

Abstract
Leishmania donovani express two nucleoside transporters of non-overlapping ligand selectivity. To evaluate the physiological role of nucleoside transporters in L. donovani, homozygous null mutants of the genes encoding the LdNT1 adenosine-pyrimidine nucleoside transporter and the LdNT2 inosine-guanosine transporter were created singly and in combination by single targeted gene replacement followed by selection for loss-of-heterozygosity. The mutant alleles were verified by Southern blotting, and the effects of gene replacement on transport phenotype were evaluated by rapid sampling transport measurements and by drug resistance profiles. The Δldnt1, Δldnt2, and Δldnt1/Δldnt2 mutants were all capable of proliferation in defined culture medium supplemented with any of a spectrum of purine nucleobases or nucleosides, except that a Δldnt2 lesion conferred an inability to efficiently salvage exogenous xanthosine, a newly discovered ligand of LdNT2. Each of the three knockout strains was viable as promastigotes and axenic amastigotes and capable of maintaining an infection in J774 and bone marrow-derived murine macrophages. These genetic studies demonstrate: 1) that L. donovani promastigotes, axenic amastigotes, and tissue amastigotes are viable in the absence of nucleoside transport; 2) that nucleoside transporters are not essential for sustaining an infection in mammalian host cells; and 3) that the phagolysosome of macrophages is likely to contain purines that are not LdNT1 or LdNT2 ligands, i.e., nucleobases. Furthermore, the Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts offer a unique genetically defined null background for the biochemical and genetic characterization of nucleoside transporter genes and cDNAs from phylogenetically diverse species and of genetically manipulated LdNT1 and LdNT2 constructs.
Keywords: Transport, transporters, purines, pyrimidines, infectivity
Abbreviations: ENT, equilibrative nucleoside transporter; LOH, loss-of-heterozygosity; PCR, polymerase chain reaction; FBS, fetal bovine serum; UTR, untranslated region; ORF, open reading frame; PBS, phosphate buffered saline; PRT, phosphoribosyltransferase
1. Introduction
Leishmania donovani is the etiologic agent of visceral leishmaniasis, a devastating and often fatal disease. Leishmania species are digenetic protozoan parasites in which the extracellular, flagellated promastigote exists in the phlebotomine sandfly vector, and the intracellular amastigote resides within the phagolysosome of macrophages and reticuloendothelial cells of the mammalian host. The current arsenal of drugs used in the treatment of leishmaniasis – or for that matter any parasitic disease – is far from ideal, and the need for more efficacious and selective anti-leishmanial drugs that exploit fundamental biochemical differences between parasite and host is acute.
Perhaps the most striking metabolic discrepancy between parasites and their mammalian hosts is the disparate mechanisms by which they generate purine nucleotides. Whereas mammalian cells synthesize purine nucleotides from amino acids and 1-carbon compounds, all of the protozoan parasites studied to date are incapable of de novo synthesis of the purine ring [1]. Thus, each genus of parasite expresses a unique complement of purine salvage enzymes that enable the parasite to scavenge preformed purines from the host milieu. The initial step in purine acquisition by the parasite involves the translocation of host purines across the parasite surface membrane, a process that is mediated by nucleoside and nucleobase transporters.
Nucleoside permeation into L. donovani is carried out by two high affinity transporters with non-overlapping ligand specificity, LdNT1 and LdNT2. LdNT1 transports adenosine and pyrimidine nucleosides, whereas LdNT2 is selective for inosine and guanosine [2,3]. Mutants genetically deficient in either LdNT1 (TUBA5) or LdNT2 (FBD5) activity have been created by negative selection, and the genes for these permeases were subsequently cloned from a cosmid library by functional rescue of these nucleoside-transport-deficient Leishmania [2–4]. Based on their primary structures and predicted membrane topologies, LdNT1 and LdNT2 belong to the equilibrative nucleoside transporter (ENT) family, although subsequent electrophysiological investigations revealed each to be an electrogenic proton symporter [5].
The TUBA5 and FBD5 parasites were originally derived after mutagenesis and selection in tubercidin, an adenosine analog, or formycin B, an inosine isomer, respectively [4]. Southern blot analyses revealed no gross gene rearrangements at the LdNT1 and LdNT2 loci in the nucleoside transport-deficient L. donovani, and Northern blot analyses indicated that the LdNT1 and LdNT2 transcripts were expressed at normal levels in TUBA5 and FBD5 cells, respectively [2,3]. Subsequent sequence analyses of the mutant alleles revealed the basis of the genetic deficiencies to be either point mutations within LdNT1 and LdNT2 that cripple the permeation mechanisms of the encoded permeases or a nonsense mutation within one ldnt2 allele [6,7]. Because of the potential for residual nucleoside transporter activity in TUBA5 and FBD5 parasites and the possibility that functionally incompetent transporters are generated or accumulate in some subcellular milieu within the nucleoside transport-deficient lines, the TUBA5 and FBD5 cells do not provide an optimal null background for transfection studies with nucleoside transporter genes or cDNAs.
Thus, to assess the consequences of a complete deficiency in LdNT1 and LdNT2 activity on the ability of L. donovani to transport, salvage, and proliferate in various purines, Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts were isolated by a combination of targeted gene replacement and selection for loss-of-heterozygosity (LOH). Mutants lacking LdNT1 could not transport adenosine but were capable of growing on all purine nucleosides and nucleobases tested. Mutants possessing a Δldnt2 lesion failed to transport inosine and xanthosine but were capable of robust growth on inosine and guanosine but not xanthosine. All three knockout strains were capable of transforming into axenic amastigotes and establishing an infection in murine macrophages. These studies establish that nucleoside transport is not essential for the viability of either parasite life cycle stage, intimates that purine nutrition within the intracellular milieu of the macrophage does not require nucleosides, and provides an ideal vehicle for future structure-function studies on LdNT1 and LdNT2 and for the expression of heterologous transporters genes and cDNAs within a genetic background completely devoid of nucleoside transport mechanisms.
2. Materials and Methods
2.1. Chemicals, materials, and reagents
[3H]inosine (38.4 Ci/mmol) and [3H]xanthosine (8.6 Ci/mmol) were purchased from Moravek Biochemicals, Inc. (Brea, CA), [3H]adenosine (40 Ci/mmol) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO), and [α-32P]dCTP (3000 Ci/mmol) was supplied by MP Biomedicals (Irvine, CA). All restriction and DNA modifying enzymes were bought from either Invitrogen (Carlsbad, CA), Gibco-BRL Life Technologies Inc. (Gaithersburg, MD), or New England Biolabs, Inc. (Beverly, MA), and the polymerase chain reaction (PCR) protocols were performed using the Advantage™ HF-2 PCR kit from BD Biosciences (Franklin Lakes, NJ). Synthetic oligonucleotides were acquired from Invitrogen. The pX63-HYG [8] and pX63-PHLEO [9] leishmanial vectors that harbor genes conferring hygromycin and phleomycin resistance, respectively, were generous gifts from Dr. Stephen M. Beverley (Washington University School of Medicine). Hygromycin, tubercidin, and formycin B were obtained from Sigma-Aldrich (St. Louis, MO). All other materials, chemicals, and reagents were of the highest purity commercially available.
2.2 Parasite cell culture
The L. donovani LdBob strain [10] was provided by Dr. Stephen M. Beverley (Washington University, St. Louis, MO). LdBob promastigotes were routinely cultured at 26 °C in purine-replete M199-based medium [10] or in Dulbecco’s modified Eagle-Leishmania (DME-L) medium [11] in which the bovine serum albumin was replaced with 10% dialyzed fetal bovine serum (FBS), 1 mM glutamine, 1 X RPMI 1640 vitamin mix, 10 μM folate, and 100 μM xanthine. Once isolated, the Δldnt1 and Δldnt2 strains were continuously cultured in 50 μg/ml hygromycin and 50 μg/ml phleomycin, respectively. The Δldnt1/Δldnt2 double knockout was maintained in both 50 μg/ml hygromycin and 50 μg/ml phleomycin. Single cell cloning of promastigotes was accomplished in semi-solid medium on plates as described [11], and axenic amastigotes were grown at 37 °C as reported [10].
2.3. Oligonucleotide primers
Primers used in the PCR-based amplification of 5′ and 3′ flanking segments of the L. donovani LdNT1 and LdNT2 loci are designated by the flank (either 5′ or 3′), the targeted locus, and orientation. The restriction sites in the PCR primers that were employed in the subcloning of the amplified products into vectors are underlined:
5′-LdNT1-Sense, 5′-AAGCTTGAGGTGCCCGCGATTGTTG-3′;
5′-LdNT1-Antisense, 5′-GTCGACGGAGAATGGGAGAAGAGAG-3′;
3′-LdNT1-Sense, 5′-TCTAGAGGATCATCGTAGCGGCGTC;
3′-LdNT1-Antisense, 5′-AGATCTAGCAGCGGGGCACAAGGTG;
5′-LdNT2-Sense, 5′-CATTAAGCTTCCCTACTTGCCTTGCTG-3′;
5′-LdNT2-Antisense, 5′-TTCAAGCTTAGTTTTAATCAGTCAGAGTAACTCAGTAAG-3′;
3′-LdNT2-Sense; 5′-TTCCCCGGGTTCTGGCCTAACTCTCTTTGTTTG-3′;
3′-LdNT2-Antisense, 5′-CTCAGATCTCAAAAAAAAAACAGGGAGAAG-3′.
2.4. DNA manipulations
Isolation of genomic DNA and Southern blotting were achieved using standard procedures [12]. Hybridization probes were the full length coding sequences for LdNT1 and LdNT2 and a probe to a portion of the 5′ untranslated region (UTR) of each open reading frame (ORF), all of which were generated by PCR and gel-purified using a QIAEX II kit (Qiagen, Valencia, CA). The plasmids from which the hybridization probes were amplified harbored the appropriate sequences that had been cloned into the TOPO-TA vector (Invitrogen™).
Overlapping and complementary mutagenic primers used to eliminate a 5′ BglII site within the 5′ flank of the LdNT1 gene were mutation primer-A, 5′-CGTGCCGTTTCGTAGATATTGTCCTCACGTCAGAC-3′, and mutation primer-B, 5′-GTCTGACGTGAGGACAATATCTACGAAACGGCACG-3′. PCR mutagenesis was accomplished as described [13].
2.5. Molecular constructs for replacement of LdNT1 and LdNT2
LdNT1 [2] and LdNT2 [3] were originally isolated from a cosmid library of L. donovani genomic DNA. 5′ and 3′ flanking regions were identified by restriction mapping, sequencing, and Southern blotting of both genomic and cosmid DNA and then amplified from genomic DNA using PCR for subcloning into targeting vectors. Standard PCR conditions (25 cycles; 94 °C for 30 s, 62 °C for 30 s, 72 °C for 60–90 s) for amplification of L. donovani genomic DNA sequences were implemented. Sense and antisense primers used in the PCR were 5′-LdNT1-Sense and 5′-LdNT1-Antisense for the 479 bp LdNT1 5′ flanking region, 3′-LdNT1-Sense and 3′-LdNT1-Antisense for the 539 bp LdNT1 3′ flanking region, 5′-LdNT2-Sense and 5′-LdNT2-Antisense for the 750 bp LdNT2 5′ flanking region, and 3′-LdNT2-Sense and 3′-LdNT2-Antisense for the 589 bp LdNT2 3′ flanking region. PCR products for the four flanking regions were separated on agarose gels, purified using a QIAEX II kit (Qiagen), digested with the appropriate restriction enzymes, and either inserted directly (LdNT2 flanks) into the germane sites of the pX63-HYG plasmid or ligated first into the TOPO-TA vector and then transferred into the relevant restriction sites within pX63-HYG. Each of the two flanks of both loci were ligated sequentially into pX63-HYG, and the gene targeting constructs were designated pX63-HYG-Δldnt1 and pX63-HYG-Δldnt2, respectively.
To create the pX63-PHLEO-Δldnt2 targeting vector, it was first necessary to mutate a Sac I restriction site within the 5′ flank of LdNT2. Overlapping mutagenic primers to eliminate the Sac I site within the LdNT2 5′ flank were 5′-CACTCGAAGTGCTTTTGTAGCTCCCATCGTGAGG-3′ and 5′-CCTCACGATGGGAGCTCCAAAAGCTCTTCGAGTG-3′, and PCR mutagenesis was accomplished as described [13]. Then, the HYG and PHLEO genes were excised from the mutagenized pX63-HYG-Δldnt2 and pX63-PHLEO vectors, respectively, by digestion with Xho I and Sac I, and the PHLEO gene was ligated into the Xho I/Sac I sites of pX63-HYG-Δldnt2 from which the HYG gene had been removed.
2.6. Transfections
Parasites were transfected by electroporation using conditions analogous to those previously reported [14]. The targeting constructs were cleaved with either Hind III and Bgl II (LdNT1) or Nhe I and Bgl II (LdNT2) to release the linear targeting fragments from pX63-HYG-Δldnt1, pX63-HYG-Δldnt2, and pX63-PHLEO-Δldnt2. Linearized targeting DNAs were designated by the plasmid from which they were cleaved without the initial letter: X63-HYG-Δldnt1 from pX63-HYG-Δldnt1, X63-HYG-Δldnt2 from pX63-HYG-Δldnt2, and X63-PHLEO-Δldnt2 from pX63-PHLEO-Δldnt2. Cells were maintained in nonselective liquid medium for 24 h after transfection and then plated on semi-solid agar under appropriate selective conditions.
2.7. Gene replacements
To isolate LdNT1/ldnt1 and LdNT2/ldnt2 heterozygotes, wild type parasites were transfected with X63-HYG-Δldnt1 or X63-HYG-Δldnt2 and selected on plates containing 50 μg/ml hygromycin B. Colonies were picked after 2-3 weeks, expanded in medium containing the selective agent, and heterozygosity was established by Southern blotting [12] using appropriate 5′ flanking probes. To obtain the Δldnt1 and Δldnt2 null mutants, the LdNT1/ldnt1 and LdNT2/ldnt2 heterozygotes were plated in the presence of 5 μM tubercidin or 1 μM formycin B, respectively. The Δldnt1/Δldnt2 double mutant was created within the Δldnt2 background by first generating the LdNT1/ldnt1/Δldnt2 clone after transfecting the Δldnt2 knockout with X63-HYG-Δldnt1 and selecting in 50 μg/ml hygromycin. The Δldnt1/Δldnt2 double knockout was then obtained from the LdNT1/ldnt1/Δldnt2 strain by selection in 5 μM tubercidin. All mutant strains created by targeted gene replacement were maintained in selective medium containing the drugs from which it and its progenitors were selected.
2.8. Transport assays
Nucleoside transport measurements were accomplished in phosphate buffered saline (PBS) using a previously described oil-stop method [15]. Rates of transport were determined for 1.0 μM [3H]adenosine (0.40 Ci/mmol), 1.0 μM [3H]inosine (0.38 Ci/mmol), and 10 μM [3H]xanthosine (0.086 Ci/mmol). The apparent Km for xanthosine uptake was calculated by determining the rate of xanthosine uptake at six different concentrations of the nucleoside.
2.9 Sensitivities to tubercidin and formycin B
The sensitivities of the Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts to tubercidin (7-deazaadenosine), a cytotoxic ligand of LdNT1 [4,16], and formycin B (7-hydroxy-3-β-D-ribofuranosylpyrazolo-[4,3-d]pyrimidine), an anti-leishmanial pyrazolopyrimidine nucleoside isomer of inosine [17], were evaluated as described [7,18]. The EC50 values are the effective concentrations of drug that inhibit growth by 50%.
2.10. Purines as nutrients
To evaluate the abilities of the Δldnt1, Δldnt2, and Δldnt1/Δldnt2 cells to grow in various purines, wild type and mutant parasites were washed several times with PBS and resuspended in purine-deplete DME-L-based medium that lacked the bovine serum albumin and purine components but that was supplemented with 10% dialyzed FBS. 1.0 ml aliquots of cells were seeded at a concentration of 5 X 104 parasites/ml and incubated for 7–10 days in this DME-L-based medium to which various purine supplements were added to a final concentration of 100 μM. Cells were enumerated visually by hemacytometer.
2.11 . Macrophage infectivity assays
Prior to infection, 2 X 105 J774 murine macrophages (ATCC, Manassas, VA) were incubated overnight in 4-well Lab-TekII® Chamber Slides (Nalge Nunc International Corp., Naperville, IL) containing 1.0 ml RPMI medium supplemented with 10% heat inactivated FBS, 4 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified 5 % CO2 atmosphere. After 16 hr, unattached macrophages were removed by washing the chamber slides twice with RPMI medium, and 2.0 X 106 stationary-phase L. donovani promastigotes were added to each chamber. After 4 hr, adherent macrophages were washed three times with RPMI to remove any remaining extracellular promastigotes, after which fresh growth medium was added at 24 and 48 hrs. The level of infection at 72 hr was assessed by staining macrophage and parasite nuclei with propidium iodide as described [19]. Parasites were detected on a Zeiss Axiovert 200M scope (Carl Zeiss Microimaging, Thornwood, NY) using a 60X oil immersion lens. The preparation and infection of murine bone marrow-derived macrophages was as reported [20]. Pictures were taken with an AxioCam MRm camera (Carl Zeiss Microimaging) and parasites enumerated visually.
3. Results
3.1. Southern blot analysis of the gene replacements
Δldnt1, Δldnt2, and Δldnt1/Δldnt2 parasites were created within LdBob, a strain of L. donovani that is capable of transformation into axenic amastigotes and that retains its capacity to infect mammalian macrophages [10]. The Δldnt1 and Δldnt2 single knockouts were each generated after a single round of targeted gene replacement followed by selection for LOH. Functional heterozygotes were first generated by homologous recombination after transfection with vectors containing 5′- and 3′-flanking regions of either LdNT1 or LdNT2 encompassing a drug resistance marker, and homozygous null mutants (Δldnt1 or Δldnt2) were then isolated from the heterozygotes by negative selection in media containing a cytotoxic transporter ligand. Each of the gene replacements within the Δldnt1/Δldnt2 double knockout was constructed by the same strategy. First, the Δldnt2 mutant was created from wild type parasites after a single round of gene replacement to create the LdNT2/ldnt2 heterozygote followed by selection in formycin B. The LdNT1/ldnt1/Δldnt2 line was generated after an additional round of gene replacement with the LdNT1 targeting vector followed by selection for LOH in tubercidin to isolate the Δldnt/Δldnt2 double knockout.
To confirm the gene replacement events in the genetically manipulated strains, Southern blotting of genomic DNA from wild type, heterozygous, and homozygous knockout parasites was performed (Fig. 1). Southern blot analysis using the LdNT1 and LdNT2 ORFs as hybridization probes revealed the loss of both wild type alleles in each of the null mutant lines, whereas the LdNT2/ldnt2 and LdNT1/ldnt1/Δldnt2 heterozygotes retained at least one wild type copy of the gene that hybridized to the radiolabelled probe. The nature of the replacements in the heterozygotes was also confirmed by probing with either the 3′ UTR from LdNT1 or the 5′ UTR from LdNT2, an analysis that was also performed after clonal isolates of the heterozygotes were initially obtained (data not shown). The hybridization signals all corresponded to the size of the restriction fragments predicted from the sequence of the LdNT1 and LdNT2 ORFs and adjacent 5′ and 3′ UTRs [2,3].
Fig. 1.
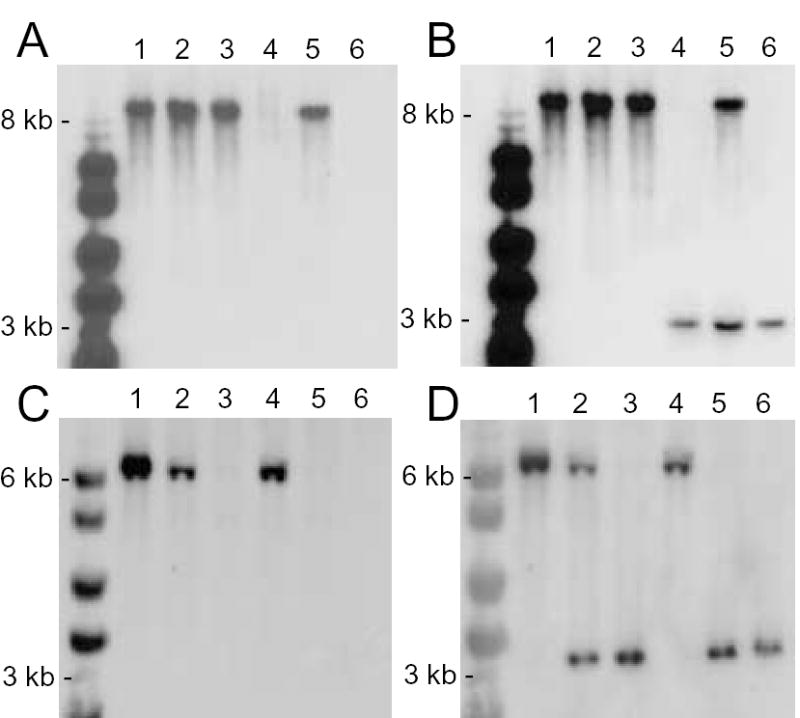
Southern blot analysis of Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts. Genomic DNA from wild type (lane 1), LdNT2/ldnt2 (lane 2), Δldnt2 (lane 3), Δldnt1 (lane 4), LdNT1/ldnt1/Δldnt2 (lane 5), and Δldnt1/Δldnt2 (lane 6) parasites was digested with Eco R1, fractionated on 0.8% agarose gels, and blotted onto nylon membranes. Blots were hybridized under high stringency conditions with probes to the LdNT1 ORF (panel A), the LdNT1 3′ UTR (panel B), the LdNT2 ORF (panel C), or the LdNT2 5′-UTR (panel D).
3.2. Transport phenotypes of the null mutants
In order to assess the phenotypic consequences of the genetic lesions, nucleoside transport assays were performed on wild type and mutant parasites using naturally occurring nucleoside ligands of the two permeases [2–4]. Wild type parasites efficiently transported exogenous [3H]adenosine and [3H]inosine, whereas the genetic lesions resulted in null transport phenotypes for ligands of the affected gene product (Fig. 2). Thus, the Δldnt1 and Δldnt1/Δldnt2 lines were unable to transport adenosine (Fig. 2, panel A), while the Δldnt2 and Δldnt1/Δldnt2 strains could not transport inosine (Fig. 2, panel B). Interestingly, adenosine transport into Δldnt2 cells was markedly up-regulated compared to wild type parasites. The mechanism for this apparent compensation is unknown.
Fig. 2.

Transport of adenosine and inosine by Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts. The abilities of wild type (•), Δldnt1 (○), Δldnt2 (▪), and Δldnt1/Δldnt2 (□) parasites to transport 1 μM [3H]adenosine (panel A) or 1 μM [3H] inosine (panel B) as a function of time was measured. Results are expressed as mean ± standard deviation (n = 2).
3.3. Drug resistance profiles of the knockout lines
Previous analyses of nucleoside transport have indicated that tubercidin and formycin B are cytotoxic ligands for LdNT1 and LdNT2, respectively [2–4]. To confirm the null transport phenotypes of the knockout lines, their sensitivities to tubercidin and formycin B were compared to that of wild type L. donovani. The effective concentrations of tubercidin that inhibited growth by 50% (EC50 value) were 957- and 3,428-fold greater for the Δldnt1 and Δldnt1/Δldnt2 strains than for wild type parasites (Table 1). Similarly, the calculated EC50 values of the Δldnt2 and Δldnt1/Δldnt2 null mutants to formycin B were 2,442- and 5,428-fold greater than the EC50 value of the wild type parasites for the drug (Table 1). The reason that the double knockout exhibits a slightly and consistently greater resistance to tubercidin and formycin B than the pertinent single knockout is not known, but the data imply that both toxic nucleoside analogs could be very low affinity ligands for the other transporter.
Table 1.
EC50 values of wild type, Δldnt1, Δldnt2, and Δldnt1/Δldnt2 promastigotes for tubercidin and formycin B. The results are averages and standard errors for two independent measurements. The relative resistance of the Δldnt1, Δldnt2, and Δldnt1/Δldnt2 cell lines to both drugs compared to wild type parasites is indicated in parentheses.
| Tubercidin (nM) | Formycin B (nM) | |
|---|---|---|
| Wild type | 9.2 ± 2.7 | 1.62 ± 0.55 |
| Δldnt1 | 8,800 ± 900 (957) | 2.33 ± 0.76 (1.44) |
| Δldnt2 | 5.33 ± 0.96 (0.58) | 3,956 ± 1389 (2442) |
| Δldnt1 /Δldnt2 | 34,300 ± 4400 (3728) | 8,793 ± 2598 (5428) |
3.4. Growth phenotype of the Δldnt1, Δldnt2, and Δldnt1/Δldnt2 promastigotes
The ability of Δldnt1, Δldnt2, and Δldnt1/Δldnt2 promastigotes to grow in various purines was compared. Whereas wild type and Δldnt1 promastigotes could grow in medium supplemented with any of the added purine nucleobases or nucleosides tested, including hypoxanthine, adenine, guanine, xanthine, and their corresponding ribonucleosides, the Δldnt2 and Δldnt1/Δldnt2 promastigotes could not proliferate in xanthosine as the sole purine nutrient (Fig. 3).
Fig. 3.

Growth phenotypes of wild type and knockout promastigotes. The abilities of wild type and knockout parasites to proliferate in purine-defined DME-L-based medium as a function of purine source is depicted.
3.5. Xanthosine transport by L. donovani
The inability of Δldnt2 and Δldnt1/Δldnt2 promastigotes to undergo a normal rate of growth in xanthosine implied that xanthosine is an heretofore undiscovered LdNT2 ligand. A comparison of xanthosine transport into wild type, Δldnt2, and Δldnt1/Δldnt2 parasites demonstrated that the uptake of xanthosine into wild type parasites was essentially absent in the two null mutants harboring the Δldnt2 lesion (Fig. 4A). Substrate saturation curves with wild type L. donovani revealed that [3H]xanthosine transport displayed Michaelis-Menten kinetics with an apparent Km value of ~43 μM (Fig. 4B).
Fig. 4.

Xanthosine transport in wild type and mutant L. donovani. The abilities of wild type (▪), Δldnt2 (▵), and Δldnt1/Δldnt2 (▽) to transport 10 μM [3H]xanthosine as a function of time were assessed (panel A). Xanthosine transport as a function of ligand concentration was also assessed (panel B). The data reported in the figure are the results of two independent experiments.
3.6. Transformation of wild type, Δldnt1, Δldnt2, and Δldnt1/Δldnt2 promastigotes to axenic and intracellular amastigotes
The ability of Δldnt1, Δldnt2, and Δldnt1/Δldnt2 promastigotes to transform into axenic amastigotes was then assessed by shifting the parasites from promastigote to axenic amastigote growth medium. All three knockout lines experienced the same morphological transformation as the wild type LdBob line and were capable of continuous growth at 37 °C and pH 5.5. The axenic amastigotes could be transformed back to the flagellated promastigotes by the appropriate shifts in medium, temperature, and pH. The nucleoside transport phenotypes of all amastigote cell lines were equivalent to their promastigote counterparts (data not shown). Therefore, purine nucleoside transport in amastigotes also proceeds via LdNT1 and LdNT2. The ability of the null transport mutants to infect murine macrophages was also assessed. Wild type, Δldnt1, Δldnt2, and Δldnt1/Δldnt2 parasites were all capable of infecting and sustaining infections of J774 murine macrophages to roughly equivalent extents (Fig. 5). Similar data were obtained with bone marrow-derived macrophages as well (data not shown).
Fig. 5.

Parasitemia of wild type and mutant lines. Infectivity assays using J774 murine macrophages were performed as described in Materials and Methods. The numbers of wild type, Δldnt1, Δldnt2, and Δldnt1/Δldnt2 parasites in J774 macrophages were enumerated visually after staining with propidium iodide. One hundred macrophages from each infection were counted. The averages and standard errors of two independent experiments are depicted.
4. Discussion
To evaluate the contributions that LdNT1 and LdNT2 play in purine salvage in both life cycle stages of the parasite, Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts were constructed within a virulent L. donovani background using well-established gene replacement approaches [8,21–23]. Although mutational approaches have previously enabled the isolation and functional characterization of LdNT1- and LdNT2-deficient L. donovani, (TUBA5 and FBD5 strains, respectively) the defective genes in the TUBA5 and FBD5 strains harbored point mutations that rendered the encoded polypeptides transport-incompetent. TUBA5 and FBD5 cells synthesize ldnt1 and ldnt2 transcripts, respectively, and transfectants encompassing the mutant alleles tagged with green fluorescent protein at the NH2-terminus produce transporter capable of targeting to the parasite cell surface [6,7]. Comparatively, therefore, the knockout lines, unlike the mutationally derived TUBA5 and FBD5 cells, provide an authentic null background to assess the role of nucleoside transporters in purine salvage, proliferation, and survival within the mammalian host. Furthermore, the knockout strains provide clean backgrounds for transfection studies of heterologous or mutant transporters that are untainted by endogenously produced mutant protein. Finally, it should be noted that the Δldnt1, Δldnt2, and Δldnt1/Δldnt2 knockouts described in this investigation were generated in an L. donovani clone that is capable of sustaining an infection in host macrophages, thereby enabling a practical test of transporter function in amastigotes. The FBD5 and TUBA5 promastigotes were isolated from an avirulent, noninfectious strain of L. donovani [4] and therefore, the requirement for LdNT1 and LdNT2 involvement in promastigote transformation to the amastigote, infectivity of mammalian host cells, and their ability to proliferate as amastigotes could not be assessed.
The nucleoside transporter gene knockouts retained the ability to transform into axenic amastigotes and to infect and proliferate within murine macrophages. Thus, Δldnt1, Δldnt2, nor Δldnt1/Δldnt2 genotypes did not confer purine auxotrophy or loss of infectivity. Therefore, neither LdNT1 nor LdNT2, alone or in combination, is essential for promastigote or amastigote viability. The ability of Δldnt1, Δldnt2, and Δldnt1/Δldnt2 parasites to establish an infection in macrophages, where they reside in the phagolysosome, strongly implies that this mammalian host cell compartment is capable of supplying purines to meet the nutritional needs of the pathogen that are not ligands of either LdNT1 or LdNT2. These purines, presumably nucleobases, are transported into the parasite by other transporters, such as LdNT3 (J. Galazka and B. Ullman, unpublished), a homolog of the LmaNT3 nucleobase transporter that has been characterized from L. major [24]. Once inside the parasite, purine nucleobases can be salvaged to the nucleotide level by three phosphoribosyltransferase (PRT) enzymes, hypoxanthine-guanine PRT, adenine PRT, and xanthine PRT, and then distributed among the internal nucleotide pools by purine interconversion enzymes.
Unsurprisingly, the transport and growth phenotypes of the Δldnt1 and Δldnt2 were similar to the previously characterized TUBA5 and FBD5 cells, respectively, [4] and were therefore not analyzed in great detail. These characterizations did however reveal one unusual and previously unobserved feature of great utility: that parasites harboring a Δldnt2 null genotype were incapable of efficient growth in xanthosine, a 6-oxypurine nucleoside, although they grew perfectly well on inosine and guanosine, two other LdNT2 ligands. Transport studies on wild type LdBob revealed xanthosine to be a relatively low affinity ligand for LdNT2 with a Km value for the nucleoside ~40-fold and 15-fold greater than those previously obtained for LdNT2 for inosine and guanosine, respectively [3]. The molecular underpinnings for the discrepant growth of Δldnt2 parasites in the three nucleoside ligands of LdNT2 are unclear, although it can be surmised that parasites and/or their culture medium is/are capable of cleaving inosine and guanosine but not xanthosine to their corresponding nucleobases. These nucleobases, hypoxanthine and guanine, can enter L. donovani through LdNT1- and LdNT2-independent mechanisms [4], e.g., LdNT3. Whether this nucleoside cleavage is catalyzed by residual mammalian purine nucleoside phosphorylase in the medium, by one of the nucleoside hydrolase activities that have been described in L. donovani [25], or some other activity will require additional dissection. The growth dependence on xanthosine, however, offers for the first time a positive selection for transport proficiency within the Δldnt2 background, further enabling a genetic analysis of transport function in these parasites.
Acknowledgments
We thank Dr. Stephen M. Beverley of Washington University School of Medicine for providing the LdBob strain for these studies. This work was supported in part by grants RO1 AI23682 and AI44138 from the National Institutes of Health (B.U.). J.B. has received financial support from the N.L.Tartar Research Fellowship from the Oregon Health & Science University. We thank Drs. Archie Bouwer, Michael Riscoe, and Jane Kelly for providing bone marrow-derived macrophages.
References
- 1.Carter NS, Rager N, Ullman B. Purine and pyrimidine transport and metabolism. In: Marr JJ, Nilsen TW, Komuniecki RW, editors. Molecular Medical Parasitology Vol. 1. Elsevier Science Ltd; London: 2003. pp. 197–223. [Google Scholar]
- 2.Vasudevan G, Carter NS, Drew ME, et al. Cloning of Leishmania nucleoside transporter genes by rescue of a transport-deficient mutant. Proceedings of the National Academy of Sciences USA. 1998;95:9873–8. doi: 10.1073/pnas.95.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter NS, Drew ME, Sanchez M, Vasudevan G, Landfear SM, Ullman B. Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. Journal of Biological Chemistry. 2000;275:20935–41. doi: 10.1074/jbc.M002418200. [DOI] [PubMed] [Google Scholar]
- 4.Iovannisci DM, Kaur K, Young L, Ullman B. Genetic analysis of nucleoside transport in Leishmania donovani. Molecular and Cellular Biology. 1984;4:1013–9. doi: 10.1128/mcb.4.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein A, Vaseduvan G, Carter NS, Ullman B, Landfear SM, Kavanaugh MP. Equilibrative nucleoside transporter family members from Leishmania donovani are electrogenic proton symporters. J Biol Chem. 2003;278:35127–34. doi: 10.1074/jbc.M306188200. [DOI] [PubMed] [Google Scholar]
- 6.Vasudevan G, Ullman B, Landfear SM. Point mutations in a nucleoside transporter gene from Leishmania donovani confer drug resistance and alter substrate selectivity. Proceedings of the National Academy of Sciences USA. 2001;98:6092–7. doi: 10.1073/pnas.101537298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galazka J, Carter NS, Bekhouche S, Arastu-Kapur S, Ullman B. Point mutations within the LdNT2 nucleoside transporter gene from Leishmania donovani confer drug resistance and transport deficiency. Int J Biochem Cell Biol. 2006;38:1221–9. doi: 10.1016/j.biocel.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 8.Cruz A, Coburn CM, Beverley SM. Double targeted gene replacement for creating null mutants. Proc Natl Acad Sci U S A. 1991;88:7170–4. doi: 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freedman DJ, Beverley SM. Two more independent selectable markers for stable transfection of Leishmania. Mol Biochem Parasitol. 1993;62:37–44. doi: 10.1016/0166-6851(93)90175-w. [DOI] [PubMed] [Google Scholar]
- 10.Goyard S, Segawa H, Gordon J, et al. An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol Biochem Parasitol. 2003;130:31–42. doi: 10.1016/s0166-6851(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 11.Iovannisci DM, Ullman B. High efficiency plating method for Leishmania promastigotes in semidefined or completely-defined medium. Journal of Parasitology. 1983;69:633–6. [PubMed] [Google Scholar]
- 12.Sambrook J, Maniatis T, Fritsch EF. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1989. [Google Scholar]
- 13.Arastu-Kapur S, Ford E, Ullman B, Carter NS. Functional analysis of an inosine-guanosine transporter from Leishmania donovani. The role of conserved residues, aspartate 389 and arginine 393. Journal of Biological Chemistry. 2003;278:33327–33. doi: 10.1074/jbc.M305141200. [DOI] [PubMed] [Google Scholar]
- 14.Robinson KA, Beverley SM. Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Molecular and Biochemical Parasitology. 2003;128:217–28. doi: 10.1016/s0166-6851(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 15.Aronow B, Kaur K, McCartan K, Ullman B. Two high affinity nucleoside transporters in Leishmania donovani. Molecular Biochemical Parasitology. 1987;22:29–37. doi: 10.1016/0166-6851(87)90066-1. [DOI] [PubMed] [Google Scholar]
- 16.Wataya Y, Hiraoka O, Sonobe Y, et al. Anti-parasite activity of nucleoside analogues in Leishmania tropica promastigotes. Nucleic Acids Symp Ser. 1984:69–71. [PubMed] [Google Scholar]
- 17.Carson DA, Chang KP. Phosphorylation and anti-leishmanial activity of formycin B. Biochem Biophys Res Commun. 1981;100:1377–83. doi: 10.1016/0006-291x(81)91976-8. [DOI] [PubMed] [Google Scholar]
- 18.Mikus J, Steverding D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue. Parasitology International. 2000;48:265–9. doi: 10.1016/s1383-5769(99)00020-3. [DOI] [PubMed] [Google Scholar]
- 19.Ralton JE, Naderer T, Piraino HL, Bashtannyk TA, Callaghan JM, McConville MJ. Evidence that intracellular beta1-2 mannan is a virulence factor in Leishmania parasites. J Biol Chem. 2003;278:40757–63. doi: 10.1074/jbc.M307660200. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez NE, Chang HK, Wilson ME. Novel program of macrophage gene expression induced by phagocytosis of Leishmania chagasi. Infect Immun. 2004;72:2111–22. doi: 10.1128/IAI.72.4.2111-2122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burchmore RJ, Rodriguez-Contreras D, McBride K, et al. Genetic characterization of glucose transporter function in Leishmania mexicana. Proc Natl Acad Sci U S A. 2003;100:3901–6. doi: 10.1073/pnas.0630165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boitz JM, Ullman B. Leishmania donovani singly deficient in HGPRT, APRT or XPRT are viable in vitro and within mammalian macrophages. Mol Biochem Parasitol. 2006 doi: 10.1016/j.molbiopara.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Boitz JM, Ullman B. A conditional mutant deficient in hypoxanthine-guanine phosphoribosyltransferase and xanthine phosphoribosyltransferase validates the purine salvage pathway of Leishmania donovani. J Biol Chem. 2006 doi: 10.1074/jbc.M600188200. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez MA, Tryon R, Pierce S, Vasudevan G, Landfear SM. Functional expression and characterization of a purine nucleobase transporter gene from Leishmania major. Mol Membr Biol. 2004;21:11–8. doi: 10.1080/0968768031000140845. [DOI] [PubMed] [Google Scholar]
- 25.Cui L, Rajasekariah GR, Martin SK. A nonspecific nucleoside hydrolase from Leishmania donovani: implications for purine salvage by the parasite. Gene. 2001;280:153–62. doi: 10.1016/s0378-1119(01)00768-5. [DOI] [PubMed] [Google Scholar]