed as being active when they provided their first samples (T = 0). All of the subjects were brought into remission (clinically assessed and had a Crohn's disease activity index [CDAI] of <150), using prednisolone (40 mg/day), and were weaned off the steroid over a 12-week period. Members of the group were also taking probiotics. Five Crohn's disease subjects (12-16) relapsed back into an active state (after clinical assessment and a CDAI of >150) and were removed from the study. In total, 49 stool samples from individuals with Crohn's disease were analyzed. A total of 18 healthy control subjects (10 men and 8 women) were also included in the analyses (number of samples = 30). This control cohort consisted of 12 individuals who were sampled once, 5 individuals who were sampled over a 3-month period, and an additional control who was sampled over 1 year. All stool samples were stored at −80°C until required for bacterial DNA extraction.
TABLE 1.
Crohn's disease subjects and time points analyzed
| Subject group | Subject codea | Age | Sex | CDAId | Probiotic | Smoker | Disease site(s) | Sample time point (wk) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Remission | 1 | 34 | M | 234 | Yes | No | Ileum | 0 | 4 | 8 | 12 | 24 |
| 2 | 37 | F | 262 | No | No | Cecum, ascending colon, transverse colon | 0 | 4 | 8 | 12 | 24 | |
| 3 | 28 | M | 295 | Yes | No | Ascending colon, transverse colon, rectum, descending colon, other (esophagus) | 0 | 4 | 8 | 12 | ||
| 4 | 48 | M | <150 | No | No | Ileum, cecum | 0 | 52 | 64 | |||
| 5 | 38 | F | 383 | No | No | Cecum, ascending colon, transverse colon | 0 | 4 | 8 | 12 | ||
| 6 | 37 | M | 308 | No | No | Transverse colon | 0 | 4 | 8 | 12 | ||
| 7 | 25 | M | 361 | Yes | No | Ileum, cecum, transverse colon, rectum, descending colon | 0 | 12 | 24 | 52 | ||
| 8 | 70 | F | 302 | No | Yes | Ileum, cecum and descending colon | 0 | 12 | ||||
| 9 | 40 | M | <150 | Yes | No | Ileum, cecum, ascending colon, transverse colon, rectum, and descending colon | 4 | 12 | ||||
| 10b | 35 | F | 181 | No | No | Ileum | 0 | 12 | ||||
| 11 | 41 | M | 247 | Yes | No | Ileum | 0 | 4 | 12 | |||
| Relapse | 12e | 54 | F | 431 | Yes | No | Ileum and cecum | 0 | 4g | |||
| 13 | 25 | M | 162 | Yes | No | Ileum, descending colon | 0 | 4 | 8g | |||
| 14 | 52 | M | 210 | No | No | Ileum and cecum | 0 | 4g | ||||
| 15 | 43 | F | 252 | No | No | Ileum and cecum | 4 | 4g | ||||
| 16 | 54 | F | 245 | Yes | No | Ileum | 0 | 4g | ||||
| Control | 17c | 50 | F | NA | No | No | Not applicable | 0 | 6 | 12 | ||
| 18 | 51 | M | NAf | No | No | Not applicable | 0 | 6 | 12 | |||
| 19 | 25 | F | NA | No | No | Not applicable | 0 | 26 | 52 | |||
| 20 | 25 | M | NA | No | Yes | Not applicable | 0 | 6 | 12 | |||
| 21 | 25 | M | NA | No | No | Not applicable | 0 | 6 | 12 | |||
| 22 | 38 | M | NA | No | No | Not applicable | 0 | 6 | 12 | |||
All individuals with Crohn's disease were on 5′-aminosalicylate except subjects 6 and 10, who were not on any medication.
All individuals were Caucasian except subject 10, who was of Afro-Caribbean ethnicity.
For cluster analysis of Crohn's disease patients versus healthy individuals, an additional 12 samples (T = 0) were obtained.
Crohn's disease activity index (T = 0).
Received augmentin for an ear infection at T = 5 days; relapse occurred at 8 weeks.
NA, not applicable.
Sample was taken when relapse occurred.
Extraction of total DNA from stool samples.
Fecal samples used were thawed on ice, and DNA was extracted using the QIAGEN QIAamp MiniStool kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions for pathogen isolation, with an initial bead-beating step of 30 s at 5,000 rpm. Extracts were treated with DNase-free RNase (100 μg/ml), and the DNA concentration was determined using a Nanodrop spectrophotometer. A total of 79 fecal extractions were performed and quantified.
PCR of partial 16S rRNA genes and DGGE to determine changes in the bacterial community.
PCR of partial 16S rRNA genes was performed using an MJ Research PTC-200 thermal cycler, and this amplification was performed for all DGGE primer sets (Table 2) on all 79 samples. Each sample was amplified in triplicate on separate occasions. PCR mixtures of 50 μl contained 1× buffer (20 mM Tris, pH 8.4, 50 mM KCl), 3 mM MgCl2, 200 μM of each deoxynucleoside triphosphate, 1.25 U of Taq polymerase (Invitrogen, United Kingdom), and 10 pmol of each primer. Appropriately diluted genomic DNA (1 ng) was added to the final PCR. The PCR conditions were as follows: 95°C for 5 min of initial denaturation, followed by 30 cycles of amplification with 95°C denaturation for 30 s, variable annealing temperature (see Table 2 for a primer pair's specific annealing temperature) for 40 s, and extension at 72°C for 1 min, with a final extension of 72°C for 5 min. PCR amplicons were separated by electrophoresis in 1% (wt/vol) agarose containing ethidium bromide (5 mg ml−1) and 1× Tris-acetate-EDTA (TAE) buffer (47) with an applied voltage of 5 V cm−1. DNA was visualized by UV illumination (302 nm).
TABLE 2.
Primers used in this study
| Primera | Sequence (5′-3′) | Specificity | Annealing temp (°C) | DGGE gradient (%)c | Reference |
|---|---|---|---|---|---|
| 968f-GCb | AAC GCG AAG AAC CTT AC | Bacterial V6-V8 region | 56 | 30-55 | 40 |
| 1401r | CGG TGT GTA CAA GAC CC | Bacterial V6-V8 region | 56 | 40 | |
| Bif164f | GGG TGG TAA TGC CGG ATG | Bifidobacterium | 66 | 45-55 | 22 |
| Bif662r-GCb | CCA CCG TTA CAC CGG GAA | Bifidobacterium | 66 | 22 | |
| Lac1f | AGC AGT AGG GAA TCT TCC A | LAB | 61 | 30-55 | 65 |
| Lac2r-GCb | ATT YCA CCG CTA CAC ATG | LAB | 61 | 65 | |
| g-Bfra-F-GCb | ATA GCC TTT CGA AAG RAA GAT | Bacteroides fragilis group | 50 | 30-50 | 33 |
| g-Bfra-R | CCA GTA TCA ACT GCA ATT TTA | Bacteroides fragilis group | 50 | 33 | |
| Sg-Clept-F | GCA CAA GCA GTG GAG T | Clostridium leptum subgroup | 50 | 30-50 | 34 |
| sg-Clept-R3-GCb | CTT CCT CCG TTT TGT CAA | Clostridium leptum subgroup | 50 | 34 | |
| g-Ccoc-F-GCb | AAA TGA CGG TAC CTG ACT AA | Clostridium coccoidessubgroup | 50 | 35-50 | 33 |
| g-Ccoc-R | CTT TGA GTT TCA TTC TTG CGA A | Clostridium coccoides subgroup | 50 | 33 |
f (forward) and r (reverse) indicate the orientation of the primers with respect to the 16S rRNA gene sequence.
GC indicates a 40-bp GC-rich sequence attached to the 5′ end of the primer: 5′-CCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCG-3′.
Denaturing acrylamide of 100% was defined as 7 M urea and 40% (vol/vol) formamide.
PCR products were separated by DGGE according to the specifications of Muyzer and colleagues (36), using the DCODE system (Bio-Rad Laboratories, United Kingdom) with the following modifications: polyacrylamide gels (dimensions, 200 mm by 200 mm by 1 mm) consisting of 8% (vol/vol) polyacrylamide (37.5:1, acrylamide-bisacrylamide) and 0.5× TAE. The denaturant gradients for each primer pair are shown in Table 2. Prior to polymerization of the denaturing gel, a stacking gel, without denaturants, was added to enable defined wells to be cast. Electrophoresis was performed for 16 h at 85 V in 0.5× TAE buffer at a constant temperature of 60°C. Gels were stained with SYBRgold according to the manufacturer's instructions. Each sample analyzed in this study was run in triplicate, and the data presented in this paper were consensus data compiled from all gels of the same sample to further minimize any error variation. Furthermore, experiments were conducted on a subset of samples to determine variation between PCR and DGGE. DNA samples from these subjects were amplified in triplicate on the same day using the same thermal cycler and cycling conditions and compared to PCR from the same DNA, amplified on the same machine but on another day. These samples were compared to each other by DGGE on the same gel, and the same PCR amplicons were run again on a different day and gel to determine gel-to-gel variation. All the DGGE profiles were compared using the method described below. Error variation due to PCR and DGGE was found to be low (between 0 to 2% difference) for a given sample and therefore unlikely to be a contributory factor in any profile or diversity differences observed in this study.
Profile analysis of DGGE patterns.
DGGE profiles were analyzed using the Gel Compare function of the Bio Numerics software program (Applied Maths, St-Martens-Latem, Belgium). Similarities between samples and their temporal stability were determined by calculating similarity indices based on the Dice similarity coefficient and the unweighted-pair group method using arithmetic averages (UPGMA). The Dice coefficient is also referred to as Sorenson's pairwise similarity coefficient (Cs) and is commonly used to compare the species composition of different ecosystems. Two identical profiles create a value of 100%, whereas two completely different profiles result in a value of 0%. Dendrograms of DGGE banding profiles were constructed to visualize any clustering patterns evident and to generate similarity matrixes for numerical and subsequent statistical analysis. All similarity results given are the Dice and UPGMA percentage similarities, since this method (band based) and the Pearson correlation coefficient (curve based) were in agreement (data not shown). Composite data sets for group-specific DGGE profiles were generated, and numerical band matching character tables were produced for export and analysis by BiodiversityPro (version 2; Scottish Association for Marine Science [http://www.sams.ac.uk]). Using the BiodiversityPro software, Simpson's, Shannon-Weaver, and Fisher's alpha ecological indices of diversity were generated. These were calculated using the following equations:
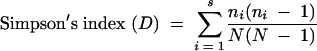 |
where ni is the total number of organisms of the ith species or band intensity and N is the total number of organisms of all species or total band intensities (57).
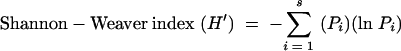 |
where s is the number of species/bands in the sample, and Pi is the proportion of species/bands for the ith species/band in the sample (7, 56).
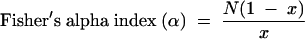 |
where x is calculated from
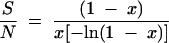 |
S is the number of species/bands, and N is the total number of individuals/bands (14, 23).
Statistical analysis.
All percentage similarities and diversity indices were analyzed to determine whether data were normally distributed (using SPSS probability plot function); for normal data unpaired Student's t tests were performed, while for nonnormally distributed data Wilcoxon's test was applied. To analyze the impact of probiotic or corticosteroid consumption on community dynamics, analysis of variance was used. Chi-square test was used to determine whether the frequency of PCR amplifications were significantly different between test cohorts for group-specific primers.
RESULTS
Analysis of the temporal stability of the dominant microbiota, Clostridium spp., Bacteroides spp., bifidobacteria, and lactic acid bacteria (LAB) of Crohn's disease patients compared to that for controls.
All test samples were analyzed using culture-independent methods in order to determine the temporal stability of the microbiota against that for a control group. The mean similarity values and variation reported for the control group are in agreement with results of previous studies of the temporal stability of the fecal microbiota in healthy individuals (62, 68). The mean similarity of DGGE profiles over time was calculated for each individual (Table 3). Since the data were shown to be normally distributed (data not shown), Student's t tests were conducted to compare the mean similarities of the dominant microbiota, Clostridium spp., Bacteroides spp., Bifidobacterium spp., and LAB of each group, and significance values are reported here. Relative diversity indices, viz., Shannon-Weaver (H′), Fisher's alpha (α), and Simpson's (D), were also calculated for each successful DGGE profile (Table 4). Analysis of variance of the impact of taking a probiotic or being administered a corticosteroid showed that there was no statistical difference between groups, and they were not treated separately for the purpose of this study.
TABLE 3.
Mean similarity values for individuals analyzed over timea
| Subject group | Subject code | No. of pairwise comparisons | % Mean similarity over time ± SD
|
|||||
|---|---|---|---|---|---|---|---|---|
| Dominant microbiota | C. leptum | C. coccoides | Bacteroides | LAB | Bifidobacterium | |||
| Remission | 1 | 10 | 69.5 ± 10.9 | 1 sample +ve | 70.5 ± 13.7 | 73.9 ± 16.9 | 64 ± 21.9 | 82.7 ± 9.9 |
| 2 | 10 | 62.3 ± 6.9 | 1 sample +ve | 79.8 ± 14.4 | NS | 46.1 ± 12.6 | 87.5 ± 9.7 | |
| 3 | 6 | 49.8 ± 14.1 | 79.9 ± 13 | 88.9 | NS | 23.3 ± 15.4 | 85.2 ± 5.6 | |
| 4 | 3 | 62.2 ± 7.8 | 51.8 ± 20.2 | 45.4 ± 5 | NS | 55.3 ± 14.6 | 82.3 ± 10.9 | |
| 5 | 6 | 56.1 ± 5.5 | 86.7 ± 7.7 | 85.8 ± 9.1 | NS | 59.3 ± 14.0 | 73 ± 9.3 | |
| 6 | 6 | 67.5 ± 9.2 | 77 ± 7.9 | 63.5 ± 26.7 | 70.8 ± 11.2 | 49.5 ± 25.0 | 81.2 ± 7.5 | |
| 7 | 6 | 42.0 ± 12.7 | 70.2 ± 12 | 66.8 ± 9.6 | 100 | 30.7 ± 11.2 | 75.4 ± 10.9 | |
| 8 | 1 | 60.3 | 75 | 54.5 | 100 | 52.3 | 100 | |
| 9 | 1 | 89.8 | 1 sample +ve | 54.5 | NS | 71.4 | 100 | |
| 10 | 1 | 69.2 | 81.8 | 66.7 | 60 | 74.8 | 71.4 | |
| 11 | 3 | 70.6 ± 11.2 | 60 ± 17.5 | 61.5 ± 2.3 | 100 | 47 ± 12.8 | 85.7 ± 7.9 | |
| Mean | 63.5 ± 12.4 | 72.8 ± 11.7 | 67.0 ± 13.5 | 84.1 ± 18 | 52.1 ± 15.6 | 84 ± 9.5 | ||
| Relapse | 12 | 1 | 30.8 | NS | 1 sample +ve | NS | 59.2 | 76.2 |
| 13 | 3 | 70.3 ± 8.4 | 46.1 ± 31 | 34 ± 5.7 | 66.9 ± 28.9 | 45.6 ± 16.9 | 100 | |
| 14 | 1 | 75 | 80 | 66.7 | 100 | 61.8 | 100 | |
| 15 | 1 | 50 | 28.6 | 70.6 | 50 | 44.4 | 100 | |
| 16 | 1 | 57.2 | 67 | 100 | 85.7 | 47.4 | 100 | |
| Mean | 56.6 ± 17.5 | 55.4 ± 22.6 | 67.8 ± 27 | 53.2 ± 32.3 | 51.7 ± 8.2 | 95.3 ± 10.6 | ||
| Control | 17 | 3 | 87.6 ± 4.8 | 83.3 ± 7.2 | 71.2 ± 7.7 | 100 | 52.6 ± 8.9 | 91.2 ± 11.5 |
| 18 | 3 | 90.9 ± 4.0 | 76.6 ± 6.9 | 75.1 ± 4.18 | 100 | 55.0 ± 10.4 | 93.9 ± 6.8 | |
| 19 | 3 | 81.1 ± 7.3 | 76.2 ± 6.8 | 66.2 ± 8.8 | 75 ± 25.3 | 60.3 ± 12.5 | 85.3 ± 8.9 | |
| 20 | 3 | 77.8 ± 23 | 77.8 ± 6.7 | 90.5 ± 3.4 | 95.1 ± 4.5 | 63.4 ± 14 | 86.7 ± 11.5 | |
| 21 | 3 | 86.5 ± 4.2 | 75.6 ± 5.3 | 65 ± 9.2 | 88.6 ± 5.6 | 75.8 ± 6.1 | 91.8 ± 14.4 | |
| 22 | 3 | 83.6 ± 3.6 | 90.5 ± 4.9 | 87.2 ± 6.1 | 91.5 ± 3.2 | 32.4 ± 17.2 | 100 | |
| Mean | 84.6 ± 4.7 | 79.9 ± 5.8 | 75.8 ± 10.7 | 91.5 ± 9.3 | 56.7 ± 14.4 | 91.5 ± 5.4 | ||
The pairwise comparison of each profile from each time point generated was calculated using Bio Numerics software. The similarity values for each individual from each pairwise comparison over time were totaled and averaged and the standard deviation calculated. When two samples were present or if only two samples gave positive PCR products, only the percentage similarity is shown and no standard deviation was calculated. Where no positive PCR signal was observed, the abbreviation NS is used. For several individuals, only one amplification was positive, and this result is presented as “1 sample +ve.”
TABLE 4.
Values of diversity indices calculated from the DGGE profilesa
| Microbial group | Diversity index value (mean ± SE)
|
|||||
|---|---|---|---|---|---|---|
| Simpson's (D)
|
Shannon-Weaver (H′)
|
Fisher's alpha (α)
|
||||
| CD | Control | CD | Control | CD | Control | |
| Dominant microbiota | 0.13 ± 0.04 | 0.07 ± 0.02 | 0.98 ± 0.13 | 1.23 ± 0.1 | 2.21 ± 0.57 | 3.78 ± 0.75 |
| C. leptum | 0.18 ± 0.09 | 0.15 ± 0.02 | 0.86 ± 0.19 | 0.86 ± 0.06 | 1.68 ± 0.65 | 1.49 ± 0.24 |
| C. coccoides | 0.29 ± 0.13 | 0.35 ± 0.13 | 0.63 ± 0.17 | 0.53 ± 0.14 | 0.96 ± 0.39 | 0.82 ± 0.25 |
| Bacteroides | 0.53 ± 0.21 | 0.32 ± 0.07 | 0.35 ± 0.15 | 0.52 ± 0.1 | 0.47 ± 0.17 | 0.62 ± 0.16 |
| Bifidobacterium | 0.31 ± 0.07 | 0.32 ± 0.11 | 0.53 ± 0.1 | 0.52 ± 0.14 | 0.7 ± 0.17 | 0.82 ± 0.25 |
| LAB | 0.19 ± 0.07 | 0.11 ± 0.04 | 0.78 ± 0.15 | 1.01 ± 0.15 | 1.4 ± 0.47 | 2.42 ± 0.78 |
Values in bold were significantly different between control and CD subjects (P < 0.05).
The temporal stability of the dominant microbiota (defined as bacterial composition of >1 to 10% of the community or >109 g−1 of feces [68]) in the control group ranged between 78% and 91%, indicating that there was very little variation in the dominant genera in this cohort of individuals, with the average stability of the control group over time being 85% ± 4.7%. This value is in agreement with the work of Vanhoutte and colleagues (88% to 96% for the V6-V8 primer profiles [62]). The average stability of Crohn's disease remission individuals was 64% ± 12.4%, and the range between individuals was 42% to 90%. Pairwise comparisons between prerelapse and relapse samples showed that on average there was a similarity of 57% ± 17.5% between the profiles, with a range of 31% to 75%. Unpaired Student's t tests performed on the mean temporal stability values of all three groups showed that all three were significantly different from each other (P < 0.005). Figure 1 shows DGGE profiles of PCR products for the V6-V8 region of the 16S rRNA gene, using primers for the dominant bacterial community in healthy individuals and those with Crohn's disease (both remission and relapse). In order to compare the relative diversity of each community, the coefficient of variance for each set of diversity indices was calculated for the control groups. The largest coefficient of variance was 10% for one individual over 12 weeks, and hence, any samples that varied by more than 10% were considered to show changes in relative diversity as measured by any of the three ecological indices.
FIG. 1.

Sample DGGE profiles of the V6-V8 regions of 16S rRNA genes from fecal samples from the Crohn's disease group and the control group. Samples are denoted by the individual code and time point; for example, 19 - 26 wks represents a sample taken from individual 19 at T = 26 weeks; mnths, months.
Dominant bacterial diversity was significantly different (P < 0.0001) between Crohn's disease and control groups, and although the average stability for remission was higher than that for relapse (64% versus 57%), the difference was not statistically significant. A notable example was sample 12, which came from the Crohn's disease group. This individual's DGGE profile was simplified after the individual went into relapse, and prerelapse and relapse samples were only 31% similar (Fig. 2); all three diversity indices also showed a reduction in overall profile complexity, with H′ changing from 0.96 to 0.72, α changing from 1.72 to 0.91, and D changing from 0.12 to 0.21 (an increase in D indicates a decrease in diversity) for prerelapse and relapse samples, respectively. However, 5 days into the study, subject 12 did receive augmentin, which can alter the host bacterial diversity (9), but the relapse occurred nearly 2 months later, we concluded that the antibiotic was not responsible for the relapse but may have altered the gut community, and this individual was removed from the analysis. By contrast, if one examines the profiles of patient 14, prerelapse and relapse, such dramatic changes in their dominant microbiota are not apparent. For individual 14, the average similarity between profiles was 75% and seemingly no major loss of diversity was observed. The diversity indices for prerelapse and relapse DGGE profiles for subject 14 were as follows: H′ = 1.06 and 1.11; α = 2.78 and 2.76; and D = 0.10 and 0.09 (Fig. 2). Other patients in the Crohn's disease relapse group showed changes in their profile similarity values, but analysis indicated that the contributory factor accounting for a low similarity was a shift in the community profile to an equally diverse but differently structured community. In other words, the number of bands and the intensities were the same, but the patterns were different.
FIG. 2.

DGGE profiles of the V6-V8 regions of 16S rRNA genes from Crohn's disease pre- and postrelapse fecal samples. The dendrogram was constructed using the Dice and UPGMA cluster method. The image shows two individuals' DGGE profiles and contrasts the differences that were detected during the relapse process. Note how the DGGE profile of individual 12 changes significantly during the relapse, while the DGGE profile of individual 14 does not alter to the same degree. The scale shown on the left represents percentage similarity between DGGE profiles. Samples are labeled as in Fig. 1.
Clostridium sp.-specific DGGE profile analysis of Crohn's disease subjects.
DGGE profiles were generated for members of the C. leptum and C. coccoides subgroups. Of the 49 samples from Crohn's disease individuals, a significant proportion did not give a positive result for the C. leptum-specific primer set, with a failure rate of 27% (P < 0.0001). Only one sample from a CD individual failed to produce a signal for the C. coccoides primer set. These primers did not fail to amplify a partial 16S rRNA gene product from any of the control samples; furthermore, as a demonstration of their robust design, they were used in a study to determine prevalence of both groups and did not fail to amplify from 46 healthy subjects (34). Those samples that were amplified and for which DGGE profiles were generated, percent similarities, diversity indices, and temporal stability were not significantly different from those for the control group.
Bacteroides fragilis subgroup-specific DGGE profile analysis of Crohn's disease subjects.
The Bacteroides fragilis subgroup DGGE profiles generated for the two groups were not significantly different. Again this set of group-specific DGGE primers failed to amplify 16S rRNA gene products from 39% of the Crohn's disease samples (P < 0.0001) but was 100% successful with DNA extracted from the control group. The relative diversity of the Bacteroides fragilis subgroup was significantly reduced from that of control samples (P < 0.0001), while its mean percent similarity over time showed no significant difference from that of the control group (Tables 3 and 4).
LAB-specific DGGE profile analysis of Crohn's disease subjects.
Lactic acid bacteria and related species were detected in all samples from which DNA was extracted. Analysis of DGGE profiles leads us to conclude that LAB profiles were specific to a host, showed high complexity, and were variable for an individual over time. Statistical analysis of the percent similarity showed no significant differences between the LAB profiles for individuals within each group. However, the diversity indices were higher for the healthy cohort than for the CD group (P < 0.05).
Bifidobacterium sp.-specific DGGE profile analysis of Crohn's disease subjects.
Bifidobacterium spp. were also detected in all samples, and the profiles ranged from simple to complex (i.e., one to six bands per lane) for each individual. Furthermore, the profiles were remarkably stable over time. The average stability of the Crohn's disease remission group was 83% ± 11%, for the Crohn's disease relapse group, 97% ± 9%, and for the control group, 90% ± 6%. No significant difference was found between the control group and either Crohn's disease group. Diversity of the Bifidobacteria between samples was also not significantly different between the groups analyzed.
Interindividual comparison of the dominant microbiota, Clostridium spp., Bacteroides spp., Bifidobacterium spp., and LAB.
Both control and disease individuals were compared at time zero for all bacterial groups in order to determine if CD subjects harbored dissimilar and distinct bacterial populations. The interindividual percent similarity for control and disease groups was only significantly different for the analysis of the dominant microbiota (P < 0.001). Comparison of dominant microbiota DGGE profiles for the control group were more similar (had a higher percentage similarity [69% ± 8.1%]) to each other than profiles from CD individuals (29% ± 8.3%). This indicates that control groups have more microbiota in common than CD individuals, who harbor microbial populations that appear to differ significantly from each other.
DISCUSSION
Utilization of culture-independent methods to analyze the microbial dynamics of ecosystems has revolutionized our view of the contribution bacteria make to their function and maintenance (1, 17). We used DGGE to generate 16S rRNA profiles of the gut microbiota and determined the temporal stability and diversity of members of several functionally important bacterial groups and how these groups varied while the Crohn's disease individual was in remission and relapse. One of the significant observations we report is that the temporal stability of species profiles from Crohn's disease remission and Crohn's disease relapse samples differed from those for the control samples. This is the first description of the instability of the bacterial community in Crohn's disease and highlights the need to take multiple samples from an individual when investigating their gut microbiota. We also found reduced community diversity with Crohn's disease, and these findings are in agreement with those of a previous study where 16S rRNA species profiles showed that the microbiota of Crohn's disease subjects is altered in active and quiescent disease (53). The profiles from the Crohn's disease remission individuals were shown to be stable over a much longer time period than previously described in the literature. However, this stability is relative and was in no way comparable to that for the control group, which shows a much greater stability and a less dynamic bacterial community. A higher stability of the bacterial community for Crohn's disease remission patients may be one factor in maintaining remission, since in relapse a more variable community was observed, but due to the low numbers it was not considered significant and needs further analysis. We also concluded that there is a reduced level of stability and a change of diversity during relapse into an active disease state. The mean percent similarity of the DGGE profiles for Crohn's disease subjects at time zero was significantly lower than that for healthy subjects in this study, and this indicates greater host specificity.
How this stability can be maintained or even initiated needs further investigation, since it would be desirable to be able to show that remission and “a stable gut” were synonymous with each other. Since the bacterial community in healthy subjects is much more stable, it follows that this trait is desirable and is a feature of a healthy gut. If we were to speculate on the biological significance of this variation, one could envisage that changes in the bacterial composition over time would impact on the functions that this community is supplying to the host. Changes in bacterial functions, such as short-chain fatty acid production, not only will impact colonocytes (66) but also can result in significant changes in numbers of other functionally important groups, such as the sulfate-reducing bacteria (4, 28), which in turn may trigger a response from the host. In addition, loss of butyrate producers, which have anti-inflammatory activity (50), may result in the host suffering greater levels of inflammation in the gut. However, a much more in-depth analysis of the hosts' metabonome would need to be undertaken in order to verify which functions were changing in this community, and this variation would need to be correlated with changes in the bacterial groups. An alternative scenario is that variations in the gut microbiota result in the host's immune system responding too strongly to the changes and thus initiating inflammation. In a healthy gut where the community is relatively stable, the immune system is constantly sampling this collection of bacteria and each time regards it as “self” and does not initiate a robust inflammatory response. However, if the diversity of microbiota was fluctuating to a greater extent than one would find in a healthy subject, the immune system may “regard” the different numbers of bacteria as a significant threat, since they are outside the normal range of variation, and initiate a response which in turn results in damage to the host. One key element in this scenario is the trigger that causes an individual's gut microbiota to change from relatively stable to unstable. A potential candidate for the trigger may be genetic, e.g., a mutation in the CARD15/NOD2 locus, but there is evidence to suggest that the coincidence of this mutation with Crohn's disease and a reduced bacterial diversity is not 100% (44). Furthermore, it is unclear which comes first, whether the change in stability of the gut microbiota triggers inflammation or vice versa.
Analysis of the C. leptum subgroup, C. coccoides subgroup, B. fragilis subgroup, Bifidobacterium spp., and LAB also revealed unexpected observations. The LAB profiles were complex, and temporal stability was low but still comparable to that of the control group. Nielsen and colleagues' analysis of the spatial distribution of LAB in biopsy samples revealed complex and variable communities with respect to sampling site and host (39). Walter and colleagues (65) previously observed the lack of stability in this bacterial group in healthy subjects and concluded that a significant proportion of the LAB populations were related to food-associated species. Indeed, the view of lactobacilli as allochthonous species in the intestinal tract has been recently proposed (60), and this classification would account for their apparent instability in the gastrointestinal tract of all our samples. Previous culture-independent investigations of the LAB in Crohn's disease have shown differing results, with loss of diversity reported (44) and no changes reported (38, 53). Thus, our observations add to the consensus of “loss of diversity” but temporally unstable and may suggest that diet needs to be more thoroughly controlled in these experiments in order to determine changes in the autochthonous LAB community.
A similar stable community was observed for the Bifidobacterium spp. for all the groups investigated, and this observation has been previously reported (31, 39, 53); however, this is the first report of stability of this group for Crohn's disease patients in remission and relapse. Culture-dependent approaches had shown that fewer Bifidobacterium spp. were recovered from subjects with active Crohn's disease (13); however, from our results we suggest that this observation is due to culturing bias and does not accurately reflect the dynamics of this group in Crohn's disease. We showed that the Bifidobacterium population was stable for all three groups, and since they constitute approximately 5% of the total community (15, 54), we concluded that this group is not responsible for changes in the dominant community profiles of individuals. Furthermore, we would question their role in maintaining Crohn's disease in remission, since no significant changes were observed in the community structure during relapse.
The key result from the C. leptum and B. fragilis group-specific analysis was the significant failure to amplify the 16S rRNA gene products from members of the Crohn's disease group. These primers have been used extensively to amplify 16S rRNA gene products and have been found to very robust; therefore, any failure to amplify a signal with them was considered a significant result. While it is accepted that negative results are difficult to verify, we are confident that the results were genuine and were not due to inhibition of the PCR by factors coextracted with the DNA from the stool samples. All the DNA samples extracted here gave positive results for the universal V6-V8, LAB, and Bifidobacterium primer sets. In the latter two cases, these bacteria are not present at the numbers we would expect for the two Clostridium groups or the Bacteroides group, which failed to be amplified for some subjects. If inhibition were the cause, we would expect it to be acting uniformly and not selectively on certain bacterial groups, and less abundant groups would also be affected. Hence, we strongly believe that these are valid observation. It is therefore conceivable that the Clostridium leptum and Bacteroides groups were present in a proportion of the initial samples at low numbers, which were below the threshold of detection for the specific primers used in this study. The inability to amplify C. leptum and Bacteroides group 16S rRNA gene products, we believe, is indicative of an underlying change in these groups in Crohn's disease individuals. Changes in numbers and diversity of the C. leptum group in Crohn's disease have been previously found by using culture-independent approaches (53) and more recently by using a metagenomic approach (19, 31).
We also observed that the Bacteroides fragilis subgroup's diversity was significantly reduced in Crohn's disease, which included both remission and relapse, in addition to a failure to amplify this group from 39% of the samples. While other groups have not reported such dramatic changes in the Bacteroidetes, we concluded that our observation was made possible due to the use of multiple samples from an individual rather than single-time-point samples. These two groups of bacteria are significant members of the gut ecosystem and play central roles in maintaining functions that are essential to gut health (2, 11). The phylum Bacteroidetes has been shown to contribute to the host's ability to degrade indigestible carbohydrates (2), while the members of the order Clostridiales have been documented as being the main producers of short-chain fatty acids, such as butyrate, in the gut (8). Thus, any changes in these keystone groups may impact on the total bacterial communities' capacity to provide beneficial functions to the host.
The role of the gastrointestinal microbiota in Crohn's disease is not fully understood, but the presence of a particular bacterial species or a component of the bacteria has been shown to be critical to onset of the disease (48). This study indicates that the microbiota of Crohn's disease subjects is unstable over time compared to that of controls and that individuals with Crohn's disease have fewer bacterial species that are shared. It is not clear whether the apparent instability and atypical community present in certain Crohn's disease patients could reflect a dysregulation of host-specific immune responses to its commensals, whether this atypical community is a contributory factor to the disease, or if the atypical microbiota are present as a result of conditions in a diseased lumen that would favor their proliferation. However, one aspect which has emerged from this work and which warrants further investigation is the role of functional redundancy in the gut ecosystem. One characteristic which seems to be shared between individuals relates to the bacterial functions relevant to the gut, for example, butyrate production. While this function may be performed by different species or even genera in different individuals, the role seems to be sufficiently important in maintaining a healthy gut and is thus found in all individuals studied (46). The common nature of these functions cannot be coincidence, since maintenance of these functions is important to the host; hence, any loss of these functions will affect not only the host but also the bacterial community. Several scenarios can be constructed which ultimately lead to situations detrimental to the host; for example, loss of short chain fatty acids synthesis results in an impact on the methanogen community and thus favors the growth of sulfate-reducing bacteria and the production of toxic hydrogen sulfite. The loss of butyrate producers may result in the loss of a potential anti-inflammatory agent (21, 64, 67), which leads to more inflammation in the gut and a possible relapse; butyrate may affect inflammation by suppressing NFκB expression (51). The observation reported here and elsewhere that the Clostridiales and Bacteroidales communities are altered in Crohn's disease may indicate that we should shift our focus to understanding the functional roles bacteria play in maintaining a healthy gut and ask whether a loss of function, rather than specific organisms, plays a role in inflammatory bowel disease.
Acknowledgments
We are grateful to Hans Heilig, Erwin Zoentendal, Ineke de Jong, Elaine Vaughan, and Hauke Smidt of Wageningen Microbiology Dept., The Netherlands, for their advice.
The authors are supported in part by Science Foundation Ireland, Higher Education Authority, and the Health Research Board.
Footnotes
Published ahead of print on 20 September 2006.
REFERENCES
- 1.Amann, R. I., W. Ludwig, and K. H. Schleifer. 1995. Phylogenetic identification and in-situ detection of individual microbial-cells without cultivation. Microb. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bäckhed, F., R. E. Ley, J. L. Sonnenburg, D. A. Peterson, and J. I. Gordon. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915-1920. [DOI] [PubMed] [Google Scholar]
- 3.Barcenilla, A., S. E. Pryde, J. C. Martin, S. H. Duncan, C. S. Stewart, C. Henderson, and H. J. Flint. 2000. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 66:1654-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belenguer, A., S. H. Duncan, A. G. Calder, G. Holtrop, P. Louis, G. E. Lobley, and H. J. Flint. 2006. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl. Environ. Microbiol. 72:3593-3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaut, M., M. D. Collins, G. W. Welling, J. Dore, J. van Loo, and W. de Vos. 2002. Molecular biological methods for studying the gut microbiota: the EU human gut flora project. Br. J. Nutr. 87:S203-S211. [DOI] [PubMed] [Google Scholar]
- 6.Boclé, J.-C., and C. Thomann. 2005. Effects of probiotics and prebiotics on flora and immunity in adults, p. 59-128. In L'Agence Française de Sécurité Sanitaire des Aliments report. L'Agence Française de Sécurité Sanitaire des Aliments, Maisons-Alfort, France.
- 7.Boon, N., W. De Windt, W. Verstraete, and E. M. Top. 2002. Evaluation of nested PCR-DGGE (denaturing gradient gel electrophoresis) with group-specific 16S rRNA primers for the analysis of bacterial communities from different wastewater treatment plants. FEMS Microbiol. Ecol. 39:101-112. [DOI] [PubMed] [Google Scholar]
- 8.Charrier, C., G. J. Duncan, M. D. Reid, G. J. Rucklidge, D. Henderson, P. Young, V. J. Russell, R. I. Aminov, H. J. Flint, and P. Louis. 2006. A novel class of CoA-transferase involved in short-chain fatty acid metabolism in butyrate-producing human colonic bacteria. Microbiology 152:179-185. [DOI] [PubMed] [Google Scholar]
- 9.de la Cochetière, M. F., T. Durand, P. Lepage, A. Bourreille, J. P. Galmiche, and J. Dore. 2005. Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J. Clin. Microbiol. 43:5588-5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doré, J., A. Sghir, G. Hannequart-Gramet, G. Corthier, and P. Pochart. 1998. Design and evaluation of a 16S rRNA-targeted oligonucleotide probe for specific detection and quantitation of human faecal Bacteroides populations. Syst. Appl. Microbiol. 21:65-71. [DOI] [PubMed] [Google Scholar]
- 11.Duncan, S. H., G. Holtrop, G. E. Lobley, A. G. Calder, C. S. Stewart, and H. J. Flint. 2004. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 91:915-923. [DOI] [PubMed] [Google Scholar]
- 12.Eckburg, P. B., E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman. 2005. Diversity of the human intestinal microbial flora. Science 308:1635-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favier, C., C. Neut, C. Mizon, A. Cortot, J. F. Colombel, and J. Mizon. 1997. Fecal beta-D-galactosidase production and Bifidobacteria are decreased in Crohn's disease. Dig. Dis. Sci. 42:817-822. [DOI] [PubMed] [Google Scholar]
- 14.Fisher, R. A., A. S. Corbet, and C. B. Williams. 1943. The relation between the number of species and the number of individuals in a random sample of an animal population. J. Anim. Sci. 12:42-58. [Google Scholar]
- 15.Franks, A. H., H. J. Harmsen, G. C. Raangs, G. J. Jansen, F. Schut, and G. W. Welling. 1998. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Appl. Environ. Microbiol. 64:3336-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garry, P., R. A. Craig, and C. S. Holling. 1998. Ecological resilience, biodiversity, and scale. Ecosystems 1:6-18. [Google Scholar]
- 17.Gray, N. D., and I. M. Head. 2001. Linking genetic identity and function in communities of uncultured bacteria. Environ. Microbiol. 3:481-492. [DOI] [PubMed] [Google Scholar]
- 18.Guarner, F., and J. R. Malagelada. 2003. Role of bacteria in experimental colitis. Best Pract. Res. Clin. Gastroenterol. 17:793-804. [DOI] [PubMed] [Google Scholar]
- 19.Hendrickson, B. A., R. Gokhale, and J. H. Cho. 2002. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. 15:79-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hugot, J. P., M. Chamaillard, H. Zouali, S. Lesage, J. P. Cezard, J. Belaiche, S. Almer, C. Tysk, C. A. O'Morain, M. Gassull, V. Binder, Y. Finkel, A. Cortot, R. Modigliani, P. Laurent-Puig, C. Gower-Rousseau, J. Macry, J. F. Colombel, M. Sahbatou, and G. Thomas. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411:599-603. [DOI] [PubMed] [Google Scholar]
- 21.Huuskonen, J., T. Suuronen, T. Nuutinen, S. Kyrylenko, and A. Salminen. 2004. Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br. J. Pharmacol. 141:874-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaufmann, P., A. Pfefferkorn, M. Teuber, and L. Meile. 1997. Identification and quantification of Bifidobacterium species isolated from food with genus-specific 16S rRNA-targeted probes by colony hybridization and PCR. Appl. Environ. Microbiol. 63:1268-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kempton, R. A., and L. R. Taylor. 1974. Log-series and log-normal parameters as diversity discriminants for the Lepidoptera. J. Anim. Sci. 43:381-399. [Google Scholar]
- 24.Lepage, P., P. Seksik, M. Sutren, M. F. de la Cochetiere, R. Jian, P. Marteau, and J. Dore. 2005. Biodiversity of the mucosa-associated microbiota is stable along the distal digestive tract in healthy individuals and patients with IBD. Inflamm. Bowel Dis. 11:473-480. [DOI] [PubMed] [Google Scholar]
- 25.Ley, R. E., D. A. Peterson, and J. I. Gordon. 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837-848. [DOI] [PubMed] [Google Scholar]
- 26.Lodes, M. J., Y. Cong, C. O. Elson, R. Mohamath, C. J. Landers, S. R. Targan, M. Fort, and R. M. Hershberg. 2004. Bacterial flagellin is a dominant antigen in Crohn disease. J. Clin. Investig. 113:1296-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loreau, M., S. Naeem, P. Inchausti, J. Bengtsson, J. P. Grime, A. Hector, D. U. Hooper, M. A. Huston, D. Raffaelli, B. Schmid, D. Tilman, and D. A. Wardle. 2001. Biodiversity and ecosystem functioning: current knowledge and future challenges. Science 294:804-808. [DOI] [PubMed] [Google Scholar]
- 28.Macfarlane, S., and G. T. Macfarlane. 2003. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 62:67-72. [DOI] [PubMed] [Google Scholar]
- 29.Mai, V., and J. G. Morris. 2004. Colonic bacterial flora: changing understandings in the molecular age. J. Nutr. 134:459-464. [DOI] [PubMed] [Google Scholar]
- 30.Mangin, I., R. Bonnet, P. Seksik, L. Rigottier-Gois, M. Sutren, Y. Bouhnik, C. Neut, M. D. Collins, J. F. Colombel, P. Marteau, and J. Dore. 2004. Molecular inventory of faecal microflora in patients with Crohn's disease. FEMS Microbiol. Ecol. 50:25-36. [DOI] [PubMed] [Google Scholar]
- 31.Manichanh, C., L. Rigottier-Gois, E. Bonnaud, K. Gloux, E. Pelletier, L. Frangeul, R. Nalin, C. Jarrin, P. Chardon, P. Marteau, J. Roca, and J. Dore. 2006. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 55:205-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marteau, P., P. Seksik, and F. Shanahan. 2003. Manipulation of the bacterial flora in inflammatory bowel disease. Best Pract. Res. Clin. Gastroenterol. 17:47-61. [DOI] [PubMed] [Google Scholar]
- 33.Matsuki, T., K. Watanabe, J. Fujimoto, Y. Miyamoto, T. Takada, K. Matsumoto, H. Oyaizu, and R. Tanaka. 2002. Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Appl. Environ. Microbiol. 68:5445-5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuki, T., K. Watanabe, J. Fujimoto, T. Takada, and R. Tanaka. 2004. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol. 70:7220-7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCann, K. S. 2000. The diversity-stability debate. Nature 405:228-233. [DOI] [PubMed] [Google Scholar]
- 36.Muyzer, G., E. C. de Waal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muyzer, G., and K. Smalla. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Leeuwenhoek 73:127-141. [DOI] [PubMed] [Google Scholar]
- 38.Mylonaki, M., N. B. Rayment, D. S. Rampton, B. N. Hudspith, and J. Brostoff. 2005. Molecular characterization of rectal mucosa-associated bacterial flora in inflammatory bowel disease. Inflamm. Bowel Dis. 11:481-487. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen, D. S., P. L. Moller, V. Rosenfeldt, A. Paerregaard, K. F. Michaelsen, and M. Jakobsen. 2003. Case study of the distribution of mucosa-associated Bifidobacterium species, Lactobacillus species, and other lactic acid bacteria in the human colon. Appl. Environ. Microbiol. 69:7545-7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nubel, U., B. Engelen, A. Felske, J. Snaidr, A. Wieshuber, R. I. Amann, W. Ludwig, and H. Backhaus. 1996. Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J. Bacteriol. 178:5636-5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogura, Y., D. K. Bonen, N. Inohara, D. L. Nicolae, F. F. Chen, R. Ramos, H. Britton, T. Moran, R. Karaliuskas, R. H. Duerr, J. P. Achkar, S. R. Brant, T. M. Bayless, B. S. Kirschner, S. B. Hanauer, G. Nunez, and J. H. Cho. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411:603-606. [DOI] [PubMed] [Google Scholar]
- 42.Orholm, M., V. Binder, T. I. Sorensen, L. P. Rasmussen, and K. O. Kyvik. 2000. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand. J. Gastroenterol. 35:1075-1081. [DOI] [PubMed] [Google Scholar]
- 43.O'Sullivan, G. C., P. Kelly, S. O'Halloran, C. Collins, J. K. Collins, C. Dunne, and F. Shanahan. 2005. Probiotics: An emerging therapy. Curr. Pharm. Des. 11:3-10. [DOI] [PubMed] [Google Scholar]
- 44.Ott, S. J., M. Musfeldt, D. F. Wenderoth, J. Hampe, O. Brant, U. R. Folsch, K. N. Timmis, and S. Schreiber. 2004. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53:685-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prindiville, T., M. Cantrell, and K. H. Wilson. 2004. Ribosomal DNA sequence analysis of mucosa-associated bacteria in Crohn's disease. Inflamm. Bowel Dis. 10:824-833. [DOI] [PubMed] [Google Scholar]
- 46.Pryde, S. E., S. H. Duncan, G. L. Hold, C. S. Stewart, and H. J. Flint. 2002. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217:133-139. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook, J., and D. Russell. 2000. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 48.Sartor, R. B. 2001. Intestinal microflora in human and experimental inflammatory bowel disease. Curr. Opin. Gastroenterol. 17:324-330. [DOI] [PubMed] [Google Scholar]
- 49.Savage, D. C. 1977. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 31:107-133. [DOI] [PubMed] [Google Scholar]
- 50.Scheppach, W., and F. Weiler. 2004. The butyrate story: old wine in new bottles? Curr. Opin. Clin. Nutr. Metab. Care 7:563-567. [DOI] [PubMed] [Google Scholar]
- 51.Segain, J. P., R. de la Bletiere, A. Bourreille, V. Leray, N. Gervois, C. Rosales, L. Ferrier, C. Bonnet, H. M. Blottiere, and J. P. Galmiche. 2000. Butyrate inhibits inflammatory responses through NFκB inhibition: implications for Crohn's disease. Gut 47:397-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seksik, P., P. Lepage, M. F. de la Cochetiere, A. Bourreille, M. Sutren, J. P. Galmiche, J. Dore, and P. Marteau. 2005. Search for localized dysbiosis in Crohn's disease ulcerations by temporal temperature gradient gel electrophoresis of 16S rRNA. J. Clin. Microbiol. 43:4654-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seksik, P., L. Rigottier-Gois, G. Gramet, M. Sutren, P. Pochart, P. Marteau, R. Jian, and J. Dore. 2003. Alterations of the dominant faecal bacterial groups in patients with Crohn's disease of the colon. Gut 52:237-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sghir, A., G. Gramet, A. Suau, V. Rochet, P. Pochart, and J. Dore. 2000. Quantification of bacterial groups within human fecal flora by oligonucleotide probe hybridization. Appl. Environ. Microbiol. 66:2263-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shanahan, F. 2002. The host-microbe interface within the gut. Best Pract. Res. Clin. Gastroenterol. 16:915-931. [DOI] [PubMed] [Google Scholar]
- 56.Shannon, C. E., and W. Weaver. 1949. The mathematical theory of communication, p. 1-144. University of Illinois Press, Urbana.
- 57.Simpson, E. H. 1949. Measurement of diversity. Nature 163:688. [Google Scholar]
- 58.Swidsinski, A., J. Weber, V. Loening-Baucke, L. P. Hale, and H. Lochs. 2005. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J. Clin. Microbiol. 43:3380-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamboli, C. P., C. Neut, P. Desreumaux, and J. F. Colombel. 2004. Dysbiosis in inflammatory bowel disease. Gut 53:1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tannock, G. W. 2004. A special fondness for lactobacilli. Appl. Environ. Microbiol. 70:3189-3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tysk, C., E. Lindberg, G. Jarnerot, and B. Floderus-Myrhed. 1988. Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 29:990-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vanhoutte, T., G. Huys, E. De Brandt, and J. Swings. 2004. Temporal stability analysis of the microbiota in human feces by denaturing gradient gel electrophoresis using universal and group-specific 16S rRNA gene primers. FEMS Microbiol. Ecol. 48:437-446. [DOI] [PubMed] [Google Scholar]
- 63.Vaughan, E. E., F. Schut, H. G. Heilig, E. G. Zoetendal, W. M. de Vos, and A. D. Akkermans. 2000. A molecular view of the intestinal ecosystem. Curr. Issues Intest. Microbiol. 1:1-12. [PubMed] [Google Scholar]
- 64.Wächtershäuser, A., and J. Stein. 2000. Rationale for the luminal provision of butyrate in intestinal diseases. Eur. J. Nutr. 39:164-171. [DOI] [PubMed] [Google Scholar]
- 65.Walter, J., C. Hertel, G. W. Tannock, C. M. Lis, K. Munro, and W. P. Hammes. 2001. Detection of Lactobacillus, Pediococcus, Leuconostoc, and Weissella species in human feces by using group-specific PCR primers and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 67:2578-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong, J. M., R. de Souza, C. W. Kendall, A. Emam, and D. J. Jenkins. 2006. Colonic health: fermentation and short chain fatty acids. J. Clin. Gastroenterol. 40:235-243. [DOI] [PubMed] [Google Scholar]
- 67.Yin, L., G. Laevsky, and C. Giardina. 2001. Butyrate suppression of colonocyte NF-κB activation and cellular proteasome activity. J. Biol. Chem. 276:44641-44646. [DOI] [PubMed] [Google Scholar]
- 68.Zoetendal, E. G., A. L. Akkermans, and W. M. deVos. 1998. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl. Environ. Microbiol. 64:3854-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]