

Abstract
More than 21 million prescriptions for warfarin are written yearly in the U.S. Despite its importance, warfarin's target, vitamin K epoxide reductase (VKOR), has resisted purification since its identification in 1972. Here, we report its purification and reconstitution. HPC4, a calcium-specific antibody that recognizes a 12-aa tag, was used to purify and identify VKOR. Partial reconstitution is achieved on the column by washing with 0.4% dioleoylphosphatidylcholine/0.4% deoxycholate. Activity is completely recovered by dialysis against a buffer containing a reducing agent but lacking dioleoylphosphatidylcholine/deoxycholate. Removal of detergent from the eluted proteins apparently facilitates liposome formation. Purified recombinant VKOR with tag is ≈21 kDa, as expected; fully active; and >93% pure. The concentration of warfarin for 50% inhibition is the same for purified protein and microsomes. It has been reported that VKOR is a multisubunit enzyme. Our results, however, suggest that a single peptide can accomplish both the conversion of vitamin K epoxide to vitamin K and vitamin K to reduced vitamin K. This purification will allow further characterization of VKOR in relation to other components of the vitamin K cycle and should facilitate its structural determination.
Keywords: purification, reconstitution, membrane protein
In animals, vitamin K is primarily known as a cosubstrate in the posttranslational conversion of glutamate to γ-carboxyglutamate (Gla). There are at least 16 Gla-containing proteins in mammals, and eight of these are related to coagulation. Vitamin K is believed to serve as a precursor to an intermediate that can abstract a proton from the 4-carbon of glutamate to form a carbanion. In turn, this carbanion is attacked by CO2 to make Gla (1). In the process, vitamin K is converted to vitamin K epoxide, which must be recycled to reduced vitamin K for the next round of reaction (2). The enzyme that catalyzes the conversion of vitamin K epoxide to vitamin K is vitamin K epoxide reductase (VKOR) (3).
VKOR has proved refractory to purification, and thus all characterizations of VKOR to date have been done with purified microsomes. Whitlon et al. (4) reported detection of VKOR activity in microsomes but not in detergent-solubilized microsomes. Fasco and coworkers reported partial purification of VKOR from rat livers (5); they used cholate to partially solubilize microsomes, followed by separation on a sucrose gradient to isolate a 200 S microsomal subfraction that contained VKOR activity. All attempts at further purification resulted in loss of enzymatic activity; however, a small protein that reacted with radioactive N-ethylmaleimide was identified in the 200 S fraction. They conjectured that this small protein was a component of a multiprotein complex that converted vitamin K epoxide to vitamin K. A further purification attempt was reported by Cain et al. (6). These authors also concluded that VKOR was a multiprotein complex, and that the two components of this complex were microsomal epoxide hydrolase and GST. However, it was later shown that a mouse knockout of microsomal epoxide hydrolase had no defect in vitamin K metabolism, and the mouse exhibited no bleeding diathesis (7).
Although both our laboratory (8) and Rost et al. (9) reported the identification of the VKOR gene in 2004, purification of its product has remained elusive. Here we report the purification of recombinant human VKOR produced from a baculovirus in insect cells.
Results
Purification of VKOR.
Because VKOR is an integral membrane protein, its solubilization from microsomes was necessary for purification. We tested 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC), CHAPS, cholate, deoxycholate, Triton X-100, and Nikkol. Preliminary experiments established that DHPC is most effective, meaning that more VKOR remained in the postmicrosomal supernatant after solubilization. The relative amount of VKOR in supernatant and pellet was measured by quantitative Western blotting (data not shown). In addition, the critical micelle concentration of DHPC is relatively high, and it is a mild detergent that helps preserve 3D protein structure and enzymatic activity (10).
Purification is presented in detail in Materials and Methods. In summary, a 27-aa tag, which included the 12-aa HPC4 tag (11) and a tandem GGS flexible linker, was attached to the N terminus of VKOR for affinity purification. In addition, the tag was also used to detect VKOR during purification, because solubilization results in loss of enzymatic activity. The monoclonal antibody that recognizes this tag is calcium-specific, binds the tag in the presence of calcium, and releases it when EDTA is added. Because it is difficult to completely solubilize membrane proteins, an extensive wash with DHPC was used to eliminate most contaminating proteins from the affinity resin. Our rationale is that if VKOR within a membrane fragment is bound to HPC4, contaminating proteins that are embedded in the same membrane fragment are washed out by DHPC. It is also important to wash the column with MgATP to remove 70-kDa heat-shock protein, which is commonly associated with denatured protein in the endoplasmic reticulum. A further wash with 0.4% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/0.4% deoxycholate resulted in partial reconstitution of VKOR activity.
Fig. 1 shows a reducing SDS-NuPAGE gel analysis with enzymatic activity superimposed. Recombinant VKOR is the main band and migrates, as expected, from its coding sequence (and added tag) at ≈21 kDa (8, 9). The enzymatic activity was measured as described in Materials and Methods. In DHPC, VKOR activity is significantly inhibited, as shown in solubilized microsomes (Fig. 1, lane 1). As demonstrated in Fig. 1, VKOR enzymatic activity is correlated with the intensity of the protein band at ≈21 kDa in the eluted fractions.
Fig. 1.

Reducing NuPAGE analysis of fractions from purification of VKOR. Fraction numbers of VKOR eluted from the HPC4 antibody column are shown on the x axis. Molecular weight markers are shown in lane 1. Lane 2, solubilized microsomes. Lane 3, column flowthrow. F = 26 μl of sample from each (1-ml) fraction was analyzed by 10% NuPAGE followed by SYPRO Ruby staining. A 20-μl sample from each fraction was tested for VKOR enzyme activity, as described in Materials and Methods. Data are presented as mean ± SD (n = 3).
Identification of Contaminating Proteins.
To identify the contaminating proteins, we purified VKOR by the method described above, except that DOPC was omitted from the elution buffer so that the purified VKOR could be further concentrated by Microcon YM-10 ultrafiltration. When higher concentrations of purified VKOR are subjected to SDS-NuPAGE, the high degree of purity is apparent. However, contaminating protein bands were still observed (Fig. 2). We estimated the purity of our purified VKOR by analyzing the intensity of each protein band stained with SYPRO ruby (contaminating bands are not visible with Coomassie blue staining) using an AlphaImager 2200 (Alpha Innotech, San Leandro, CA). According to these calculations, VKOR is 92–97% of the purified material, depending on the definition of the best-fit line (data not shown).
Fig. 2.
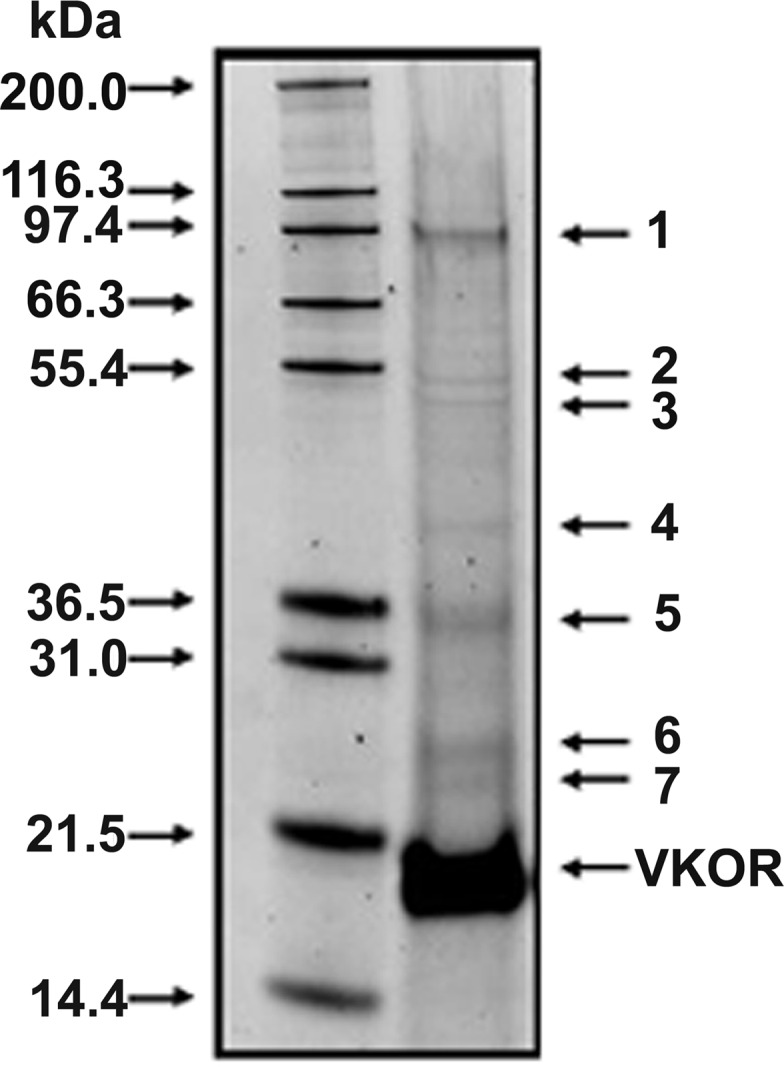
Estimation of purity and identification of contaminating proteins in purified VKOR. Purified VKOR was subjected to a 10% reducing SDS-NuPAGE gel and stained by SYPRO Ruby. Molecular weight markers are shown in the left lane, and purified VKOR is shown in the right lane. The contaminating proteins were excised and subjected to mass spectrometry for identification. 1, sarco(endo)plasmic reticulum-type calcium ATPase; 2, mitochondrial ATP synthase α-subunit; 3, mitochondrial ATP synthase β-subunit; 4, ribosome-associated protein p40; 5 and 6, Autographa californica nucleopolyhedrovirus protein; 7, ADP-ribosylation factor.
Contaminating proteins (Fig. 2, bands 1–7) were analyzed by mass spectroscopy. We were able to identify seven contaminating proteins. Among the most prominent proteins, band 1 is sarco(endo)plasmic reticulum-type calcium ATPase, and bands 5 and 6 are nucleopolyhedrovirus proteins. The other identified proteins, numbers 2, 3, and 4, are α- and β-subunits of mitochondrial ATP synthase and ribosome-associated protein p40, respectively. It is apparent that none of the identified contaminants are likely to affect VKOR activity or to act as components of a multisubunit enzyme.
Reconstitution of Purified VKOR.
Addition of any detergent we tested resulted in decreased VKOR activity compared with microsomes alone. Our previous topology experiments led us to predict that the active site is within the membrane, and that an intact membrane is needed for VKOR activity (12). Therefore, we attempted to establish an environment mimicking the membrane to regain activity. To perform this experiment, we dialyzed purified VKOR against a buffer containing a reducing agent such as Tris(hydroxypropyl)phosphine (THP) but neither DOPC nor deoxycholate. Using these conditions, deoxycholate is removed by dialysis, but DOPC remains with the protein in the dialysis bag and presumably forms membrane-like structures. Subsequently, we collected samples at different times and compared turnover numbers. By turnover number, we are referring to the rate of reaction divided by the enzyme concentration at saturating KO and DTT, under conditions described in Materials and Methods.
Fig. 3 compares the turnover number of purified VKOR to the turnover number of VKOR in microsomes. Before dialysis, the turnover number of purified VKOR is slightly lower than its turnover number in microsomes (Fig. 3, bar 2). After 24-h dialysis to remove deoxycholate, the solution becomes turbid, presumably indicating formation of membrane-like structures. In the presence of 4 mM THP, enzymatic activity increases with dialysis time and reaches a maximum by 48 h with a turnover number of vitamin K formation at 0.24 sec−1, approximately three times higher than in the starting microsomes (Fig. 3, bars 3–6).
Fig. 3.

Reconstitution of VKOR activity. Purified VKOR was dialyzed against buffer A containing 4 mM THP, and samples were taken at different time points. Enzyme activities were measured and are presented as turnover number (mol Vit K produced/mol VKOR/sec). Bar 1, microsomes; bar 2, purified VKOR before dialysis; bars 3–6, purified VKOR dialyzed for 5, 24, 48, or 96 h against buffer A with 4 mM THP, respectively. Data are presented as mean ± SD (n = 3).
Effects of THP and Liposome Formation.
We performed another experiment to elucidate the importance of detergent removal compared with the effect of the reducing agent both in microsomes (Fig. 4, bars 1–4) and purified VKOR (Fig. 4, bars 5–9). Bar 1 shows the activity of microsomes before the addition of detergent. If the deoxycholate-containing elution buffer is added to microsomes, there is some loss of activity, probably because of the presence of detergent in the elution buffer (bar 2). It is not only the detergent that causes loss of activity, however, because dialysis to remove the detergent does not result in the return of activity to the level measured in microsomes (bar 3). Adding THP to microsomes after dialysis causes an increase in turnover number to a value greater than that seen in the starting microsomes (bar 4). Bars 5–9 depict results with purified VKOR. Bar 5 represents VKOR eluted with 0.4% DOPC/0.4% deoxycholate and diluted 10-fold for enzyme activity assays. For bar 6, elution was done as for bar 5, but 4 mM THP was added after elution, and the sample was incubated for 24 h before the assay. Bars 7–9 represent results with purified VKOR dialyzed for 96 h against buffer A. Removal of detergent alone does not restore activity (bar 7). If after dialysis the sample is incubated an additional 24 h with 4 mM THP, there is a significant increase in turnover number (bar 8). The activity of VKOR is also increased by dialysis against buffer A containing THP (bar 9). However, neither incubation with THP without dialysis (bar 6) nor dialysis without THP (bar 7) results in increased VKOR turnover number. Surprisingly, dialysis followed by addition of THP or dialysis in the presence of THP results in a higher turnover number than is found in the starting microsomes (bars 8 and 9). Thus, both removal of detergent and the presence of a strong reductant, such as THP, are crucial for reconstitution of VKOR activity, both in microsomes and with the purified protein.
Fig. 4.

The effect of dialysis on activity; removal of deoxycholate compared with addition of THP. All samples have the same amount of VKOR. Bars 1–4 represent microsomes, and bars 5–9 represent purified VKOR. Bar 1, microsomes. Bar 2, microsomes in 0.4% DOPC/0.4% deoxycholate. Bar 3, microsomes in 0.4% DOPC/0.4% deoxycholate dialyzed to remove deoxycholate. Bar 4, microsomes in 0.4% DOPC/0.4% deoxycholate dialyzed to remove deoxycholate then incubated 24 h with 4 mM THP. Bars 5–9, purified VKOR. Bar 5, purified VKOR in 0.4% DOPC/0.4% deoxycholate. Bar 6, purified VKOR incubated overnight with 4 mM THP before assay. Bar 7, purified VKOR dialyzed for 96 h against buffer A. Bar 8, purified VKOR dialyzed for 96 h against buffer A then further incubated with 4 mM THP at 4°C for 24 h. Bar 9, purified VKOR dialyzed against buffer A containing 4 mM THP for 96 h. Data are presented as mean ± SD (n = 3).
Warfarin Inhibition.
We previously suggested that VKOR can convert both vitamin K epoxide to vitamin K and vitamin K to vitamin KH2 (13). In microsomes and purified protein, the vitamin K epoxide to vitamin K conversion showed similar sensitivity of warfarin inhibition (Fig. 6). We also tested the purified enzyme for its ability to convert vitamin K to vitamin KH2. We demonstrated that purified VKOR can accomplish the vitamin K to KH2 reaction with a turnover number of 0.016 sec−1 (Fig. 5). For purified VKOR, warfarin inhibition of the conversion of vitamin K to vitamin KH2 is more sensitive than the conversion of vitamin K epoxide to vitamin K (Fig. 6).
Fig. 6.

Inhibition of VKOR by warfarin. The reaction was performed by using microsomes or purified VKOR that had been dialyzed in the presence of THP. Various concentrations of warfarin were added to both microsomes and purified VKOR. Both microsomes (●) and purified VKOR (■) were used to study the vitamin K epoxide to vitamin K reaction, whereas purified VKOR (□) alone was used to study the vitamin K to vitamin KH2 reaction. The activity was determined as described in Materials and Methods. The results are represented as percent VKOR activity without warfarin (n = 3).
Fig. 5.

Conversion of Vitamin K to vitamin KH2 by VKOR. The reaction was performed by using purified VKOR that had been dialyzed in the presence of THP. VKOR activity is represented as nmol KH2 produced per 60 min. Bar 1, DTT with elution buffer as background control. Bar 2, purified VKOR after dialysis against buffer A with 4 mM THP. Data are presented as mean ± SD (n = 3).
Discussion
In retrospect, the difficulties in purifying VKOR are obvious. All of the detergents that we used to solubilize VKOR also rendered it inactive. One possible reason for this is that the active site (14–16) is embedded in the membrane and may require the membrane structure for activity (12). For this reason, we attached tags at either the N or C terminus to allow purification of VKOR and also to detect its presence during purification. The presumption was that the activity could be recovered after purification. In this study, we used the construct with the N-terminal tag; however, the turnover numbers of purified VKOR for both constructs were similar (data not shown). This suggests that the tag had minimal effect on enzymatic activity.
We tried a number of detergents to determine which were most suitable for solubilization. Western blot analysis was used to select the detergent that left the most VKOR in the supernatant after a 150,000 × gavg centrifugation of solubilized microsomes. For solubilization, DHPC was the most effective. Although VKOR activity is nil in the presence of DHPC, after binding to the HPC4 affinity column and extensive washing, activity can be recovered by changing the wash buffer to 0.4% DOPC/0.4% deoxycholate. After elution with EDTA in this buffer, activity is recovered (the enzyme must be diluted at least 10-fold into the reaction mix to measure activity), and the activity is similar to that in microsomes (Fig. 3).
There have been reports that VKOR is a multisubunit enzyme (6); thus, one of the major questions concerning VKOR is, can a single polypeptide accomplish this reaction? To clarify this issue, we tried two approaches. The first was to measure the turnover number of VKOR in microsomes and compare it to the turnover number of purified VKOR. We measured the turnover number in microsome suspensions, because detergents cause activity loss. If VKOR were a multisubunit enzyme, then purification of the product predicted by the identified cDNA (9) would be expected to have a much-reduced turnover number. If an essential component were lost or even much reduced but still present as a contaminant, the turnover number of VKOR in microsomes would be much greater than that of the purified enzyme. However, the turnover number of the dialyzed reduced purified enzyme was actually approximately three times higher than that of VKOR in microsomes (Fig. 3). This increased turnover number provides strong evidence that the single VKOR gene, recently identified by molecular biology techniques (8, 17), codes for the enzyme that converts vitamin K epoxide to vitamin K.
We also tried to eliminate the possibility that any of the minor contaminants seen in our VKOR preparations are related to VKOR function. We concentrated the purified protein and overloaded SDS-NuPAGE gels to identify contaminating proteins. There are several contaminating proteins; however, they are <7% of total purified protein by weight and certainly much less on a molar basis, because VKOR is smaller than any of the contaminants (Fig. 2). Moreover, none of the identified contaminating proteins, sarcoplasmic reticulum-type calcium ATPase, mitochondrial ATP synthase α- and β-subunits, ribosome-associated protein p40, and nucleopolyhedrovirus protein from the expression vector are likely to contribute to VKOR activity. These contaminants can be minimized by extensive washing but with more extensive washing, VKOR is also lost from the Ca2+-dependent monoclonal antibody to human protein C (HPC4 antibody) column.
The increase in turnover number in the purified protein compared with microsomes was somewhat surprising. The active site of VKOR has been proposed to be cysteine residues 132 and 135, and these must be in their reduced form to catalyze the vitamin K epoxide to vitamin K reaction (14–16). Therefore, one possibility for the further increase in turnover number upon dialysis against buffer containing a reducing agent (DTT or THP) is that these bonds may be in a partially oxidized state and may be converted into the fully active reduced state upon exposure to reducing agent. Another possibility for the increase in turnover number after dialysis with reducing agent is that the insect cells are making more enzyme than can be properly folded. In our case, ≈3% of microsomal protein is recombinant human VKOR. If improper disulfide bonds are formed, they may be rearranged during dialysis in the presence of the reducing agent.
Not only is the turnover number of purified VKOR higher than that measured in microsomes; it is also ≈10-fold higher than that of the γ-glutamyl carboxylase (18). Given that VKOR and carboxylase are integral membrane proteins, and that in vitro assays are far removed from actual in vivo conditions, which at the present are unknown, the VKOR turnover number appears to be adequate for the vitamin K cycle. At this time, we are not able to comment on the report that the chaperonin calumenin is associated with both VKOR and the γ-glutamyl carboxylase and has a role in the vitamin K cycle (19). However, our results argue against the notion that a multicomponent enzyme is required for VKOR activity.
Based on our results, the only activity not provided by purified VKOR is an enzyme to reduce the disulfide form of VKOR to the active reduced form. In our assays, as well as almost all previous work on VKOR, this presumed enzyme is replaced by DTT. The presumed enzyme that re-reduces VKOR in vivo is still not known but may be analogous to thioredoxin. In fact, Silverman and Nandi demonstrated that Escherichia coli thioredoxin and thioredoxin reductase can stimulate the VKOR reaction in microsomes (20).
We published that VKOR can dramatically increase the fraction of carboxylated factor X produced in HEK 293 cells (13). These results have been confirmed by Wallin's and Berkner's laboratories (21, 22). Because vitamin K is fed to the cells, it seemed reasonable that VKOR was converting vitamin K epoxide to vitamin K and vitamin K to vitamin KH2. Therefore, we tested purified VKOR for its vitamin K to vitamin KH2 activity. Our results are clear but preliminary. One difficulty is that the enzyme that converts the oxidized cysteines back to their reduced active forms of VKOR is still not known; therefore, we use DTT in the reaction mix to catalyze the reaction. The problem is that DTT also reduces vitamin K to vitamin KH2. To overcome this obstacle, it was necessary to lower the concentration of the vitamin K substrate so that the chemical conversion by DTT was negligible compared with the enzymatic conversion by VKOR. We have repeated the assay multiple times and are confident that VKOR can catalyze the vitamin K to vitamin KH2 reaction. However, we have not yet optimized the reaction. We used the same conditions as for the vitamin K epoxide to vitamin K, so the turnover number is nominal. The observation that VKOR conversion of vitamin K to vitamin KH2 is also inhibited by warfarin confirms the necessity of another enzyme such as DT-diaphorase to carry out this conversion when using vitamin K to treat patients with warfarin overdose.
In summary, we have shown that VKOR may be purified as a single peptide, and that this peptide can accomplish the conversion of both vitamin K epoxide to vitamin K and vitamin K to vitamin KH2. Little is known about this reaction mechanism or that of warfarin binding. This purification will allow further characterization and understanding of the VKOR reaction mechanism and its interactions. Perhaps most importantly, the final preparation of active enzyme is very similar to conditions needed to form 2D crystals for electron diffraction studies.
Materials and Methods
Chemicals.
All chemicals were reagent grade. The baculovirus viral DNA was purchased from Novagen (Madison, WI). The Sf9 (Spodoptera frugiperda) insect cells were obtained from the Lineberger Cancer Center at the University of North Carolina. The HPC4 (11) antibody affinity resin was kindly provided by Charles T. Esmon (Oklahoma Medical Research Foundation, Oklahoma City, OK). Protease inhibitor mixture (10 ×) contained 5 μg/ml leupeptin, 7 μg/ml pepstatin A, 20 μg/ml aprotinin (Roche, Boulder, CO). DOPC, and DHPC were from Avanti Polar Lipids (Alabaster, AL). 3-(N-morpholino)-propanesulfonic acid (Mops) was from RPI (Mt. Prospect, IL). N-[Tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid, protein standard BSA, warfarin (3-(α-acetonylbenzyl)-4-hydroxycoumarin), deoxycholate, and β-hydroxytoluene were from Sigma (St. Louis, MO). The reducing agent THP was from Calbiochem (San Diego, CA). SnakeSkin dialysis tubing was from Pierce (Rockford, IL). PVDF and Microcon YM-10 centrifuge filter devices were from Millipore (Bedford, MA). SYPRO ruby protein gel stain reagent was from Molecular Probes (Eugene, OR). A vitamin K internal standard, 2-methyl-3(3,7,11,15,19-pentamethyl-2-eicosenyl)-1,4-naphthalenedione (K1(25)) was from GL Synthesis, Worcester, MA. Vitamin K epoxide was kindly provided by Paul Dowd (University of Pittsburgh, Pittsburgh, PA). Vitamin K1 (phytonadione) was from Abbot (Chicago, IL). ECL Western blotting detection reagents were from Amersham Biosciences (Piscataway, NJ).
Cloning and Construction of Expression Vector.
The cDNA encoding human VKOR was subcloned into the pVL1392 vector (8). An in-frame coding sequence coding for EDQVDPRLIDGK (GGS)5 was added to the N or C terminus of VKOR, and the turnover numbers were similar for the two constructs. In this manuscript, the construct with the N-terminal tag was used. The HPC4 tag is underlined; the GGS repeats function as flexible linkers to minimize potential structural perturbation from the affinity tag. The modified cDNA for human VKOR was then subcloned into the BglII and EcoR I sites of the insect cell expression vector pVL1392. The protein was expressed in baculo-virus-infected Sf 9 cell, as described (8).
Preparation of Microsomes.
Microsomes from cells overexpressing VKOR were prepared as described (23). The microsomal pellet was resuspended in 25 mM 3-(N-morpholino)-propanesulfonic acid (pH 7.5)/150 mM NaCl containing 1× protease inhibitor mixture and 30% glycerol then flash-frozen in liquid nitrogen before storing at −80°C.
Purification of VKOR.
Microsomes were diluted to a final protein concentration of 2 mg/ml total protein in 25 mM 3-(N-morpholino)-propanesulfonic acid (pH 7.5)/150 mM NaCl/20% glycerol (buffer A) plus 40 mM DHPC and 4 mM THP. After incubation at 4°C for 1 hr, the solubilized microsomes were centrifuged at 150,000 × gavg for 45 min in a Beckman 70 Ti rotor. Buffer A was made 1× protease inhibitor mixture and used to dilute supernatant to a final concentration of 8 mM DHPC. Calcium chloride was added to the supernatant to 2 mM and mixed with the HPC4 resin. The mixture was incubated at 4°C overnight with gentle rotation. The resin was poured into a column and washed with 10 column volumes of buffer A containing 15 mM DHPC, 10 mM MgATP, 2 mM CaCl2, and 4 mM THP. The resin was then washed with 10 column volumes of reconstitution buffer (0.4% DOPC/0.4% deoxycholate/2 mM CaCl2/4 mM THP in buffer A). The bound proteins were eluted with buffer A containing 0.4% DOPC/0.4% deoxycholate/10 mM EDTA/4 mM THP then stored at 4°C.
Reconstitution by Dialysis.
A further increase in VKOR activity was achieved by dialysis against buffer A in the presence of 4 mM THP at 4°C. One liter of dialysis buffer was used for each milliliter of purified VKOR, and dialysis was carried out for 96 h with one buffer change. Another set of experiments was performed by dialyzing against the same buffer but in the absence of the reducing agent THP. At the end of 96-h dialysis, 4 mM THP was added to the dialyzed sample and incubated overnight at 4°C. The reconstituted protein was stored at 4°C.
Quantitation of VKOR.
The concentration of purified VKOR was determined by densitometry analysis. The proteins were separated on reducing SDS-NuPAGE 10% Bis-Tris gel, stained with SYPRO ruby gel stain and visualized by a transilluminator at 300 nm. Concentrations of BSA and VKOR were adjusted so that the signal detected by densitometry was linear. Purified VKOR, whose concentration had been measured as above, was used as a standard for determining VKOR concentrations in microsomes. For this quantification, microsomal proteins were separated by using a reducing SDS-NuPAGE gel and then transferred to a PVDF membrane. The membrane was treated with anti-HPC4 monoclonal antibody followed by a horseradish peroxidase-conjugated secondary antibody and then visualized by the enhanced chemiluminescence Western blot reagent. The data were analyzed by an AlphaImager 2200 (Alpha Innotech, San Leandro, CA).
VKOR Activity Assay.
VKOR enzymatic activity was assayed (24, 25) with the following modifications. Twenty microliters of microsomes or purified protein was added to 180 μl of buffer B (25 mM N-[Tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid, pH 8.6/150 mM NaCl/30% glycerol). Eight microliters of 5 mM vitamin K epoxide in isopropanol was added as substrate, and 5 μl of 200 mM DTT was added to start the reaction. The reaction mixture was incubated in the dark at 30°C for 20 min, and the reaction was stopped by adding 500 μl of 0.05 M AgNO3:isopropanol (5:9) followed by adding 500 μl of 1.5 μM K125 in hexane as an internal standard. The mixture was vortexed for 1 min, followed by brief centrifugation to separate the phases. Four hundred microliters of the upper organic phase was transferred to a 1.8-ml brown vial and dried with a gentle stream of N2. Four hundred microliters of the HPLC mobile phase [acetonitrile:isopropanol:water (100:7:2)] was added to dissolve the dried sample. The sample was analyzed by HPLC (Agilent 1100, Agilent Technologies, Austin, TX) on a C-18 column (Vydac, St. Louis, MO; catalog no. 218TP54). Vitamin K production was detected by UV detector at 248 nm.
Vitamin K Reductase Activity Assay.
The ability of VKOR to convert vitamin K to KH2 was assayed similarly, except that 2 μl of vitamin K1 (10 mg/ml) was added as a substrate, and 8 pmol of purified enzyme was used. The total reaction volume was 200 μl. The reaction was carried out for 1 hr. At its completion, 500 μl of β-hydroxytoluene was added to stabilize the reduced vitamin KH2, and the product was analyzed by HPLC by using fluorescence detector at excitation wavelength 243 nm and emission wavelength at 430 nm. β-Hydroxytoluene does not cause reduction of vitamin K. Controls for background KH2 formation from DTT were done in the absence of enzyme.
Warfarin Inhibition.
Twenty microliters of microsomes or purified protein was added to 180 μl of buffer B. Various concentrations of warfarin were added to the samples and preincubated on ice for 10 min. VKOR activity from vitamin K epoxide to vitamin K and vitamin K to vitamin KH2 was assayed as described above.
Acknowledgments
We thank Yeu-Fann Stafford, Lee J. Pedersen, Paul Modrich, David Straight, and Sheue-Mei Wu for critical reading of this manuscript. Funding for this work was provided by National Institutes of Health and National Heart, Lung, and Blood Institute Grant 5-R01-HL077740-01.
Abbreviations
- VKOR
vitamin K epoxide reductase
- DHPC
1,2-dihexanoyl-sn-glycero-3-phosphocholine
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- THP
Tris(hydroxypropyl)phosphine.
Footnotes
The authors declare no conflict of interest.
References
- 1.Dowd P, Hershline R, Ham SW, Naganathan S. Science. 1995;269:1684–1691. doi: 10.1126/science.7569894. [DOI] [PubMed] [Google Scholar]
- 2.Bell RG, Matschiner JT. Nature. 1972;237:32–33. doi: 10.1038/237032a0. [DOI] [PubMed] [Google Scholar]
- 3.Willingham AK, Matschiner JT. Biochem J. 1974;140:435–441. doi: 10.1042/bj1400435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitlon DS, Sadowski JA, Suttie JW. Biochemistry. 1978;17:1371–1377. doi: 10.1021/bi00601a003. [DOI] [PubMed] [Google Scholar]
- 5.Lee JJ, Principe LM, Fasco MJ. Biochemistry. 1985;24:7063–7070. doi: 10.1021/bi00346a007. [DOI] [PubMed] [Google Scholar]
- 6.Cain D, Hutson SM, Wallin R. J Biol Chem. 1997;272:29068–29075. doi: 10.1074/jbc.272.46.29068. [DOI] [PubMed] [Google Scholar]
- 7.Miyata M, Kudo G, Lee YH, Yang TJ, Gelboin HV, Fernandez-Salguero P, Kimura S, Gonzalez FJ. J Biol Chem. 1999;274:23963–23968. doi: 10.1074/jbc.274.34.23963. [DOI] [PubMed] [Google Scholar]
- 8.Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Nature. 2004;427:541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 9.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, et al. Nature. 2004;427:537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 10.Kessi J, Poiree JC, Wehrli E, Bachofen R, Semenza G, Hauser H. Biochemistry. 1994;33:10825–10836. doi: 10.1021/bi00201a033. [DOI] [PubMed] [Google Scholar]
- 11.Stearns DJ, Kurosawa S, Sims PJ, Esmon NL, Esmon CT. J Biol Chem. 1988;263:826–832. [PubMed] [Google Scholar]
- 12.Tie JK, Nicchitta C, von Heijne G, Stafford DW. J Biol Chem. 2005;280:16410–16416. doi: 10.1074/jbc.M500765200. [DOI] [PubMed] [Google Scholar]
- 13.Sun YM, Jin DY, Camire RM, Stafford DW. Blood. 2005;106:1–5. doi: 10.1182/blood-2005-06-2495. [DOI] [PubMed] [Google Scholar]
- 14.Goodstadt L, Ponting CP. Trends Biochem Sci. 2004;29:289–292. doi: 10.1016/j.tibs.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Wajih N, Sane DC, Hutson SM, Wallin R. J Biol Chem. 2005;280:10540–10547. doi: 10.1074/jbc.M413982200. [DOI] [PubMed] [Google Scholar]
- 16.Rost S, Fregin A, Hunerberg M, Bevans CG, Muller CR, Oldenburg J. Thromb Haemost. 2005;94:780–786. doi: 10.1160/TH05-02-0082. [DOI] [PubMed] [Google Scholar]
- 17.Rost S, Fregin A, Koch D, Compes M, Muller CR, Oldenburg J. Br J Haematol. 2004;126:546–549. doi: 10.1111/j.1365-2141.2004.05071.x. [DOI] [PubMed] [Google Scholar]
- 18.Lin PJ, Straight DL, Stafford DW. J Biol Chem. 2004;279:6560–6566. doi: 10.1074/jbc.M312239200. [DOI] [PubMed] [Google Scholar]
- 19.Wajih N, Sane DC, Hutson SM, Wallin R. J Biol Chem. 2004;279:25276–25283. doi: 10.1074/jbc.M401645200. [DOI] [PubMed] [Google Scholar]
- 20.Silverman RB, Nandi DL. Biochem Biophys Res Commun. 1988;155:1248–1254. doi: 10.1016/s0006-291x(88)81274-9. [DOI] [PubMed] [Google Scholar]
- 21.Wajih N, Hutson SM, Owen J, Wallin R. J Biol Chem. 2005;280:31603–31607. doi: 10.1074/jbc.M505373200. [DOI] [PubMed] [Google Scholar]
- 22.Hallgren KW, Qian W, Yakubenko AV, Runge KW, Berkner KL. Biochemistry. 2006;45:5587–5598. doi: 10.1021/bi051986y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu SM, Morris DP, Stafford DW. Proc Natl Acad Sci USA. 1991;88:2236–2240. doi: 10.1073/pnas.88.6.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thijssen HH, Baars LG, Vervoort-Peters HT. Br J Pharmacol. 1988;95:675–682. doi: 10.1111/j.1476-5381.1988.tb11692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallin R, Martin LF. J Clin Invest. 1985;76:1879–1884. doi: 10.1172/JCI112182. [DOI] [PMC free article] [PubMed] [Google Scholar]