

Abstract
We report experiments that infer a radical reorientation of tyrosine-phosphorylated parallel STAT1 dimers to an antiparallel form. Such a change in structure allows easy access to a phosphatase. With differentially epitope-tagged molecules, we show that the two monomers of a dimer remain together during dephosphorylation although they most likely undergo spatial reorientation. Extensive single amino acid mutagenesis within crystallographically established domains, manipulation of amino acids in an unstructured tether that connects the N-terminal domain (ND) to the core of the protein, and the demonstration that overexpressed ND can facilitate dephosphorylation of a core molecule lacking an ND all support this model: When the tyrosine-phosphorylated STAT1 disengages from DNA, the ND dimerizes and somehow assists in freeing the reciprocal pY–SH2 binding between the monomers of the dimer while ND ⋅ ND dimerization persists. The core of the monomers rotate allowing reciprocal association of the coiled:coil and DNA-binding domains to present pY at the two ends of an antiparallel dimer for ready dephosphorylation.
Keywords: STAT1 domain, reorientation, dephosphorylation
The STAT proteins are one of several different families of latent cytoplasmic transcription factors that are activated in response to polypeptide ligands binding to their cognate cell surface receptors (Brivanlou and Darnell 2002). When these protein families were first discovered, it was natural that attention focused on the details of their activation and nuclear accumulation, their DNA binding, and their target genes. However, the extent and duration of activation of these factors and how their activity is restrained and therefore balanced are crucially important to understanding their physiologic function. In the case of the seven STAT proteins, all of which are activated by phosphorylation on a single tyrosine by a very wide array of cytokines and growth factors (Levy and Darnell 2002), several different classes of proteins that restrain STAT activity have been documented. (1) Cytoplasmic phosphatases can dephosphorylate activated receptors (for review, see Levy and Darnell 2002). (2) Suppressors of cytokine signaling proteins, SOCS, themselves induced by activated STATs, bind to receptors to prohibit further activation of STATs (for review, see Alexander and Hilton 2004). (3) Proteins termed PIAS (proteins that inhibit active STATs) can block activated STATs from binding to DNA (for review, see Shuai and Liu 2005). The physiologic importance of these several classes of inhibitors is proven because genetic loss of these proteins, induced in mice and naturally occurring in human tumors (for review, see Darnell 2002), results in heightened STAT function that leads to disease.
The final event in inactivation of activated STATs is tyrosine dephosphorylation, and much less is known about this crucial step. Much of what is known pertains to STAT1. First, in the interferon (IFN)-γ dependent cycle of phosphorylation–dephosphorylation, there is no loss of STAT1 protein (Haspel et al. 1996). Blockage of STAT1 dephosphorylation by phosphatase inhibition leads to nuclear accumulation of all the tyrosine-phosphorylated STAT1 and its subsequent persistence (Haspel and Darnell 1999). A nuclear tyrosine phosphatase, TC45 (one of two proteins encoded by the TCPTP gene), is capable of dephosphorylating STAT1 (and also STAT3), and cells lacking TC45 exhibit persistent STAT1 phosphorylation after IFN-γ treatment (ten Hoeve et al. 2002). Restoration of TC45 restores the inactivation of STAT1.
Crystallographic structures have provided many of the details of the activation–inactivation of the STATs (Becker et al. 1998; Chen et al. 1998; Mao et al. 2005; Neculai et al. 2005). The tyrosine-phosphorylated core (residues ∼130–712) of STAT1 and STAT3 bind DNA as dimers held together through reciprocal phosphotyrosine–SH2 (pY–SH2) interactions with no other protein contacts between the DNA-bound dimers. The structure of the N-terminal domains (ND; amino acids 1–130) of STAT1 and STAT4 were solved separately (Vinkemeier et al. 1998; Mao et al. 2005) and are compact structures of a series of helical segments. The ND is attached to the core of a monomer molecule by ∼20 flexible (∼12 unstructured) amino acids we refer to as a tether. The domains in the core from the N to C terminus are a coiled:coil domain (CCD), a DNA-binding domain (DBD), a structural linker domain (LD), and an SH2 domain (see Fig. 1). An unstructured transcription activation domain of 38 amino acids at the C terminus completes the molecule. Because the SH2 domains are at the same end of the tyrosine-phosphorylated, DNA-bound structure, we refer to it as “parallel.” How a phosphatase gains access to the two tyrosine phosphates each buried in an –SH2 pocket is not clear.
Figure 1.
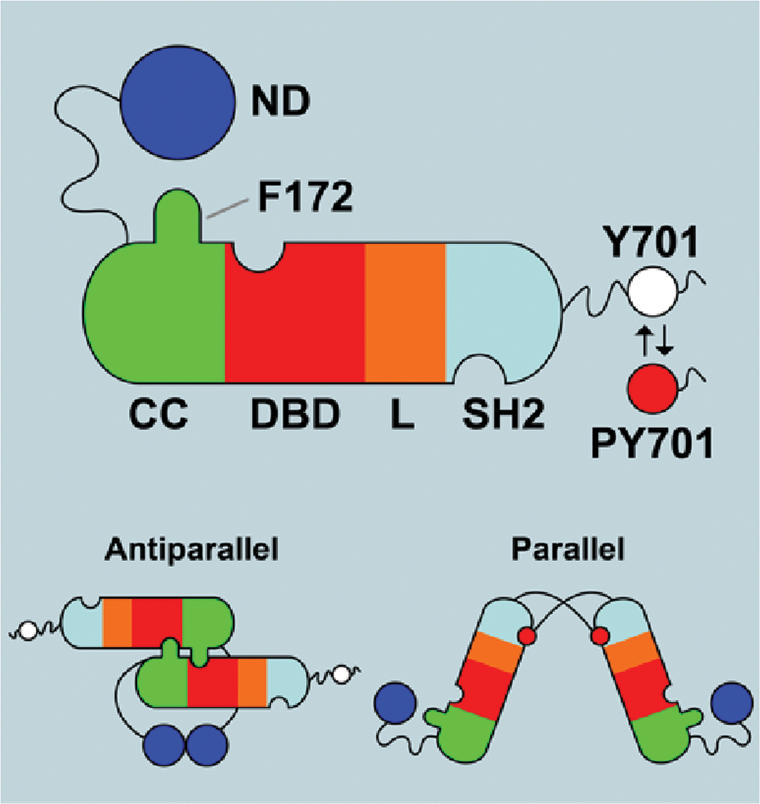
Schematic of salient features of STAT1 structure. The domains are N-terminal domain (ND), coiled:coil domain (CC), DNA-binding domain (DBD), linker domain (L), and SH2 domain (SH2). The ND is shown tied to the CC through a flexible tether (not to scale), and the residues C-terminal to the –SH2 include the Y701, which is phosphorylated (red dot) when the molecule is activated. The C-terminal region is also flexible as indicated by the wavy black line. At the bottom of the figure are diagrams of the parallel and antiparallel structures supported by crystallographic results (Chen et al. 1998; Mao et al. 2005). Notable is the F172 residue that is important in the antiparallel structure.
Recently the solution of the nonphosphorylated STAT1 (Mao et al. 2005) and STAT5 (Neculai et al. 2005) structures revealed the same core structure within each monomer as in the phosphorylated STAT1 and STAT3, and the nonphosphorylated core monomers were dimerized but oriented oppositely in space leaving the SH2 domains at opposite ends of an “antiparallel” dimer (Fig. 1). The monomers in this antiparallel configuration were aligned such that a reciprocal interaction, CCD to DBD, was seen between the two monomers.
Prior to the solution of the nonphosphorylated dimeric STAT structures, accumulation from several laboratories of data using biophysical measurements and coprecipitation of epitope-tagged molecules (Lackmann et al. 1998; Novak et al. 1998; Ndubuisi et al. 1999; Braunstein et al. 2003) showed that full-length nonphosphorylated STAT1 and STAT3 were mostly dimeric. Furthermore, Murphy and colleagues (Ota et al. 2004) found that interaction between the NDs of each monomer in a dimer was necessary for dimerization of nonphosphorylated, full-length molecules of STAT1, STAT3, STAT4, STAT5, and STAT6. The same residues in the ND interface that were known from crystallography and mutagenesis to be important for dimerization of the isolated NDs (Vinkemeier et al. 1998; Chen et al. 2003) were also important for the ND-mediated dimerization of full-length molecules (Ota et al. 2004).
Thus, three protein interfaces have been recognized between portions of STAT molecules: reciprocal pY–SH2 in the parallel, pY molecules, and ND:ND and reciprocal DBD:CCD in the antiparallel, unphosphorylated dimers. We tested by limited mutagenesis the importance of these latter two interfaces in the dimerization of nonphosphorylated STAT1 and in the activation–inactivation cycle of STAT1 in IFN-γ-treated cells (Zhong et al. 2005). STAT1 protein bearing mutations that disrupt the ND:ND dimer (F77A and/or L78A) or a mutation in a residue (F172W) that appeared in the crystal structure to be involved in the coiled:coil/DBD interactions no longer formed nonphosphorylated dimers. In addition, these mutants in two different domains exhibited the same surprising phenotype-persistent phosphorylation in IFN-treated cells and resistance as pure protein to in vitro dephosphorylation by TC45 tyrosine phosphatase. Strehlow and Schindler (1998) had earlier found that substituting the ND of STAT2 or STAT5 for the ND of STAT1 also gave the persistent phosphorylation phenotype of the STAT1 chimera. And Shuai et al. (1996) found that deletion of the first 60 residues of the ND of STAT1 led to persistent phosphorylation. On the basis of all of these observations, we proposed (Zhong et al. 2005) that after leaving DNA, a phosphoSTAT1 dimer still held together by ND:ND interaction even after pY–SH2 disjunction might undergo a reorientation in space from parallel to antiparallel thus exposing the pY as a better target of a phosphatase. Here we test several predictions of this model by extensive further mutagenesis and biochemical experiments, all of which suggest that spatial mobility of the ND on its tether is required for parallel-to-antiparallel reorientation of the phosphorylated core to allow efficient dephosphorylation and that the ND has a necessary role in promoting the dephosphorylation.
Results
Predictions of model
The model of dephosphorylation of the phosphoSTAT1 dimer dependent on reorientation from the parallel to the antiparallel form before phosphatase action makes several predictions and suggests several testable points. (1) The two monomers of a STAT1 phosphodimer should remain associated after dephosphorylation. (2) The ND is attached to the CCD by a flexible ∼20-amino-acid tether that is believed to be too short to allow the two NDs of the dimer to interact when the phosphodimer is bound to DNA (Chen et al. 1998). We hypothesize that upon leaving the DNA, the phosphodimer might be flexible enough to allow the two NDs, attached by their tethers to the STAT1 core, to dimerize as the first step in the pathway to dephosphorylation. We supposed it is the length and not the sequence of the tether that matters in this proposed ND contact. (A phosphorylated Dictyostelium STAT not bound to DNA, but capable of binding DNA, did in fact crystallize as an extended rod-like structure, proving flexibility presumably in the SH2 region of that molecule [Soler-Lopez et al. 2004].) (3) The dissociation of the two reciprocal pY–SH2 bonds of a phosphodimer occurs by an unknown means. The possibility exists that the ND dimer binds to the core of the two subunits of the phosphodimer to assist in promoting dissociation of the pY–SH2 interaction. (4) If the pY–SH2 interactions of a phosphodimer are simultaneously disrupted but the ND:ND dimers on their tethers remain associated, the core of the two STAT subunits might be free to rotate, allowing interaction between the CCD:DBD motifs stabilizing the antiparallel form of the still phosphorylated dimer. As earlier noted, this proposed interaction requires F172, which appears to bind a pocket involving Q340Q408G384. Mutations of the residues in this pocket should prevent stable spatial reorientation to the antiparallel form, thus causing persistent phosphorylation as did the F172W mutation.
The monomers of a STAT1 phosphodimer remain associated after dephosphorylation by TC45
In early experiments (Shuai et al. 1994) using artificially long and wild-type STAT1, we found that phosphodimers of either type (long or wild type) did not exchange partners during incubation unless forced apart with homologous phosphotyrosine-containing peptides. To test whether dephosphorylation of a STAT1 phosphodimer in vitro led to dissociation of the monomeric subunits so that partner interchange might occur, we prepared differentially epitope-tagged STAT1 phosphoprotein (Myc · His on one construct and Flag on the other). Cells (293T) were transfected with two vectors expressing either STAT1 tagged with Flag epitope or the Myc ⋅ His epitope and treated with interferon (IFN)-γ. Differentially tagged phosphodimers (Fig. 2A) were purified in three steps (a Ni++ column for Myc · His, an antibody column for Flag, and a substrate-bound oligonucleotide containing a STAT1-binding sequence; see Materials and Methods) to ensure the collection of only phosphodimers containing one Myc · His-tagged and one Flag-tagged monomer. The purified preparation (Fig. 2B) complexed to DNA could be shifted in electrophoretic mobility shift assay (EMSA) analysis by either a Myc or a Flag antibody and could be “double shifted” with both antibodies. Therefore all the DNA-binding dimers contained both Myc · His and Flag epitopes. When this preparation, capable of binding DNA, was dephosphorylated by TC45, all the DNA-binding capacity was removed (because, of course, only STAT1 phosphodimers bind DNA) (Fig. 2C,D, lanes 3,4, top panels). After dephosphorylation, the preparations were again selected by attachment to and elution from Ni++ NTA agarose and subjected to Western blot analysis with anti-Flag antibody to see if the Myc · His and Flag epitopes remained together. The dephosphorylation was carried out both with the tagged purified STAT1 alone (Fig. 2C,D, lanes 3) or with added nontagged STAT1β in approximately an equal amount (Fig. 2C, lane 4) or a 10-fold excess to the tagged STAT1 (Fig. 2D, lane 4). If during dephosphorylation the monomers of a phosphodimer dissociate, the Myc ⋅ His and Flag epitopes that are on separate STAT1 monomers should be momentarily separated before again forming nonphosphorylated dimers. So about half would be lost as mixed tagged molecules (presumably reassociating as Myc ⋅ Myc or Flag ⋅ Flag dimers). If excess nontagged molecules were present during a hypothetical dissociation, the number of mixed tagged nonphosphorylated dimers should be drastically reduced. However, when the dephosphorylated preparations were again selected on the Ni++ column (the Myc ⋅ His affinity column), the amount of Flag epitope recovered was the same as before dephosphorylation (Fig. 2C,D, lanes 4, bottom panel). This experiment strongly supports the conclusion that during dephosphorylation there is no disruption of the dimers into monomers and reassociation into nonphosphorylated dimers. Since crystallography shows that the phosphodimer is a parallel structure and the nonphosphorylated molecule (at least the core) is antiparallel, the experiment suggests a realignment in space during dephosphorylation leaving nonphosphorylated antiparallel dimers.
Figure 2.

Monomers of a STAT1 phosphodimer remain associated during dephosphorylation. (A,B) Purification of STAT1-Flag:STAT1-Myc phosphodimers. 293T cells were cotransfected with expression constructs for Flag-tagged STAT1 and Myc-tagged STAT1. After IFN-γ induction, whole-cell extracts were subjected to a three-step affinity-purification scheme (see Materials and Methods). Material was evaluated by EMSA with a M67 probe before purification (A) and after purification (B) using anti-Flag and anti-Myc antibodies for shifting STAT1–DNA complexes. (C,D) Continued association of STAT1 monomers in a dimer after in vitro dephosphorylation with GST-TC45. Purified doubly tagged STAT1 phosphodimers alone (C,D, lanes 1,3) or as mixture with purified nontagged phospho STAT1β (C,D, lanes 2,4) were incubated for 60 min with purified tyrosine phosphatase TC45. (Top panels) Tyrosine phosphorylation of STAT1 proteins was monitored before and after TC45 treatment by EMSA. (Bottom panels) The presence of Flag-Myc heterodimers before and after dephosphorylation was assayed by pull-down with Ni-NTA agarose and immunoblot detection of coimmunoprecipitated proteins with anti-Flag antibodies. Note that in C approximately equal amounts of doubly tagged STAT1 (lane 2, upper band) and nontagged STAT1β (lane 2, lower band) were used, whereas in D 10-fold as much nontagged STAT1β was included to assay for a possible dissociation and reassociation of tagged dimers.
Importance of the tether in dephosphorylation
In the crystal structures of the ND and of the core of STAT1, the last hydrophobic anchor residue of the ND is 116 and the first such residue of the core is 142. Within this flexible region, residues 123–135 are unstructured. Thus this region of the molecule can be considered a “tether” (Gokhale and Khosla 2000) connecting the ND to the body of the molecule (Chen et al. 1998; Vinkemeier et al. 1998). We explored the contribution of this region to the STAT activation:inactivation cycle by shortening the tether, or by changing the sequence (Fig. 3A).
Figure 3.

Persistent phosphorylation of STAT1 tether mutants. (A) Amino acid sequence of STAT1 tether and tether mutants. (B) Dephosphorylation of STAT1 and STAT1 mutants. U3A cells that lack STAT1 were left untransfected (U3A) or transfected for 24 h with vectors encoding wild-type STAT1 (WT), STAT1 lacking the first 135 amino acids (Δ135), or other mutant STAT1 proteins: F172W, Δ17T, Δ12T, Δ7T; as indicated in A. Parallel samples of cells were treated or not treated with IFN-γ for 30 min, and IFN-γ for 30 min followed by the tyrosine kinase inhibitor staurosporine for an additional 45 min. Total cell extracts were prepared and Western blotted with anti-pY STAT1 (top) and anti-STAT1 antibodies (bottom). (C) Time course of dephosphorylation in staurosporine-treated cells. U3A cells were complemented with wild-type STAT1 or tether mutants 12TCR (12 central amino acids reversed, shown in red in A) or Δ12TC (12 central amino acids deleted). Expressed STAT1 proteins were IFN-γ activated for 30 min followed by treatment with staurosporine for the indicated times (30, 60, 90, 120 min). Immunoblot analysis of whole-cell extracts as in B. (D) Same assay as in B with wild-type STAT1 or mutants Δ12T and MetH-T in which residues 121–141 of STAT1 are replaced with 21 amino acids of human methionine synthase (shown in green in A).
Vectors encoding wild-type or mutant protein were transfected into U3A cells that lack STAT1 followed by IFN-γ treatment for 30 min and staurosporine treatment for 45 min. This kinase inhibitor immediately blocks further tyrosine phosphorylation, enabling a ready analysis of the removal of pre-existing STAT pY (Haspel et al. 1996; Zhong et al. 2005). An antibody specific for STAT1 pY showed that pY disappeared from wild-type STAT1 virtually completely during staurosporine treatment (Fig. 3B). We previously reported that the mutation (F172W) that blocks nonphosphorylated STAT1 dimerization has a persistent phosphorylation phenotype, and this mutant was used as a control (Zhong et al. 2005). Our assumption has been that the role of an unstructured tether might be to allow the ND of each monomer of the phosphorylated STAT1 to bind to its opposite ND when the phosphoSTAT1 is not bound to DNA. If this were the first step in dephosphorylation, deletion of the ND should block dephosphorylation. This is, indeed, the case: The Δ135 mutant protein (lacking the first 135 residues of STAT1) is phosphorylated normally but resists dephosphorylation after staurosporine treatment (Fig. 3B). When only seven amino acids (residues 136–142; mutant Δ7T) were removed from the tether, phosphorylation and dephosphorylation were the same as in wild-type protein (Fig. 3B). However, when 12 or 17 amino acid residues (Δ12T or Δ17T) were removed, phosphorylation was normal, but dephosphorylation persisted essentially unchanged after staurosporine treatment (Fig. 3B). When the 12 central amino acids in the tether between residues 121 and 132 were reversed in their order compared with the wild-type sequence (12TCR), normal phosphorylation and dephosphorylation were observed (Fig. 3C). After 120 min of staurosporine treatment, the protein with 12 central amino acids deleted (Δ12TC) remained phosphorylated, while the wild-type protein and the protein with the central 12 amino acids reversed in the tether (12TCR) had been largely dephosphorylated in 30 min and completely dephosphorylated after 60 min of staurosporine treatment (Fig. 3C). Finally, we replaced 21 residues of the STAT1 tether with 21 amino acids from human methionine synthase (MetH-T) (Fig. 3D). These MetH-T amino acids serve as a linker in this enzyme to accommodate domain movement during the methyl transfer reactions of the methionine synthesis cycle and bear no sequence relatedness to the STAT1 tether (Bandarian et al. 2001; Evans et al. 2004). STAT1 with the MetH-T insertion was phosphorylated and dephosphorylated like wild-type STAT1. Thus it is the length of the STAT1 tether that is important to promote dephosphorylation, not the sequence.
The ND dimer plays a direct role in dephosphorylation
Since the Δ135 mutant remains persistently Y phosphorylated for hours after IFN-γ treatment, we tested whether the ND expressed separately from the Δ135 molecule would have an effect on dephosphorylation of Δ135. U3A cells were transfected both with vectors encoding Δ135 and either the wild-type ND or the ND mutant F77A,L78A or a vector encoding the ND of STAT5b (amino acids 1–129). Strehlow and Schindler have shown that a chimera of STAT1 with the ND of STAT5 exhibits persistent phosphorylation (Strehlow and Schindler 1998).
In representative experiments of several similar experiments (Fig. 4A,B), we found that the STAT1 wild-type ND was capable of causing the phosphoΔ135 to be dephosphorylated, while the STAT5b ND was not capable of causing dephosphorylation. The effect of STAT1 ND F77A/L78A mutant ranged from ∼50% to 75% dephosphorylation. Thus we are not able to conclude absolutely whether dimerization of the ND is obligatory, but the effect of wild-type ND in promoting dephosphorylation was consistent in all experiments. (The ND preparations were tagged with HA, and Western blotting indicated approximately equal expression of the various ND molecules in Fig. 4A.) In Figure 4B the apparent expression of STAT5b ND greatly exceeds that of the STAT1 wild-type ND, and still the STAT5b ND does not promote dephosphorylation of the Δ135 STAT1. Therefore we conclude there is definitely a direct and specific facilitating role of the soluble wild-type ND in dephosphorylation of the Δ135 mutant, suggesting that in the intact STAT1 molecule the ND must play a similar role.
Figure 4.

Expression of free STAT1-ND but not STAT5b-ND rescues the phenotype of mutant Δ135. (A) Coexpression of Δ135 with STAT1 ND constructs in U3A cells. Cells were transfected with indicated combinations of Δ135 and empty vector, Δ135 and HA-tagged wild-type ND, or Δ135 and HA-tagged ND-F77/L78A, which prevents ND dimerization. After IFN-γ activation followed by staurosporine treatment for indicated times (in minutes), total cell extracts were subjected to immunoblot analysis. (Bottom panel) Expression of NDs was detected with anti-HA antibodies. (B) Coexpression of Δ135 with ND of STAT1 and STAT5b. Preparation of samples as in A with staurosporine treatment for 45 min. Experiments were carried out in duplicate.
Importance of the DBD-binding pocket
We next turned to the importance of the CC/DBD interaction first detected in the crystal structure of the nonphosphorylated STAT1 dimer (Mao et al. 2005). Crystallography of the nonphosphorylated STAT1 core showed that residue F172 apparently inserted its side chain into a pocket within the DBD of the other molecule of the dimer (Fig. 5A). And the mutation F172W resulted in a persistent phosphorylation phenotype (Zhong et al. 2005). To solidify the suggestion about the importance of this interaction in maintaining the antiparallel structure as a basis for persistent phosphorylation, we made the following mutations: Q340A or Q340W, G384A or G384W, and Q408A or Q408W to potentially change the nature of the pocket. All six mutations of the three “pocket” residues resulted in proteins exhibiting the persistent phosphorylation phenotype (Fig. 5B,C, data for G384W and Q408A not shown). That is, the proteins were tyrosine-phosphorylated after IFN-γ treatment but resisted dephosphorylation after staurosporine treatment. As a test that these mutations did not affect the other properties of the STAT1 protein, activated STAT1 Q340A, G384A, and Q408W were shown to bind DNA (Fig. 5D).
Figure 5.

Mutations of the pocket for residue F172 in the DBD cause persistent phosphorylation. (A) Shown is the crystallographically determined structure of amino acids in DBD of STAT1 that form a pocket in which the F172 residues of the CCDs interact. (B,C) STAT1 proteins with indicated mutations that were designed to disrupt the CCD/DBD dimer interface were expressed in U3A cells. After activation with IFN-γ for 30 min and staurosporine treatment for 45 min, total cell extracts were analyzed by immunoblotting with anti-pY STAT1 (top) and anti-STAT1 antibodies (bottom). (D) DNA binding of activated pocket mutants. Total cell extracts prepared as in B and C were subjected to EMSA analysis with an M67 probe.
Nuclear entry of proteins exhibiting persistent phosphorylation phenotypes
Since previous indications suggested that dephosphorylation of STAT1 after IFN-γ treatment occurred in the nucleus (Haspel and Darnell 1999; ten Hoeve et al. 2002), we tested the nuclear accumulation of wild-type and several of the mutant proteins. After transfection of U3A cells with wild-type or mutant constructs (Δ135, Q340A), either whole-cell extracts or nuclear extracts were prepared after 30 min treatment with IFN-γ and a 45-min treatment with staurosporine. The whole-cell extracts (Fig. 6A) repeated earlier results: Wild-type protein was activated at 30 min and dephosphorylated after staurosporine. Both mutants, however, showed persistent phosphorylation after staurosporine (Fig. 6A). Nuclear extracts showed that entry into the nucleus of phosphoSTAT1 occurred similarly for wild-type and mutant proteins and the staurosporine-resistant phosphoprotein was nuclear. In a second experiment, U3A cells were transfected as above with vectors encoding Δ135, Q340A, or wild-type protein and activated by IFN-γ treatment for 30 min, and staurosporine treatment was carried out for 15, 30, 45, and 60 min. In this experiment, both cytoplasmic extracts and nuclear extracts were examined (Fig. 6B). The wild-type phosphoSTAT1 was present in both the cytoplasm and nucleus before staurosporine treatment and was gone from the cytoplasm within 15 min of staurosporine treatment and gradually declined in the nucleus after that. The pSTAT-Q340A was also divided between nucleus and cytoplasm before staurosporine treatment and declined more slowly in the cytoplasm. In this overexpression scenario, the pY Δ135 was equally distributed between nucleus and cytoplasm throughout the staurosporine treatment but was clearly not dephosphorylated in either compartment. Thus we conclude that the persistent dephosphorylation phenotype of the mutant proteins is not due to a failure to enter the nucleus.
Figure 6.

Nuclear entry of persistently phosphorylated STAT1 mutants. (A) U3A cells transfected with expression constructs for wild-type STAT1 and mutants Δ135 and Q340A were treated with IFN-γ for 30 min and staurosporine for 45 min. Whole-cell extracts (WCE) and nuclear extracts (NE) were prepared in parallel from identical samples by dividing cells evenly before lysate preparation. Samples were subjected to immunoblotting with antibodies to pY STAT1 and STAT1. (B) Flow of activated STAT1 proteins upon staurosporine addition. U3A cells expressing wild-type STAT1 or mutant proteins Δ135 and Q340A were treated with IFN-γ for 30 min followed by staurosporine treatment for the indicated times (in minutes). Cytoplasmic (Cy) and nuclear extracts (NE) were analyzed by immunoblotting.
Discussion
We propose that a fairly radical set of mobile domain changes is required for the efficient enzymatic dephosphorylation of the pY of STAT1 (Fig. 7). The proposal stems initially from the knowledge of two crystallographic structures of STAT1 core: (1) a tyrosine-phosphorylated STAT1 dimer bound to DNA that has reciprocal pY–SH2 interactions at one end of the dimer—a parallel structure; and (2) a nonphosphorylated antiparallel structure with SH2 domains at opposite ends of the dimer (Chen et al. 1998; Mao et al. 2005). We know that TC45 is required to dephosphorylate STAT1 in the nucleus to recycle the STAT1 back to the cytoplasm (ten Hoeve et al. 2002). We demonstrate here that, in fact, monomers of a phosphodimer do remain associated after in vitro dephosphorylation, consistent with rotation of the body of the molecule without dissociation during dephosphorylation.
Figure 7.

Model for STAT1 dephosphorylation using crystal structures of ND and core of STAT1. The domains and icons for STAT1 are given in Figure 1. Note that the red dot at Y701 indicates phosphorylation. First consider the parallel phosphorylated dimer (pSTAT dimer) bound to DNA. Disengagement from DNA is shown in the second panel; flexibility of the free pSTAT dimer allows contact between NDs because they are on an ∼60 Å tether (Chen et al. 1998). The ND, possibly by contacting the body of the pSTAT, induces pY–SH2 disengagement. The dimer still held together by the ND/ND interface allows rotation of the monomers so that CC/DBD interaction can occur, presenting the pY at 701 for dephosphorylation. Exit to the cytoplasm of the antiparallel nonphosphorylated dimer then occurs. In the cytoplasm, nonphosphorylated STAT1 is shown as either a monomer or dimer and can likely be tyrosine-phosphorylated in either mode. STAT4 may be obligatorily a dimer in order to be phosphorylated at the receptor (Ota et al. 2004).
Mutagenesis in this and the previous paper (Zhong et al. 2005) provides strong evidence that the proposed ND dimerization after STAT1 phosphodimer disengagement from DNA is required for subsequent steps. Experiments clearly show that the length but not the sequence of the tether connecting the ND to the CCD is important for dephosphorylation. The flexibility of the tether presumably allows for ND dimerization. Furthermore, we show here that a wild-type dimeric ND expressed in cells can, albeit less efficiently than an attached ND, promote dephosphorylation of the phosphorylated STAT1 lacking an ND (Δ135; i.e., 136–750) that otherwise is resistant to dephosphorylation. Neither mutant ND protein nor a STAT5 ND is capable of inducing dephosphorylation. These experiments suggest an active role by the ND dimer in binding to the core, perhaps promoting pY–SH2 disengagement. Subsequent rotation of the two bodies of the dimer could allow CCD/DBD interaction dependent on the F172W association with the Q340, G384, Q408 pocket stabilizing the antiparallel structure, providing an accessible substrate for TC45. Mutations of the pocket residues plus the F172W mutant leading to persistent phosphorylation strongly support this idea. The dephosphorylated antiparallel dimer would then remain intact and exit to the cytoplasm, and Braunstein et al. (2003) found the majority of nonphosphorylated cytoplasmic STAT1 to be dimeric.
The structures of the monomer cores of STAT1 are the same in the parallel or antiparallel form, so allosteric change in the core structure may not be required for dephosphorylation (Mao et al. 2005). Large-scale movement of domains, however, would be. Movement of domains without necessary allosteric changes in modular proteins is a well-established phenomenon (Gokhale and Khosla 2000; George and Heringa 2003; Josi et al. 2005), although all such previously documented changes operate over much shorter distances than the ∼150 Å reorientation we propose for STAT1. For example, in one of the earliest crystallographically illustrated cases, the two lobes of the catalytic subunit of cyclic AMP-activated protein kinase are connected by a short five-amino-acid linker (for review, see Cox et al. 1994). The enzyme crystallizes in both an open and closed conformation in which the linker allows rotation of the small lobe away from the large, changing access of substrate to the active site at the base of the cleft between the two lobes. Other examples in which the spatial change in domains is greater is exemplified by methionine synthase (Bandarian et al. 2001; Evans et al. 2004). This large enzyme consists of four modules. The third module (Cob domain) binds cobalamin, the methyl carrier that ultimately converts homocysteine to methionine. In so doing the Cob subunit must visit physically the two separated sites that bind first N5-methyltetrahydrofolate and then homocysteine to deliver a methyl group to make methionine. These events require large-scale (∼50 Å) movements of the two modules that bind homocysteine and N5-methyltetrahydrofolate. The folate and homocysteine domains have a linker that plays a structural role in holding these two modules in a fixed configuration, but the linker between these two modules and the Cob domain is between 20 and 30 amino acids long in different methionine synthases and has no structural role. However, the tether is required to allow variable positioning of the Cob domain during methionine synthesis. It is 21 amino acids from this linker in the human enzyme that we used in place of the STAT1 tether that allowed normal dephosphorylation of STAT1.
Other instances of domain movement and the role of linkers in protein biochemistry have been reviewed (Khosla and Harbury 2001). One particularly instructive case is the α-subunit of Escherichia coli RNA polymerase that consists of two domains (N + C) joined by a flexible linker capable of stretching to 44 Å. This C-domain of the α-subunit can bind DNA weakly and can also bind activators of transcription. Because of the flexibility of the linker, the positioning of the C-domain-binding site on the DNA and the activator DNA-binding sites can vary considerably in their arrangements (for review, see Gourse et al. 2000) and still result in increasing transcription.
It is also worth noting that dephosphorylation of tyrosine residues is critical in balancing signaling through many pathways, and how phosphatases gain access to pY residues buried in SH2 cavities is largely unknown except for Src-like proteins. In the case of Src kinase itself (for review, see Harrison 2003), phosphorylation of the C-terminal tyrosine, residue 527, renders the enzyme inactive because the pY 527 is buried in an SH2 pocket, changing the confirmation of the enzyme. PTP1α, a tyrosine phosphatase capable of dephosphorylating Y527 and activating Src is a multidomain protein that can itself be tyrosine-phosphorylated (Zheng et al. 2000). The pY residue on PTP1α can dislodge (exchange with) the pY527 of Src, leaving the catalytic domains of PTP1α free to dephosphorylate the Src p527, reverting the enzyme to its active conformation.
In the case of STAT1 (and of the STATs, in general), it is physiologically important to restrain activity, and since there are two reciprocal pY–SH2 interactions producing tight binding, it seems logically possible that an evolved mechanism was required to interrupt the otherwise reluctantly separated pY–SH2 state. It is known that some other STATs once activated are also eventually dephosphorylated, and it is also known that the STAT5 core forms the antiparallel structure. Finally, it should be noted that STAT1 can be phosphorylated as a monomer (Fig. 7) and presumably must do so when activated by IFN-α, where the principle activated molecule is a STAT1/STAT2 dimer. However, STAT4 is not phosphorylated except as a dimer as shown by Ota et al. (2004). It will be important to investigate also other members of the STAT family for the physical details that occur during dephosphorylation.
Materials and methods
Cell culture, reagents, and antibodies
U3A and 293T cells were cultivated in DMEM (Gibco) supplemented with 10% cosmic calf serum (HyClone) and penicillin/streptomycin (Gibco). Human IFN-γ (R&D Systems) was used at a final concentration of 5 ng/mL; staurosporine (Calbiochem) was used at a final concentration of 500 nM. Monoclonal antibody to the STAT1 C terminus was from Transduction Laboratories. Polyclonal antiserum specific for STAT1 phosphotyrosine 701 and monoclonal antibody specific for the HA epitope were purchased from Cell Signaling Technology. The Flag epitope was detected with monoclonal antibody M2 from Sigma; monoclonal anti-Myc antibody was from Santa Cruz Biotechnology.
Affinity purification of phosphorylated STAT1-Flag:STAT1-mycheterodimers
293T cells grown on 100-mm plates were transfected with 4 μg of each differentially tagged STAT1α expression construct (C-terminal Flag epitope in pRc/CMV and C-terminal myc-His double tag in pcDNA3.1/Myc-His) using Lipofectamine reagent (Invitrogen). Twenty-four hours after transfection, ∼2 × 108 to 3 × 108 cells were treated with IFN-γ for 30 min, followed by preparation of whole-cell lysates in lysis buffer (50 mM Tris at pH 7.6, 150 mM NaCl, 0.1% Triton X-100, fresh Protease Inhibitor Cocktail [Calbiochem]). Lysates were subjected to a three-step affinity-purification procedure, taking advantage first of the Myc-His tag that binds selectively to Ni-NTA-agarose (Invitrogen). Material eluted from these beads with imidazole was subjected to DNA affinity chromatography using biotinylated oligonucleotides from the Ly6E gene promoter (5′-CATGT TATGCATATTCCTGTAAGTGCATG-3′) (Shuai et al. 1993). Bound phosphorylated STAT1 dimers were eluted with 800 mM NaCl and subsequently purified on M2 agarose (Sigma) to obtain STAT1-Flag:STAT1-Myc-His-phosphorylated heterodimers.
Purified material (50 ng) was incubated with recombinant GST-TC45 for 60 min at 30°C as described (Zhong et al. 2005). Reactions were carried out with and without either 50 ng or 500 ng purified nontagged phosphorylated STAT1β purified according to Chen et al. (1998). After in vitro phosphatase assays, half of the reaction was analyzed by EMSA with 0.06 pmol of 32P-labeled M67 probe (Zhong et al. 2005); the other half was selected by and eluted from Ni-NTA-agarose and subjected to immunoblotting with anti-Flag antibody.
Plasmids and mutagenesis
Mammalian expression constructs for STAT1 tether mutations and DBD pocket mutations were generated by QuikChange site-directed mutagenesis (Stratagene) using human STAT1α in pRc/CMV as template. All mutants were DNA sequenced over the entire reading frame. STAT1 tether mutations Δ7T, Δ12T, and Δ17T were generated by deleting seven, 12, and 17 amino acids from the C terminus starting at residue 142 (Δ136–142 in Δ7T, Δ131–142 in Δ12T, and Δ126–142 in Δ17T). Mutation Δ12TC was obtained by deleting amino acids 121–132, and mutation 12TCR was obtained by reversing the sequence of amino acids 121–132. To obtain mutant MetH-T, amino acids 121–141 of STAT1 were replaced with amino acids 639–659 of human methionine synthase (MetH; GenBank accession no. U73338). Mutagenesis for this construct was done in two steps. First, amino acids 121–141 of STAT1 were replaced with amino acids 639–649 of MetH. The PCR product was used as the template in a second PCR reaction to introduce the remaining 10 amino acids of MetH (amino acids 650–659). The cDNA sequence of MetH was modified at five positions without changing the encoded amino acid sequence in order to prevent secondary structure formation of the oligonucleotides. The following point mutations in the CCD or DBD were prepared: F172W, Q340W, Q340A, G384W, G384A, Q408W, and Q408A. The expression construct for mouse STAT1 lacking the first 135 amino acids (Δ135) was a gift from Robert Schreiber (Washington University, St. Louis, MO). The N domains of human STAT1 (1–131) and mouse STAT5b (1–129) were amplified by PCR and inserted into pRc/CMV. Both constructs were generated with C-terminal HA-epitope. The STAT1 ND construct was used to introduce mutation F77A/L78A by site-directed mutagenesis. STAT5b cDNA used for ND amplification was a gift from Christian Schindler (Columbia University, New York).
Transfection assays and extract preparation
Transient transfections were carried out in six-well plates. Approximately 2.5 × 105 U3A cells were transfected with 0.5–2.0 μg of expression constructs for STAT1 mutants using Lipofectamine reagent. The DNA amount of different expression constructs was adjusted empirically to yield approximately equal expression of different proteins. In case of coexpression experiments, 1.5 × 105 U3A cells were transfected with 100 ng of Δ135 DNA along with 2 μg of DNA for STAT1-ND or 500 ng of DNA for STAT5b-ND. Total DNA was adjusted to 2 μg with empty vector. After 24 h, cells were treated with IFN-γ for 30 min, followed by staurosporine treatment for 45 min or as indicated in the staurosporine time course. Preparation of cytoplasmic, nuclear, and whole-cell extracts has been described (Zhong et al. 2005). In some experiments, nuclear extracts and whole-cell extracts were prepared in parallel from the same plate by collecting the cells with trypsin and splitting them evenly before lysis. Cell extracts were analyzed by SDS-PAGE and immunoblotting with anti-pY STAT1 antibody. The same membrane was reprobed with anti-STAT1 antibody.
EMSA
The DNA-binding activity of STAT1 mutants was assayed with 32P-labeled M67 probe. Probe was prepared by end-labeling of double-stranded oligonucleotide with [γ-32P]ATP and T4-polynucleotide kinase. Twenty micrograms of whole-cell extracts from untreated, IFN-γ stimulated, or IFN-γ stimulated/staurosporine-treated cells were incubated with 0.015 pmol of probe in binding buffer (20 μL) containing 10 mM HEPES (pH 7.9), 40 mM KCl, 6% glycerol, 0.5% Nonidet P-40 (NP-40), 1 mM DTT, and 1 μg of poly(dI-dC). Reaction mixtures were incubated for 20 min at 30°C, loaded onto a 4% polyacrylamide gel (acrylamide/bisacrylamide 37.5:1) in 0.5× Tris-borate-EDTA, and electrophoresed at 130 V for 2 h at room temperature. The gel was dried and exposed to X-ray film.
Acknowledgments
We thank Lois Cousseau for preparation of the manuscript. This work was supported by NIH grants AI32489 and AI34420 to J.E.D. and GM068566 to X.C.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1485406
References
- Alexander, W.S., Hilton, D.J. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Bandarian, V., Pattridge, K.A., Lennon, B.W., Huddler, D.P., Matthews, R.G., Ludwig, M.L. Domain alternation switches B12-dependent methionine synthase to the activation conformation. Nat. Struct. Biol. 2001;9:53–56. doi: 10.1038/nsb738. [DOI] [PubMed] [Google Scholar]
- Becker, S., Groner, B., Muller, C.W. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Braunstein, J., Brutsaert, S., Olson, R., Schindler, C. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 2003;278:34111–34140. doi: 10.1074/jbc.M304531200. [DOI] [PubMed] [Google Scholar]
- Brivanlou, A.H., Darnell, J.E., Jr. Signal transduction and the control of gene expression. Science. 2002;295:813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- Chen, X., Vinkemeier, U., Zhao, Y., Jeruzalmi, D., Darnell, J.E., Jr., Kuriyan, J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- Chen, X., Bhandari, R., Vinkemeier, U., Van den Akker, F., Darnell, J.E., Jr., Kuriyan, J. A reinterpretation of the dimerization interface of the N-terminal domains of STATs. Protein Sci. 2003;12:361–365. doi: 10.1110/ps.0218903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, S., Radzio-Andzelm, E., Taylor, S.S. Domain movements in protein kinases. Curr. Opin. Struct. Biol. 1994;4:893–901. doi: 10.1016/0959-440x(94)90272-0. [DOI] [PubMed] [Google Scholar]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Evans, J.C., Huddler, D.P., Hilgers, M.T., Romanchuk, G., Matthews, R.G., Ludwig, M.L. Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc. Natl. Acad. Sci. 2004;101:3727–3734. doi: 10.1073/pnas.0308082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, R.A., Heringa, J. An analysis of protein domain linkers: Their classification and role in protein folding. Protein Eng. 2003;15:871–879. doi: 10.1093/protein/15.11.871. [DOI] [PubMed] [Google Scholar]
- Gokhale, R.S., Khosla, C. Role of linkers in communication between protein modules. Curr. Opin. Chem. Biol. 2000;4:22–27. doi: 10.1016/s1367-5931(99)00046-0. [DOI] [PubMed] [Google Scholar]
- Gourse, R.L., Ross, W., Gaal, T. UPs and downs in bacterial transcription initiation: The role of the α subunit of RNA polymerase in promoter recognition. Mol. Microbiol. 2000;37:687–695. doi: 10.1046/j.1365-2958.2000.01972.x. [DOI] [PubMed] [Google Scholar]
- Harrison, S.C. Variation on an Src-like theme. Cell. 2003;112:737–740. doi: 10.1016/s0092-8674(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Haspel, R.L., Darnell, J.E., Jr. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc. Natl. Acad. Sci. 1999;96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel, R.L., Salditt-Georgieff, M., Darnell, J.E., Jr. The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends on a protein tyrosine phosphatase. EMBO J. 1996;15:6262–6268. [PMC free article] [PubMed] [Google Scholar]
- Josi, A.K., Witkowski, A., Berman, H.A., Zhang, L., Smith, S. Effect of modification of the length and flexibility of the acyl carrier protein–thioesterase interdomain linker on functionality of the animal fatty acid synthase. Biochemistry. 2005;44:4100–4107. doi: 10.1021/bi047856r. [DOI] [PubMed] [Google Scholar]
- Khosla, C., Harbury, P.B. Modular enzymes. Nature. 2001;409:247–252. doi: 10.1038/35051723. [DOI] [PubMed] [Google Scholar]
- Lackmann, M., Harpur, A.G., Oates, A.C., Mann, R.J., Gabriel, A., Meutermans, W., Alewood, P.F., Kerr, I.M., Stark, G.R., Wilks, A.F. Biomolecular interaction analysis of IFN γ-induced signaling events in whole-cell lysates: Prevalence of latent STAT1 in high-molecular weight complexes. Growth Factors. 1998;16:39–51. doi: 10.3109/08977199809017490. [DOI] [PubMed] [Google Scholar]
- Levy, D., Darnell, J.E., Jr. STATs: Transcriptional control and biologic impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Mao, X., Ren, Z., Parker, G.N., Sondermann, H., Pastorello, M.A., Wang, W., McMurray, J.S., Demeler, B., Darnell, J.E., Jr., Chen, X. Structural basis of unphosphorylated STAT1 association and receptor binding. Mol. Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Ndubuisi, M.I., Guo, G.G., Fried, V.A., Etlinger, J.D., Sehgal, P.B. Cellular physiology of STAT3: Where’s the cytoplasmic monomer? J. Biol. Chem. 1999;274:25499–25509. doi: 10.1074/jbc.274.36.25499. [DOI] [PubMed] [Google Scholar]
- Neculai, D., Neculai, A.M., Verrier, S., Straub, K., Klumpp, K., Pfitzner, E., Becker, S. Structure of the unphosphorylated STAT5a dimer. J. Biol. Chem. 2005;280:40782–40787. doi: 10.1074/jbc.M507682200. [DOI] [PubMed] [Google Scholar]
- Novak, U., Ji, H., Kanasundaram, V., Simpson, R., Paradiso, L. STAT3 forms stable homodimers in the presence of divalent cations prior to activation. Biochem. Biophys. Res. Commun. 1998;247:558–563. doi: 10.1006/bbrc.1998.8829. [DOI] [PubMed] [Google Scholar]
- Ota, N., Brett, T.J., Murphy, T.L., Fremont, D.H., Murphy, K.M. N-domain-dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat. Immunol. 2004;5:208–215. doi: 10.1038/ni1032. [DOI] [PubMed] [Google Scholar]
- Shuai, K., Liu, L. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- Shuai, K., Ziemiecki, A., Wilks, A.F., Harpur, A.G., Sadowski, H.B., Gilman, M.Z., Darnell, J.E., Jr. Polypeptide signaling to the nucleus through tyrosine phosphorylation of JAK and STAT proteins. Nature. 1993;366:580–583. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- Shuai, K., Horvath, C.M., Tsai Huang, L.H., Qureshi, S.A., Cowburn, J., Darnell, J.E., Jr. Interferon activation of the transcription factor Stat91 involves dimerization through SH2–phosphotyrosyl peptide interactions. Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- Shuai, K., Liao, J.Y., Song, M.M. Enhancement of antiproliferative activity of γ interferon by the specific inhibition of tyrosine dephosphorylation of Stat1. Mol. Cell. Biol. 1996;16:4932–4941. doi: 10.1128/mcb.16.9.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Lopez, M., Petosa, C., Fukuzawa, M., Ravelli, R., Williams, J.G., Muller, C.W. Structure of an activated Dictyostelium STAT in its DNA-unbound form. Mol. Cell. 2004;13:791–804. doi: 10.1016/s1097-2765(04)00130-3. [DOI] [PubMed] [Google Scholar]
- Strehlow, I., Schindler, C. Amino-terminal signal transducer and activator of transcription (STAT) domains regulate nuclear translocation and STAT deactivation. J. Biol. Chem. 1998;273:28049–28056. doi: 10.1074/jbc.273.43.28049. [DOI] [PubMed] [Google Scholar]
- ten Hoeve, J., Ibarrra-Sanchez, M., Fu, Y., Zhu, W., Tremblay, M., David, M., Shuai, K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkemeier, U., Moarefi, I., Darnell, J.E., Jr., Kuriyan, J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279:1048–1052. doi: 10.1126/science.279.5353.1048. [DOI] [PubMed] [Google Scholar]
- Zheng, X.M., Resnick, R.J., Shalloway, D. A phosphotyrosine displacemnt mechanism for activation of Src by PTPα. EMBO J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, M., Henriksen, M.A., Takeuchi, K., Schaefer, O., Liu, B., Hoeve, J.T., Ren, Z., Mao, X., Chen, X., Shuai, K., et al. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation–inactivation cycle. Proc. Natl. Acad. Sci. 2005;102:3966–3971. doi: 10.1073/pnas.0501063102. [DOI] [PMC free article] [PubMed] [Google Scholar]