

Summary
Mice with a null mutation of the presenilin 1 gene (Psen1-/-)die during late intrauterine life or shortly after birth and exhibit multiple CNS and non-CNS abnormalities, including cerebral hemorrhages and altered cortical development. The cellular and molecular basis for the developmental effects of Psen1 remain incompletely understood. Psen1 is expressed in neural progenitors in developing brain, as well as in postmitotic neurons. We crossed transgenic mice with either neuron-specific or neural progenitor-specific expression of Psen1 onto the Psen1-/- background. We show that neither neuron-specific nor neural progenitor-specific expression of Psen1 can rescue the embryonic lethality of the Psen1-/- embryo. Indeed neuron-specific expression rescued none of the abnormalities in Psen1-/- mice. However, Psen1 expression in neural progenitors rescued the cortical lamination defects, as well as the cerebral hemorrhages, and restored a normal vascular pattern in Psen1-/- embryos. Collectively, these studies demonstrate that Psen1 expression in neural progenitor cells is crucial for cortical development and reveal a novel role for neuroectodermal expression of Psen1 in development of the brain vasculature.
Keywords: Cortical development; CNS hemorrhages; Familial Alzheimer’s disease; Neural progenitor cells; Presenilin 1 (PS1, Psen1); Transgenic mice; Vascular development
Introduction
Alzheimer’s disease (AD) is a degenerative disorder of the central nervous system (CNS). Although most cases occur sporadically, in some families the disease is inherited in an autosomal dominant fashion, referred to as familial AD (FAD). Mutations in the presenilin 1 (PSEN1) gene are the most commonly recognized cause of early onset FAD and more than 100 mutations in the PSEN1 gene have been linked to FAD (Lleo et al., 2004). Mutations in a related gene, presenilin 2, cause FAD in a more limited number of cases (Cruts et al., 1998).
In adult brain, Psen1 is expressed primarily in neurons (Elder et al., 1996), although neural progenitor cells in adult hippocampus express Psen1 (Wen et al., 2002a) and its expression can be induced in reactive astrocytes (Cribbs et al., 1996), including those surrounding senile plaques (Weggen et al., 1998). Developmentally, Psen1 expression is found as early as the preimplantation embryo (Jeong et al., 2000) and Psen1 is prominently expressed in neural progenitor cells in the ventricular zone of embryonic rodents (Moreno-Flores et al., 1999) and humans (Kostyszyn et al., 2001). Mice with a null mutation of the Psen1 gene (Psen1-/-) die during late intrauterine life or shortly after birth, and exhibit multiple CNS and non-CNS abnormalities, including cerebral hemorrhages and altered cortical development (Hartmann et al., 1999; Shen et al., 1997; Wong et al., 1997).
The cellular and molecular basis for the developmental effects of Psen1 remain incompletely understood. In particular, the role that Psen1 plays in different cell types remains unknown. Recently, we generated transgenic mice with either neuron-specific (Wen et al., 2002b) or neural progenitor-specific expression of Psen1 (this paper). We show by crossing these transgenes onto the Psen1-/- background that neither neuron-specific nor neural progenitor-specific expression of Psen1 rescues the embryonic lethality of the Psen1-/- embryo. Indeed, neuron-specific expression ameliorates none of the abnormalities in the Psen1-/- embryo. However, Psen1 expression in neural progenitors rescues cortical development in Psen1-/- embryos, including normalizing cortical lamination patterns and eliminating the cerebral hemorrhages.
Materials and methods
Transgenic and Psen1-/-mice
Transgenic mice expressing human Psen1 under the control of the neuron specific enolase (NSE) promoter/enhancer regions have been previously described (Wen et al., 2002b; Wen et al., 2004b). A transgene allowing expression of human Psen1 in neural progenitor cells (NesPsen1) was constructed in the vector gIITKlacZ (Zimmerman et al., 1994), which contains the nestin intron 2 neural specific enhancer upstream of a 160 bp thymidine kinase (tk) minimal promoter followed by an artificial intron, cloning site and SV40 polyadenylation signal. The lacZ gene was replaced with a human Psen1 wild-type cDNA (Elder et al., 1996). A Nestin/Cre recombinase transgene (NesCrenls) was prepared by cloning the nestin-tk promoter/enhancer from gIITKlacZ into the plasmid pOG231 (O’Gorman et al., 1997), which places the nestin-tk promoter/enhancer upstream of an 0.2 kb synthetic intron followed by a Cre-coding sequence containing a nuclear localization sequence and a polyadenylation signal. A Cre reporter transgene was generated by replacing the lacZ sequences in the plasmid pcAct-XstopXnZ (obtained from Drs Eric Mercer and David Anderson, Howard Hughes Medical Institute, Caltech, USA) with an enhanced green fluorescent protein (EGFP) cDNA (Clontech, Palo Alto, CA, USA). This transgene (cActXstopXEGFP) includes the 2.1 kb chicken β-actin promoter along with an additional 1 kb containing the β-actin exon 1, intron 1 and 5′ untranslated sequence from exon 2, while downstream of exon 2 it contains a translation ‘stop’ cassette sequence (Lakso et al., 1992) flanked by 34 bp loxP sites and the EGFP cDNA.
Transgenic mice were produced by pronuclear injection using C57Bl/6J ×C3H (B6C3) as a source of fertilized eggs. Genotypes were determined by PCR on DNA isolated from tail biopsies or from regions of the embryos or yolk sac. The NesPsen1 transgene was identified with primers homologous to the tk promoter (5′CACGCAGATGCAGTCGGG3′) and the human Psen1 cDNA (5′GTGTTCTCCTCCAGGCCAAG3′) that yield a 287 bp product. Primers to the Cre cDNA (5′GTCGAGCGATGGATTTCCGTCT3′ and 5′GCTTGCATGATCTCCGGTATT3′) were used to identify a 274 bp product from the NesCrenls transgene. cActXStopXEGFP transgenic mice were identified with the primers 5′CGTAAACGGC-CACAAGTTCAG3′ and 5′ATGCCGTTCTTCTGCTTGTCG3′ that amplify a 420 bp product from the EGFP cDNA. Lines were maintained by breeding transgenic animals to C57Bl/6 wild-type mice.
The Z/EG transgenic line (Novak et al., 2000) was obtained from Jackson laboratories (Bar Harbor, MA, USA; stock name Tg(ACTB-Bgeo/GFP); stock number 003920). Psen1-/- mice were obtained from Dr Huntington Potter (University of South Florida, Tampa FL, USA) and are those generated by Shen et al. (Shen et al., 1997). These animals were provided on a mixed genetic background and have been maintained by breeding to C57Bl/6 wild-type mice. Genotypes were determined as described by Shen et al. (Shen et al., 1997). Owing to the mixed genetic background of both the Psen1-/- and the Psen1 transgenic animals, non-transgenic littermates were used as controls in all studies
Animals were housed on 12-hour light/dark cycles and received food and water ad libitum. All procedures were approved by the Mount Sinai School of Medicine Institutional Animal Care and Use Committee and were in conformance with the National Institutes of Health ‘Guide for the Care and Use of Laboratory Animals’.
Western blotting
Western blotting was performed as previously described (Wen et al., 2002b).
BrdU injections
5′-bromo-2′-deoxyuridine (BrdU; Sigma, St Louis, MO, USA) was dissolved in 0.9% NaCl at a concentration of 10 mg/ml and sterilized through an 0.45 μm filter. Mice received one intraperitoneal injection (50 μg/gm of body weight).
Histology and immunohistochemistry
Timed pregnant female mice were euthanized with carbon dioxide. The day of vaginal plug was designated as E0.5. Embryos were fixed in 4% paraformaldehyde overnight and embedded in paraffin wax. Brains were cut into 6-8 μm coronal or horizontal sections. Adult animals were perfused transcardially and the tissues processed as previously described (Wen et al., 2002b).
Histological sections were stained with Cresyl Violet and a standard set of at least five sections per embryo was examined. For horizontally sectioned embryos, a series centered through the lateral ventricle above the level of the caudate/putamen was used. A series centered at the level of the medial septal nucleus was examined in coronally sectioned embryos.
For immunohistochemical staining sections were blocked with PBS/0.1% Triton X-100/5% goat serum (TBS-TGS) for 30 minutes and primary antibodies were applied in TBS-TGS at room temperature overnight. The primary antibodies used were Rat 401, a mouse monoclonal anti-rat nestin (1:500, Chemicon, Temecula, CA, USA); CS56, a mouse monoclonal anti-chondroitin sulfate proteoglycan (1:400, Sigma); G10, a mouse monoclonal anti-reelin (1:500, Chemicon); a rabbit polyclonal anti-EGFP (1:1500, Molecular Probes, Eugene, OR, USA); a rabbit polyclonal anti-von Willebrand factor (1:200, Sigma); and a rabbit polyclonal anti-fibronectin (1:400, Sigma). Immunofluorescence staining was detected with species-specific Alexa Fluor secondary antibody conjugates (1:400, Molecular Probes) applied for 2 hours; sections were mounted using Gel/Mount (Biomeda, Foster City, CA, USA). BrdU immunohistochemistry was performed as previously described (Wen et al., 2002b).
Vascular staining with Griffonia simplicifolia isolectin B4 was performed using methods similar to those described in Ashwell (Ashwell, 1991). Deparaffinized sections were incubated for 20 minutes at 90°C in 10 mM Na citrate buffer (pH 8.6) and allowed to cool to room temperature. After blocking with TBS containing 0.02% Triton X-100, 1 mM MgCl2, 1 mM CaCl2 and 1 mM MnCl2 for 1 hour at room temperature, biotinylated Isolectin B4 (Sigma, 10 μg/ml in the above buffer) was added and sections incubated overnight at 4°C. To detect lectin binding, sections were incubated with Streptavidin-Alexa 488 (Molecular Probes, 1:300 diluted in TBS) for 2 hours at room temperature. When Isolectin B4 staining was combined with immunohistochemistry, lectin staining was performed after the immunostaining.
Images were collected using a Zeiss Axiophot microscope (Zeiss, Thornwood, NY, USA) or a Nikon Eclipse E400 connected to a DXC-390 CCD camera (Nikon, Melville, NY, USA). Images were color balanced and merged using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA, USA).
When spontaneous EGFP fluorescence was imaged and combined with aquaporin immunohistochemistry, Vibratome sections (200 μm) were immunostained with a polyclonal anti-aquaporin 4 antibody (1:100, Chemicon) followed by Texas Red-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA). Spontaneous EGFP fluorescence and Texas Red immunofluorescence were imaged with a Radiance 2000 MP system (Bio-Rad, Hercules, CA, USA) equipped with a Mira 900F Ti: sapphire laser (tuned to 860 nm) and a Verdi 10-W pump laser (Coherent, Santa Clara, CA, USA).
In situ hybridization
E12.5-E13.5 embryos were immersion fixed for 2 hours in 4% paraformaldehyde dissolved in diethylpyrocarbonate (DEPC)-treated PBS, equilibrated sequentially in 10%, 20% and 30% sucrose and embedded in OCT compound (Tissue Tek, Elkhart, IN, USA). Cryosections (15 μm) were cut, air dried for 20 minutes and fixed in 4% paraformaldehyde in PBS-DEPC. Sections were treated with 1 μg/ml proteinase K for 5 minutes at room temperature and fixed in paraformaldehyde for an additional 20 minutes. After several washes with PBS-DEPC, sections were acetylated with acetic anhydride in the presence of triethanolamine. The sections were then prehybridized for 2 hours in 50% formamide, 5 × SSC, 5 × Denhardt’s solution, 250 μg/ml yeast RNA, 500 μg/ml herring sperm DNA and hybridized at 55°C for 16 hours with 400 ng/ml of a heat denatured (80°C) digoxigenin-labeled antisense RNA probe complementary to a 260 bp sequence in the human PSEN1 3′ untranslated region (nucleotides 1854-2114 in GenBank Accession Number BC011729). Probes were labeled by random incorporation of digoxigenin-labeled deoxyuridine triphosphate using a commercially available kit (Roche, Indianapolis, IN, USA). Slides were washed for 1 hour in 0.2 × SSC at 70°C and subsequently with 50 mM Tris-HCl (pH 8.0), 0.15 M NaCl (TBS) at room temperature. After blocking with 10% heat-inactivated goat serum in TBS at room temperature, sections were incubated overnight with a 1:250 dilution of anti-digoxigenin antibodies at 4°C (Roche). Following several washes with TBS, slides were equilibrated in alkaline phosphatase buffer [0.1 M Tris-HCl (pH 9.5), 0.1 M NaCl, 50 mM MgCl2, 0.01% Tween-20, 0.25 mg/ml levamisole] for 30 minutes followed by staining with 0.4 mg/ml nitro tetrazolium blue chloride, 0.19 mg/ml 5-bromo-4-chloro-3-indolyl-phosphate in the same solution for 72 hours at 4°C. E16.5 embryos were hybridized in an identical manner except that the brains were dissected and frozen directly in OCT compound without prior fixation. Additionally, the proteinase K digestion step was omitted and the hybridization was performed at 60°C.
Results
Generation of transgenic mice expressing human presenilin 1 in neural progenitor cells under the control of the rat nestin intron 2 enhancer
Nestin is an intermediate filament that is expressed in neural progenitor cells of the developing CNS (Frisen et al., 1998; Lendahl et al., 1990; McKay, 1997). To express Psen1 selectively in neural progenitor cells, we used the 257 bp enhancer from the second intron of the rat nestin gene that has been used to drive expression of heterologous genes in neural progenitor cells in transgenic mice in both embryonic and adult CNS (Aoki et al., 2000; Josephson et al., 1998; Zimmerman et al., 1994). Like endogenous nestin, expression of these transgenes ceases as progenitor cells differentiate into neurons or glial cells.
In the present studies, two lines (9 and 14) with similar patterns of transgene expression were used. Western blotting of E12.5 brain with a human specific anti-Psen1 antibody showed that in both lines the human Psen1 protein could be detected in transgenic brain while no signal was found in non-transgenic controls (Fig. 1). In addition, western blotting with a Psen1 antibody that reacts with both the mouse and human proteins gave an approximately threefold higher signal in the transgenic embryos (Fig. 1). To establish the cell type specificity of transgene expression, we performed in situ hybridization on E13.5 embryos using a human Psen1-specific cRNA probe. In nestin-Psen1 line 9 there was prominent human Psen1 expression in the ventricular zone of the telencephalon as expected for a nestin-driven transgene (see Fig. S1 in the supplementary material; see Fig. 8).
Fig. 1.
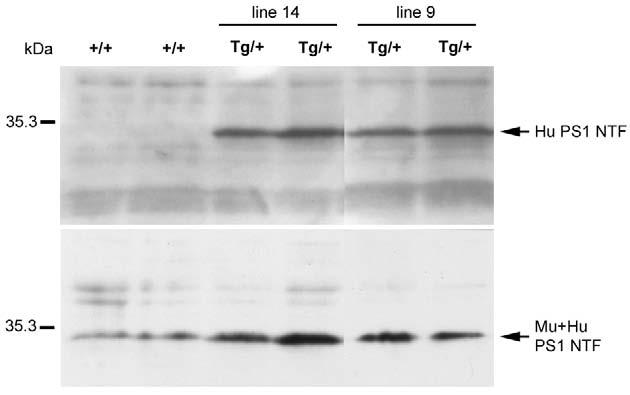
Expression of human PSEN1 protein in transgenic mice under the control of the nestin intron 2 enhancer. Western blotting was performed on brain extracts from E12.5 nestin-Psen1 transgenic (Tg/+) embryos from line 9 and line 14, as well as non-transgenic controls (+/+). Blots were probed with NT.1 (upper panel) an antibody that recognizes the 30 kDa N-terminal fragment (NTF) of the transgenic human (Hu) PSEN1 but not the endogenous mouse (Mu) Psen1 (Wen et al., 2002b). The two wild-type (+/+) embryos are immunonegative. The lower panel was probed with a rabbit polyclonal antibody 222 that recognizes both the mouse and human NTFs (Wen et al., 2002b). Antibody 222 was raised against amino acids 2-12 of the human NTF, a region that contains one mismatch between the mouse and human proteins (Wen et al., 2002b). Thus, the apparent elevation in total Psen1 (Mu + Hu) in the transgenic (Tg/+) embryos may in part reflect a higher affinity of this antibody for the human protein.
Fig. 8.

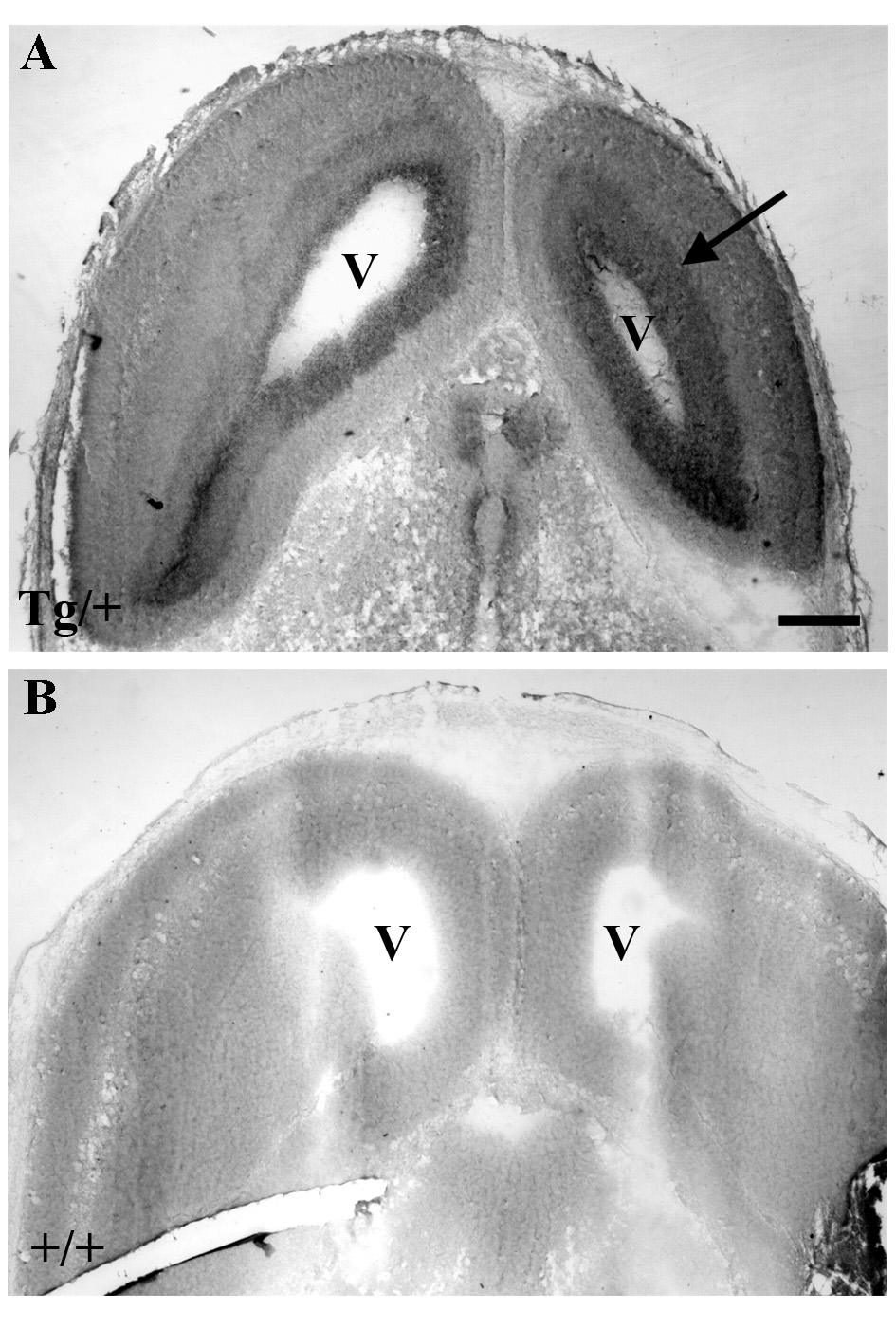
Lack of detectable transgene expression in cephalic mesenchyme by in situ hybridization with a human-specific Psen1 cRNA. Shown are horizontal sections through the telencephalon of an E12.5 nestin-Psen1 transgenic embryo from line 9 (A,B,D) or a non-transgenic littermate control (C,E) embryo hybridized with a human specific Psen1 probe. (A) A section through the anterior telencephalon. The lateral ventricle (LV) and interhemispheric fissure (IH) are indicated. There is prominent hybridization in the brain parenchyma. Hybridization in the ventricular zone from a transgenic (Tg) and non-transgenic (+/+) embryo is shown in B and C. The transgenic embryo is strongly hybridized. (D) Hybridization at the juncture between the brain parenchyma and the overlying cephalic mesenchyme in the interhemispheric fissure from a transgenic embryo. (E) A comparable region from a non-transgenic embryo (+/+) is shown to indicate the level of background staining. An arrow indicates the border between the brain parenchyma (showing hybridization product in D but not in E) and the cephalic mesenchyme. Scale bar: 20 μm for A; 50 μm for B-E.
Neither neural progenitor nor neuron specific expression of Psen1 rescues the embryonic lethality of Psen1-/-mice
Hemizygous nestin-Psen1 transgenic mice were indistinguishable from wild-type littermates with regard to behavior and physical appearance. To determine whether selective expression of Psen1 in neural progenitor cells could rescue the embryonic lethality of Psen1-/- animals, we bred the nestin-Psen1 transgene onto the Psen1-/- background. For this purpose, hemizygous transgenic/heterozygous Psen1+/- animals were created and crossed with Psen1+/- mice. From such matings one out of eight offspring should be Psen1-/- if rescue is possible. We were not able to rescue live born Psen1-/- animals from matings with either nestin-Psen1 line 9 or 14 in over 40 pups generated from these matings (P=0.004 that no Psen1-/- would be born).
Previously, we generated transgenic mice that express human wild-type Psen1 in a neuron specific fashion under control of the neuron specific enolase (NSE) promoter/enhancer regions (Wen et al., 2002b; Wen et al., 2004b). The Psen1 transgene in these lines was widely expressed in neurons in adult brain (Wen et al., 2002b). We also determined the expression pattern in developing brain by in situ hybridization. As shown in Fig. 2, in E16.5 brain the transgenic human Psen1 in NSE-Psen1 line 30 is widely expressed in neurons in the developing cortex but not in the ventricular zone, suggesting that transgene expression begins in the earliest stages of neuronal differentiation but is not present in neural progenitor cells. Thus, the NSE driven transgene seemed optimal to determine the effect of Psen1 expression in the earliest developing postmitotic neurons in the absence of neural progenitor expression.
Fig. 2.

Expression pattern of an NSE-Psen1 transgene. (A,B) Horizontal sections through the lateral telencephalon of an E16.5 NSE-Psen1 transgenic (A) or non-transgenic littermate control (B) embryos hybridized with a human-specific PSEN1 probe. In the transgenic embryo there is prominent hybridization in the intermediate zone (IZ), cortical plate (CP) and marginal zone (MZ), and an absence of signal in the ventricular zone (VZ). Scale bar: 50 μm.
To determine if neuron-specific expression of Psen1 could rescue the embryonic lethality of Psen1-/- mice we bred NSE-Psen1 line 30 onto the Psen1-/- background. Again, we were unable to rescue live born Psen1-/- mice in over 20 pups born from these matings (P=0.06). Thus, neither neuron-specific nor neural progenitor-specific expression of Psen1 can rescue the embryonic lethality of the Psen1-/- mouse.
Selective expression of Psen1 in neural progenitor cells rescues the cerebral hemorrhages and normalizes the appearance of the developing neocortex in Psen1-/- embryos
As we could not rescue live born Psen1-/- animals with either nestin- or NSE-driven transgenes we next examined the status of the embryos. By E12.5 Psen1-/- embryos possess a generally hypomorphic caudal body with a kinked tail and by E18.5 a disorganized cortical laminar structure and almost invariably gross cerebral hemorrhages (Shen et al., 1997; Wong et al., 1997). Therefore, we examined E12.5 and E18.5 embryos from matings of hemizygous transgenic/heterozygous Psen1+/- with Psen1+/- animals. As shown in Fig. 3, E18.5 embryos with the nestin-Psen1 transgene on the Psen1-/- background still possessed a hypomorphic caudal body with a kinked tail, suggesting that the nestin driven transgene does not rescue the defects in somite segmentation in Psen1-/- animals (Wong et al., 1997). However, none of the embryos with the nestin-Psen1 transgene on the Psen1-/- background evidenced gross cerebral hemorrhages that are nearly always present in pure Psen1-/- embryos at E18.5 (Fig. 3).
Fig. 3.
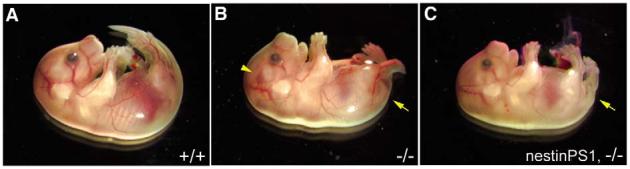
A nestin-Psen1 transgene rescues the cerebral hemorrhages but not the caudal defects in the Psen1-/- embryo. (A-C) E18.5 wild-type (A), Psen1-/- (B)embryos and an embryo with the nestin-Psen1 transgene on the Psen1-/-background (C). The Psen1-/- embryo has an abnormal tail region (arrow) and a cerebral hemorrhage (arrowhead). By contrast, the embryo with the nestin-Psen1 transgene on the Psen1-/- background still exhibits the caudal defects (arrow) but no cerebral hemorrhages.
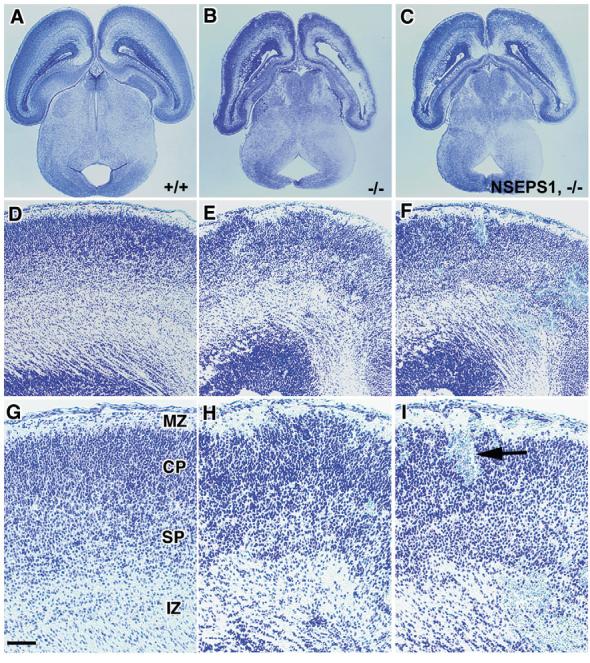
Because defects in cortical development occur in Psen1-/- mice (Hartmann et al., 1999; Shen et al., 1997), we examined the histology of the telencephalon in crosses of the nestin-Psen1 transgene onto the Psen1-/- background. Cresyl Violet-stained sections of E12.5 and E18.5 embryos are shown in Figs 4 and 5. At E12.5 (Fig. 4), the telencephalon consists of a ventricular zone (VZ) and preplate (PP), and there were no obvious differences between Psen1+/+, Psen1+/-, Psen1-/- mice or animals with the nestin-Psen1 transgene on the Psen1+/+, Psen1+/- or Psen1-/- backgrounds. At E18.5, wild-type telencephalon shows a recognizable laminar structure (Fig. 5). By contrast, Psen1-/- animals show hemorrhages and a general disorganization of the laminar structure of the cortex, even in areas that are free of hemorrhage. In many cortical areas, Psen1-/- embryos also exhibited cellular ectopias in the marginal zone (Fig. 5) as well as the leptomeningeal gaps and thickening first described by Hartmann et al. (Hartmann et al., 1999).
Fig. 4.
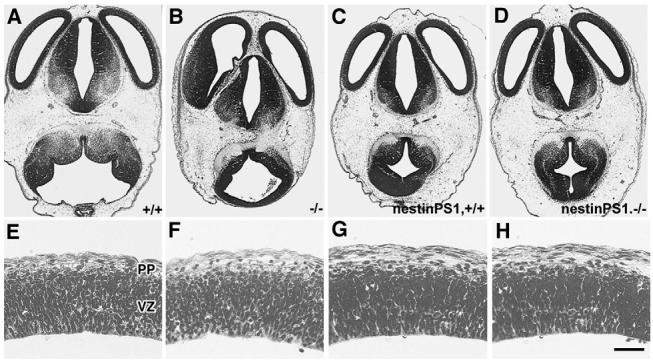
Normal appearance of the developing cortical plate in E12.5 embryos with or without Psen1. Cresyl Violet-stained horizontal sections from E12.5 wild-type (A,E) and Psen1-/-(B,F) embryos, as well embryos with the nestin-Psen1 transgene on the Psen1+/+(C,G) and Psen1-/- backgrounds (D,H). (E-H) Higher power views through the lateral wall of the telencephalon. The ventricular zone (VZ) and preplate (PP) are indicated. No differences were apparent between any of the genotypes. Scale bar: 200 μm for A-D; 25 μm for E-H.
Fig. 5.

Rescue of cerebral hemorrhages and cortical lamination defects in Psen1-/- embryos with a nestin-Psen1 transgene. Cresyl Violet-stained coronal sections of E18.5 embryos from wild-type (A,F,K), Psen1+/- (B,G,L), Psen1-/- (C,H,M), nestin-Psen1 transgene on Psen1-/- background (D,I,N) and nestin-Psen1 transgene on Psen1+/+ background (E,J,O). (F-O) Higher power views through the lateral telencephalon. In the E18.5 embryos, the wild-type embryo shows a clear lamination pattern with a recognizable ventricular zone (VZ), intermediate zone (IZ), subplate (SP), cortical plate (CP) and marginal zone (MZ). The Psen1-/- animal has multiple areas of hemorrhage (one indicated by arrow in M) and lacks distinct cortical layers. A cortical ectopia in the Psen1-/- embryo is indicated by an arrowhead (M). By contrast, the embryo with the nestin-Psen1 transgene on the Psen1-/- background, as well as the Psen1+/- and the embryo with the nestin-Psen1 transgene on the Psen1+/+ background, are normal in appearance. Scale bars: 500 μm in A-E; 100 μm in F-J; 50 μm in K-O.
None of six embryos with the nestin-Psen1 transgene on Psen1-/- background derived from four separate litters showed microscopic evidence of hemorrhage and the cortical lamination pattern appeared identical to wild-type embryos (Fig. 5). Rescued embryos also showed no heterotopias in the marginal zone and the leptomeninges showed neither the gaps nor fibrosis seen in Psen1-/- embryos (see Fig. S2 in the supplementary material). Likewise, the previously described loss of chondroitin sulfate proteoglycan staining in the marginal zone (Hartmann et al., 1999) was normalized (see Fig. S3 in the supplementary material), and reelin immunostaining (see Fig. S4 in the supplementary material) showed that Cajal-Retzius cells were as easily identified in the marginal zone of rescued animals as in wild-type embryos, showing no evidence for the loss of these cells that occurs in latter stage Psen1-/- embryos (Hartmann et al., 1999; Kilb et al., 2004; Wines-Samuelson et al., 2005). Indeed, except for pure Psen1-/- embryos, there were no differences between wild-type embryos and any of the genotypes that were generated in these crosses (i.e. Psen1+/- or nestin-Psen1 transgene on mouse Psen1+/+, Psen1+/- or Psen1-/- backgrounds) Thus, neural progenitor specific expression of Psen1 is sufficient to normalize cortical lamination patterns as well as prevent cerebral hemorrhages in Psen1-/- embryos.
An NSE driven transgene rescues neither the cerebral hemorrhages nor the cortical lamination defects found in Psen1-/-mice
We also examined brains from embryos with the NSE-Psen1 transgene on the Psen1-/- background. As with the nestin-Psen1 transgene, the NSE driven transgene failed to rescue the hypomorphic caudal body and kinked tail of Psen1-/- animals (data not shown). In addition, E18.5 embryos with the NSE-Psen1 transgene on the Psen1-/- background had gross cerebral hemorrhages that were evident microscopically to the same degree found in pure Psen1-/- embryos (Fig. 6), as well as cellular ectopias in the marginal zone and cortical lamination defects that were identical to those found in Psen1-/- mice (Fig. 6). Thus, expression of Psen1 in postmitotic neurons using an NSE promoter rescues none of the defects found in Psen1-/-embryos.
Fig. 6.
Failure of an NSE-Psen1 transgene to rescue the brain pathology in Psen1-/- mice. Shown are Cresyl Violet-stained horizontal sections of E18.5 embryos that are wild type (A,D,G), Psen1-/- (B,E,H) or have the NSE-Psen1 transgene on Psen1-/- background (C,F,I). (D-I) Higher power views of the lateral cerebral wall. The embryo with the NSE-Psen1 transgene on the Psen1-/- background exhibits multiple areas of hemorrhage (one indicated by arrow), as well as disrupted cortical lamination in a pattern that is indistinguishable from that of the Psen1-/-embryo. Cortical layers (MZ, CP, SP, IZ) are indicated as in Fig. 5. Scale bar: 500 μm in A-C; 100 μm in D-F; 50 μm in G-I.
Neural progenitor specific expression of Psen1 normalizes cell migration patterns in the telencephalon in Psen1-/-embryos
Neural progenitor cells migrate prematurely into the developing neocortex of Psen1-/- embryos (Handler et al., 2000; Yuasa et al., 2002). We therefore determined whether neural progenitor specific expression of Psen1 could rescue cell migration defects. Initially, we examined the BrdU labeling patterns 2 hours after injections into E12.5 embryos. There were no discernable differences in labeling patterns between the various genotypes (see Fig. S5 in the supplementary material), indicating that at E12.5 the progenitor population in the ventricular zone is still grossly intact in the Psen1-/- embryo and that this pattern is not affected by overexpression of Psen1 in neural progenitor cells.
Next, we injected BrdU at E12.5 and examined embryos at E18.5. As shown in Fig. 7, the majority of BrdU-labeled cells in a Psen1+/+ embryo were concentrated in the deeper layers of the cortical plate, a pattern expected for a wild-type animal. In Psen1-/- embryos BrdU labeled cells were spread diffusely throughout the presumptive cortical plate, similar to the cell migration defects observed by others (Handler et al., 2000; Yuasa et al., 2002). By contrast, BrdU labeled cells in embryos with the nestin-Psen1 transgene on the Psen1-/- background were concentrated in the deeper layers of the cortical plate as in wild-type animals. Embryos with the nestin-Psen1 transgene on the Psen1+/+ background or Psen1+/- mice also had BrdU labeling patterns that were similar to wild-type embryos. These effects have been observed in at least three embryos for each genotype, in each case the embryos coming from more than one litter, and have been observed in embryos from crosses of both the nestin-Psen1 line 9 and line 14 onto the Psen1-/- background.
Fig. 7.

Rescue of cell migration defects in Psen1-/- embryos with a nestin-Psen1 transgene. Pregnant females were given one dose of BrdU at E12.5 and the embryos were collected at E18.5 and immunostained with anti-BrdU antibodies. Shown are BrdU-immunostained sections through the lateral cerebral wall of E18.5 embryos from wild-type (A), Psen1+/- (B), Psen1-/- (C), nestin-Psen1 transgene on Psen1-/- background (D) and nestin-Psen1 transgene on Psen1+/+ background (E). BrdU labeling is dispersed in the Psen1-/- embryo and normalized by the nestin-Psen1 transgene. Cortical layers (MZ, CP, SP, IZ, VZ) are indicated as in Fig. 5. Scale bar: 100 μm.
The nestin intron 2 enhancer is not active in vascular endothelial cells or vascular progenitor cells
The brain vascular system develops from mesodermally derived angioblasts that invade the brain from overlying mesenchymal tissue (Harrigan, 2003). Additional blood vessels form via sprouting or splitting of initially established vessels (Harrigan, 2003). Thus, rescue of the cerebral hemorrhages is somewhat surprising given that the nestin intron 2 enhancer is not regarded as active in mesoderm (Zimmerman et al., 1994).
As leakage of the transgene into mesodermal- or possibly neural crest-derived cephalic mesenchymal cells would provide a trivial explanation for the rescue of the cerebral hemorrhages, we re-examined transgene expression pattern in E12.5 brain by in situ hybridization, with particular attention as to whether the transgenic human PSEN1 was expressed in the overlying cephalic mesenchyme. As expected, strong hybridization was seen within the brain parenchyma of the transgenic specimen, with the signal being especially prominent in cells surrounding the ventricular zone (Fig. 8). Owing to the resolution of the digoxigenin detection system, it was difficult to determine unequivocally whether hybridization product was present in endothelial cells within brain parenchyma. However, no hybridization was apparent in any region of the overlying cephalic mesenchyme, including blood vessels and perivascular cells within this structure (Fig. 8) indicating that the transgene is not expressed in mesoderm or in neural crest-derived elements that migrate into this region (Etchevers et al., 1999).
We have also generated transgenic mice expressing Cre recombinase under the control of the nestin intron 2 enhancer/tk promoter (NesCrenls mice). Cre expression in these lines has been verified by both western blotting and by immunohistochemistry on E13.5 mouse brain (data not shown). Because low-level transgene expression could be missed by in situ hybridization, as an additional test of nestin intron 2 enhancer activity, we crossed NesCrenls transgenic mice to two lines of Cre reporter mice, a technique that should be sensitive to even low levels of nestin intron 2 enhancer activity. NesCrenls line 27 mice were firstly crossed with a Cre/loxP reporter line cActXstopXEGFP44. This line serves as an indicator of Cre expression in that Cre removes a loxP flanked stop cassette sequence and activates EGFP expression. Spontaneous EGFP fluorescence was imaged and combined with immunohistochemistry for aquaporin 4, which is expressed in astrocytic foot processes that surround blood vessels and thus serves as an effective vascular marker (Verkman, 2002). EGFP expression from the hippocampal CA1 region of an adult double transgenic mouse is shown in Fig. 9, where prominent expression of EGFP is seen in CA1 pyramidal cells while no EGFP expression is seen in the aquaporin outlined vessels.
Fig. 9.
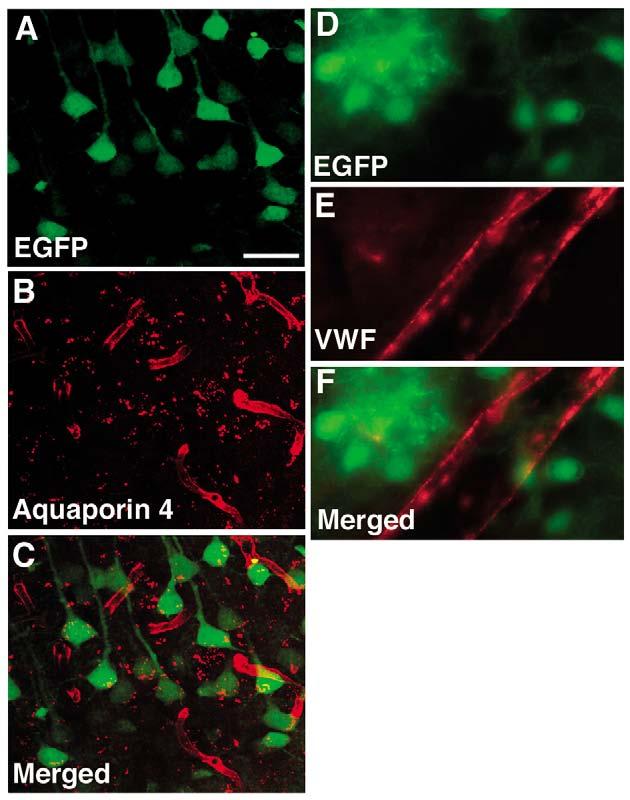
Lack of the nestin intron 2 enhancer activity in vascular progenitor cells or endothelial cells. NesCrenls line 27 was crossed with a Cre/loxP reporter line cActXstopXEGFP44. The hippocampal CA1 region of an adult double transgenic mouse is shown in A-C. Spontaneous EGFP fluorescence (A) was imaged and combined with immunohistochemistry for aquaporin 4 (B). EGFP is prominently expressed in the pyramidal cells but not in the aquaporin 4 outlined vessels (merged image is shown in C). (D-F) Combined immunohistochemical staining is shown for EGFP (D), and von Willebrand factor (E) on an adult brain from a NesCrenls line 27 mouse crossed to the Z/EG reporter line (merged image is shown in F). Immunostaining in D-F was performed on sections cut from tissue perfusion fixed with paraformaldehyde. A large penetrating vessel is visible at the cortical surface. There is no EGFP labeling of the von Willebrand factor-stained vessel. Scale bar: 50 μm for A-C; 10 μm for D-F.
As an independent test of nestin intron 2/tk promoter activity, we also crossed NesCrenls lines 27 and 13 mice with the Z/EG reporter line (Novak et al., 2000). In this line, Cremediated recombination removes a lacZ gene and activates expression of an EGFP reporter under the control of CMV/chicken β-actin promoter. These mice have been used in several recent studies to follow Cre-mediated excision in both early embryonic and adult tissues (Guo et al., 2002; Malatesta et al., 2003; Novak et al., 2000). Crosses with both NesCrenls lines gave similar results in showing widespread EGFP activation in adult brain but no activation in or around brain vessels. Immunohistochemical staining for EGFP combined with staining for von Willebrand factor as an endothelial cell marker (Costa et al., 2001) is shown in Fig. 9, where EGFP labeled cells can be seen to the left and right of a large penetrating vessel at the cortical surface, while no EGFP expression is apparent in the von Willebrand factor stained vessel. In these same sections, we have also been unable to detect EGFP activation in either aquaporin 4- or fibronectin-(Peters and Hynes, 1996) labeled vessels (data not shown). Thus, collectively, these studies show that the nestin intron 2 enhancer is not active in vascular endothelial cells or in vascular progenitor cells at any stage of development.
Selective expression of Psen1 in neural progenitor cells restores a normal vasculature in Psen1-/- embryos
Cerebral hemorrhages can occur in Psen1-/- embryos as early as E12.5 but are unusual at this age, while at E18.5 intracerebral as well as intraventricular hemorrhages are invariably present. To investigate how Psen1 expression in neural progenitor cells might influence vascular development, we labeled vascular endothelial cells in Psen1-/- and wild-type embryos with Griffonia simplicifolia isolectin B4 (Ashwell, 1991). In E18.5 Psen1-/- brains, lectin-stained vessels in the cerebral hemispheres appeared distorted and more lobular than vessels in wild-type brains (Fig. 10). Such changes were evident even in areas without macroscopic or microscopic hemorrhage, and were also present in vessels in the ganglionic eminence (data not shown). In E12.5 embryos, differences between vessels in Psen1-/- and wild-type control brains were more subtle. However, vessels in both the cortical plate (Fig. 10) and the ganglionic eminence (data not shown) of Psen1-/- brain appeared enlarged compared with wild type. These changes at both E12.5 and E18.5 were, however, completely normalized in embryos with the nestin-Psen1 transgene on the Psen1-/- background (Fig. 10).
Fig. 10.

Rescue of brain vascular abnormalities in embryos with the nestin-Psen1 transgene on the Psen1-/- background. (A-C) Staining with isolectin B4 was performed on paraffin-embedded E12.5 embryos from wild-type (A) and Psen1-/- (B)embryos, and embryos with the nestin-Psen1 transgene on the Psen1-/- background (C). Sections through the lateral telencephalon are shown. Vessels in the Psen1-/-embryo are enlarged. (D-L) Isolectin staining (E,H,K) was combined with nestin immunostaining (D,G,J) on paraffin-embedded E18.5 embryos from wild-type (D-F) and Psen1-/- (G-I) embryos, and embryos with the nestin-Psen1 transgene on the Psen1-/- background (J-L). Merged images are shown in F,I,L. Regions along the cortical surface of the lateral telencephalon are illustrated. The lectin-stained vessels in the Psen1-/- brain are distorted and lobular appearance, and this pattern is normalized by the nestin-Psen1 transgene. The pattern of nestin staining is altered in the Psen1-/- embryo. Scale bar: 100 μm for A-C; 50 μm for D-L.
During cerebral angiogenesis, endothelial cells form close contacts with perivascular mural cells or pericytes (Beck and D’Amore, 1997), and once within the neuroectoderm, endothelial/pericyte complexes make close contacts with brain cells, including radial glia (Bass et al., 1992). To address whether defective interactions between neuroepithelial cells and developing vessels might be occurring in Psen1-/-embryos, we used nestin immunostaining as a marker of neuroepithelial cells and isolectin B4 staining as a vascular marker. Nestin staining in E12.5 Psen1-/- telencephalon appeared similar to wild-type embryos, with many radially distributed processes extending throughout the telencephalic wall (data not shown). At E18.5, nestin staining in wild-type controls continued to show many radially distributed processes extending throughout the full thickness of the cerebral hemispheres (Fig. 10) with some nestin-stained processes traversing near cerebral vessels. By contrast, in E18.5 Psen1-/-animals, nestin staining was largely absent in the cerebral hemispheres with at most sparse fibers that failed to extend through the full thickness of the cerebral wall, a pattern that was evident even in areas that lacked cerebral hemorrhages. Interestingly, the nestin staining that was present in Psen1-/-brains was concentrated around and possibly within distorted cerebral vessels. Similar patterns were observed in both the cerebral hemispheres (Fig. 10) and the developing caudate/putamen (data not shown) of Psen1-/- embryos. However, in embryos with the nestin-Psen1 transgene on the Psen1-/- background, many radially directed processes were nestin stained, and these processes associated with brain vessels in a pattern that was identical to wild-type controls (Fig. 10).
Discussion
Psen1-null mutant mice die during late intrauterine life or shortly after birth (Shen et al., 1997; Wong et al., 1997). The embryos are small and exhibit a generally hypomorphic caudal body with a kinked tail, as well as skeletal malformations. Abnormalities in the tail region can be seen as early as E9.5, with the somites being indistinct and largely absent in the most caudal region (Wong et al., 1997). By E15.5, the vertebral column is shortened and fails to undergo proper segmentation, resulting in fused vertebrae. Some embryos have midline defects in the body wall and cranial vault (Hartmann et al., 1999). The telencephalon appears relatively normal at E12.5. However, by E14.5 to E15.5 homozygous mutants exhibit abnormalities in cortical and subcortical development and, by E18.5, invariably display CNS hemorrhages (Handler et al., 2000; Louvi et al., 2004; Shen et al., 1997). The cellular basis for these defects remains largely unexplained. A prior study (Nakajima et al., 2003), reported abnormal vascular dilation in Psen1-/- embryos, similar to that observed here, as well as reduced capillary sprouting and abnormal vascular morphologies, including endothelial cell multiplication, apoptosis and necrosis. Although one study has suggested that neural progenitor cells differentiate prematurely in Psen1-/- mice (Handler et al., 2000), a more recent study has cast doubt on the generality of this conclusion (Wen et al., 2004a).
We show that selective expression of Psen1 in neural progenitors fails to rescue the embryonic lethality of Psen1-/- mice, as well as failing to rescue the hypomorphic caudal body malformations. However within brain, the nestin-Psen1 transgene rescued the cortical lamination defects found in Psen1-/- mice, normalized cell migration patterns (Handler et al., 2000; Yuasa et al., 2002) and eliminated the CNS hemorrhages. By contrast neuron-specific expression rescued neither the embryonic lethality nor any of the histological abnormalities in the brain. The ability of a nestin-driven transgene to rescue the cortical abnormalities in Psen1-/- mice documents a central role for Psen1 expression in neural progenitor cells. More surprising is the ability of the transgene to rescue the cerebral hemorrhages, because the brain vascular system develops from mesodermally derived angioblasts that invade brain from overlying leptomeninges (Marin-Padilla, 1985).
Several studies have suggested that vascular endothelial cells, including embryonic blood vessels in brain as well as pericytes and periendothelial cells may express nestin based on immunostaining with antibodies such as Rat 401 (Alliot et al., 1999; Mokry and Nemecek, 1998; Mokry and Nemecek, 1999; Tohyama et al., 1992). However, we found no evidence by in situ hybridization of transgene expression in the overlying cephalic mesenchyme, including blood vessels contained within this region and there was also no evidence for activity of the nestin intron 2 enhancer in endothelial cells or vascular progenitor cells when this enhancer was used to drive Cre recombinase and crossed to Cre reporter lines. Thus, leakage of the transgene into cephalic mesenchymal elements, whether mesodermal or neural crest derived does not appear to be the basis for the rescue of the vascular abnormalities. The failure of the nestin-Psen1 transgene to rescue the caudal defects in the Psen1-/- mouse also argues against mesodermal expression. Several other studies have also failed to detect vascular activity of the nestin intron 2 enhancer in transgenic mice when it was used to drive EGFP directly (Filippov et al., 2003; Kawaguchi et al., 2001; Mignone et al., 2004; Yamaguchi et al., 2000).
In addition, another study (Wines-Samuelson et al., 2005) recently reported the production of a conditional knockout of Psen1 in neural progenitor cells by crossing a nestin-Cre transgene onto a floxed Psen1 background. Although the cortical phenotype in these mice was milder than a pure Psen1-null mutant, the conditional knockout mice exhibited widespread intracranial hemorrhages that were thought to be a proximal cause of postnatal death in these animals. This study is thus consistent with our results suggesting that neural progenitor specific expression of Psen1 is necessary for the development of a competent vasculature.
Collectively, these results argue that Psen1 is essential for neural-derived signals that are necessary for formation of a normal vasculature. The disturbed nestin staining pattern in Psen1-/- embryos points to an abnormal association of neuroepithelial cells with brain blood vessels. In this context, two recent studies have also reported abnormal radial glial development in Psen1-/- embryos (Louvi et al., 2004; Wines-Samuelson et al., 2005). At the molecular level, it remains unclear how Psen1 expression in neural progenitor might mediate this effect. Psen1 influences a variety of pathways known to be important developmentally (Koo and Kopan, 2004). Yet whatever the mechanisms of action of Psen1, the studies described here show that Psen1 expression in neural progenitor cells is sufficient to rescue completely the brain abnormalities found in Psen1-/- mice, including the unexpected rescue of the vascular defects. As such, the combination of nestin-Psen1 transgenic mice and the Psen1-null mutant provide a model for dissecting the specific molecular pathways that Psen1 influences in neural progenitor cells.
Supplementary Material
Expression pattern of a nestin-Psen1 transgene. (A,B) Horizontal sections through the telencephalon of an E13.5 nestin-Psen1 transgenic (A) or non-transgenic littermate control (B) embryo hybridized with a human-specific PSEN1 probe. There is prominent hybridization in the ventricular zone (indicated by the arrow) surrounding the lateral ventricles (V) and an absence of signal in the non-transgenic specimen. Scale bar: 200 μm.
{kind=link}
Rescue of leptomeningeal abnormalities in Psen11–/– embryos with a nestin-Psen1 transgene. Cresyl Violet-stained horizontal sections from the lateral telencephalon of E18.5 embryos that are wild type (A), Psen11–/– (B) or have the nestin-Psen1 transgene on the Psen11–/– background (C). The leptomeninges are marked with asterisks. The Psen1–/–embryo displays the leptomeningeal thickening (B), which is normalized in the embryo with the nestin-Psen11–/– transgene on the Psen11–/– background (C). Scale bar: 50 μm.
{kind=link}
Restoration of chondroitin sulfate proteoglycan expression in the marginal zone of Psen11–/– embryos by a nestin-Psen1 transgene. Immunohistochemical staining for CSPG (red) combined with a DAPI nuclear stain (blue) through the lateral cerebral wall of E18.5 embryos that are wild type (A) or Psen11–/– (B), or express the nestin-Psen1 transgene on the Psen11–/– background (C). The marginal zone (MZ) and cortical plate (CP) are indicated. CSPG immunoreactivity is lost in the Psen11–/– marginal zone (B) and restored when the nestin-Psen1 transgene was bred onto the Psen11–/– background. An ectopia in the marginal zone of the Psen11–/– brain is indicated by an arrow (B). Scale bar: 50 μm.
{kind=link}
Normal appearance of Cajal-Retzius cells in Psen11–/– embryos expressing a nestin-Psen1 transgene. Reelin immunostained sections through the lateral cerebral wall of E18.5 embryos that are wild type (A,D) or Psen11–/– (B,E), or have the nestin-Psen1 transgene on the Psen11–/– background (C,F). Cajal-Retzius cells (indicated by arrows) are stained in the marginal zone of all three embryos. (D-F) Higher power views of reelin-stained cells. Cajal-Retzius cells appeared modestly depleted in the Psen11–/– embryos but normal in number when the nestin-Psen1 transgene was bred onto the Psen11–/– background. Scale bars: 50 mm in A-C; 10 μm in D-F.
{kind=link}
Normal patterns of BrdU labeling in E12.5 embryos with or without Psen1. Pregnant females mice received one injection of BrdU 2 hours prior to sacrifice. BrdU immunostaining of horizontal sections from wild type (A,B), Psen1+/– (C,D), Psen1–/– (E,F), nestin- Psen1 transgene on Psen11–/– background (G,H) and nestin-Psen1 transgene on Psen1 background (I,J) are shown. (B,D,F,H,J) Higher power images of labeling through the lateral cerebral wall. The preplate (PP) and ventricular zone (VZ) are indicated in B. No differences were apparent between any of the genotypes. Scale bar: 80 μm for A,C,E,G,I; 10 mm for B,D,F,H,J.
{kind=link}
Footnotes
This work was supported by grants from the National Institutes of Health (AG20139, AG21305, MH70603, AG23599, AG02219 and AG05138). Transgenic mice were generated through the Mount Sinai School of Medicine Mouse Genetics Shared Resource Facility. We thank Dr Paul Matthews for supplying the NT.1 antibody and Dr Nikolaos Robakis for antibody 222. We also thank Drs Ron McKay, Stephen O’Gorman, Eric Mercer and David Anderson for providing plasmids, and Dr Huntington Potter for making the Psen1-/- mice available. P.R.H. is the Regenstreif Professor of Neuroscience. R.D.G. is a recipient of a Young Investigator Award from the National Alliance for Research in Schizophrenia and Affective Disorders (NARSAD).
References
- Alliot F, Rutin J, Leenen PJ, Pessac B. Pericytes and periendothelial cells of brain parenchyma vessels co-express aminopeptidase N, aminopeptidase A, and nestin. J. Neurosci. Res. 1999;58:367–378. [PubMed] [Google Scholar]
- Aoki Y, Huang Z, Thomas SS, Bhide PG, Huang I, Moskowitz MA, Reeves SA. Increased susceptibility to ischemia-induced brain damage in transgenic mice overexpressing a dominant negative form of SHP2. FASEB J. 2000;14:1965–1973. doi: 10.1096/fj.00-0105com. [DOI] [PubMed] [Google Scholar]
- Ashwell K. The distribution of microglia and cell death in the fetal rat forebrain. Dev. Brain Res. 1991;58:1–12. doi: 10.1016/0165-3806(91)90231-7. [DOI] [PubMed] [Google Scholar]
- Bass T, Singer G, Slusser J, Liuzzi FJ. Radial glial interaction with cerebral germinal matrix capillaries in the fetal baboon. Exp. Neurol. 1992;118:126–132. doi: 10.1016/0014-4886(92)90029-p. [DOI] [PubMed] [Google Scholar]
- Beck L, Jr, D’Amore PA. Vascular development: cellular and molecular regulation. FASEB J. 1997;11:365–373. [PubMed] [Google Scholar]
- Costa C, Harding B, Copp AJ. Neuronal migration defects in the Dreher (Lmx1a) mutant mouse: role of disorders of the glial limiting membrane. Cereb. Cortex. 2001;11:498–505. doi: 10.1093/cercor/11.6.498. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Chen LS, Cotman CW, LaFerla FM. Injury induces presenilin-1 gene expression in mouse brain. NeuroReport. 1996;7:1773–1776. doi: 10.1097/00001756-199607290-00016. [DOI] [PubMed] [Google Scholar]
- Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum. Mol. Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- Elder GA, Tezapsidis N, Carter J, Shioi J, Bouras C, Li H-C, Johnston JM, Efthimiopoulos S, Friedrich VL, Robakis NK. Identification and neuron specific expression of the S182/presenilin I protein in human and rodent brains. J. Neurosci. Res. 1996;45:308–320. doi: 10.1002/(SICI)1097-4547(19960801)45:3<308::AID-JNR13>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Etchevers HC, Couly G, Vincent C, Le Douarin NM. Anterior cephalic neural crest is required for forebrain viability. Development. 1999;126:3533–3543. doi: 10.1242/dev.126.16.3533. [DOI] [PubMed] [Google Scholar]
- Filippov V, Kronenberg G, Pivneva T, Reuter K, Steiner B, Wang LP, Yamaguchi M, Kettenmann H, Kempermann G. Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Mol. Cell. Neurosci. 2003;23:373–382. doi: 10.1016/s1044-7431(03)00060-5. [DOI] [PubMed] [Google Scholar]
- Frisen J, Johansson CB, Lothian C, Lendahl U. Central nervous system stem cells in the embryo and adult. Cell Mol. Life Sci. 1998;54:935–945. doi: 10.1007/s000180050224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yang W, Lobe CG. A cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis. 2002;32:8–18. doi: 10.1002/gene.10021. [DOI] [PubMed] [Google Scholar]
- Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127:2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- Harrigan MR. Angiogenic factors in the central nervous system. Neurosurgery. 2003;53:639–660. doi: 10.1227/01.neu.0000079575.09923.59. [DOI] [PubMed] [Google Scholar]
- Hartmann D, De Strooper B, Saftig P. Presenilin-1 deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasia similar to human type 2 lissencephaly. Curr. Biol. 1999;9:719–727. doi: 10.1016/s0960-9822(99)80331-5. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Kim HS, Chang KA, Geum DH, Park CH, Seo JH, Rah JC, Lee JH, Choi SH, Lee SG, et al. Subcellular localization of presenilins during mouse preimplantation development. FASEB J. 2000;14:2171–2176. doi: 10.1096/fj.99-1068com. [DOI] [PubMed] [Google Scholar]
- Josephson R, Muller T, Pickel J, Okabe S, Reynolds K, Turner PA, Zimmer A, McKay RD. POU transcription factors control expression of CNS stem cell-specific genes. Development. 1998;125:3087–3100. doi: 10.1242/dev.125.16.3087. [DOI] [PubMed] [Google Scholar]
- Kawaguchi A, Miyata T, Sawamoto K, Takashita N, Murayama A, Akamatsu W, Ogawa M, Okabe M, Tano Y, Goldman SA, et al. Nestin-EGFP transgenic mice: visualization of the self-renewal and multipotency of CNS stem cells. Mol. Cell. Neurosci. 2001;17:259–273. doi: 10.1006/mcne.2000.0925. [DOI] [PubMed] [Google Scholar]
- Kilb W, Hartmann D, Saftig P, Luhmann HJ. Altered morphological and electrophysiological properties of Cajal-Retzius cells in cerebral cortex of embryonic presenilin-1 knockout mice. Eur. J. Neurosci. 2004;20:2749–2756. doi: 10.1111/j.1460-9568.2004.03732.x. [DOI] [PubMed] [Google Scholar]
- Koo EH, Kopan R. Potential role of presenilin-regulated signaling pathways in sporadic neurodegeneration. Nat. Med. 2004;10:S26–S33. doi: 10.1038/nm1065. [DOI] [PubMed] [Google Scholar]
- Kostyszyn B, Cowburn RF, Seiger A, Kj AA, Sundstrom E. Expression of presenilin-1 and notch-1 receptor in human embryonic CNS. Neuroscience. 2001;103:885–898. doi: 10.1016/s0306-4522(01)00045-8. [DOI] [PubMed] [Google Scholar]
- Lakso M, Sauer B, Mosinger B, Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl. Acad. Sci. USA. 1992;89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS-1 mutations. Am. J. Geriatr. Psychiat. 2004;12:146–156. doi: 10.1097/00019442-200403000-00006. [DOI] [PubMed] [Google Scholar]
- Louvi A, Sisodia SS, Grove EA. Presenilin 1 in migration and morphogenesis in the central nervous system. Development. 2004;131:3093–3105. doi: 10.1242/dev.01191. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Gotz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Early vascularization of the embryonic cerebral cortex: Golgi and electron microscopic studies. J. Comp. Neurol. 1985;241:237–249. doi: 10.1002/cne.902410210. [DOI] [PubMed] [Google Scholar]
- McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- Mokry J, Nemecek S. Angiogenesis of extra- and intraembryonic blood vessels is associated with expression of nestin in endothelial cells. Folia Biol. (Praha) 1998;44:155–161. [PubMed] [Google Scholar]
- Mokry J, Nemecek S. Cerebral angiogenesis shows nestin expression in endothelial cells. Gen. Physiol. Biophys. 1999;18:25–29. [PubMed] [Google Scholar]
- Moreno-Flores MT, Medina M, Wandosell F. Expression of presenilin 1 in nervous system during rat development. J. Comp. Neurol. 1999;410:556–570. [PubMed] [Google Scholar]
- Nakajima M, Yuasa S, Ueno M, Takakura N, Koseki H, Shirasawa T. Abnormal blood vessel development in mice lacking presenilin-1. Mech. Dev. 2003;120:657–667. doi: 10.1016/s0925-4773(03)00064-9. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- O’Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germline of mice, but not in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, Hynes RO. Fibronectin isoform distribution in the mouse. I. The alternatively spliced EIIIB, EIIIA, and V segments show widespread codistribution in the developing mouse embryo. Cell Adhes. Commun. 1996;4:103–125. doi: 10.3109/15419069609010766. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Tohyama T, Lee VM, Rorke LB, Marvin M, McKay RD, Trojanowski JQ. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumor cells. Lab. Invest. 1992;66:303–313. [PubMed] [Google Scholar]
- Verkman AS. Aquaporin water channels and endothelial cell function. J. Anat. 2002;200:617–627. doi: 10.1046/j.1469-7580.2002.00058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Diehlmann A, Buslei R, Beyreuther K, Bayer TA. Prominent expression of presenilin-1 in senile plaques and reactive astrocytes in Alzheimer’s disease brain. NeuroReport. 1998;9:3279–3283. [PubMed] [Google Scholar]
- Wen PH, Friedrich VL, Jr, Shioi J, Robakis NK, Elder GA. Presenilin-1 is expressed in neural progenitor cells in the hippocampus of adult mice. Neurosci. Lett. 2002a;318:53–56. doi: 10.1016/s0304-3940(01)02485-5. [DOI] [PubMed] [Google Scholar]
- Wen PH, Shao X, Shao Z, Hof PR, Wisniewski T, Kelley K, Friedrich VL, Jr, Ho L, Pasinetti GM, Shioi J, et al. Overexpression of wild type but not an FAD mutant presenilin-1 promotes neurogenesis in the hippocampus of adult mice. Neurobiol. Dis. 2002b;10:8–19. doi: 10.1006/nbdi.2002.0490. [DOI] [PubMed] [Google Scholar]
- Wen PH, De Gasperi R, Gama Sosa MA, Elder GA. Neural progenitor cells do not differentiate prematurely in presenilin-1 null mutant mice. Neurosci. Lett. 2004a;371:249–254. doi: 10.1016/j.neulet.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, et al. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp. Neurol. 2004b;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Wines-Samuelson M, Handler M, Shen J. Role of presenilin-1 in cortical lamination and survival of Cajal-Retzius neurons. Dev. Biol. 2005;277:332–346. doi: 10.1016/j.ydbio.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. NeuroReport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- Yuasa S, Nakajima M, Aizawa H, Sahara N, Koizumi K, Sakai T, Usami M, Kobayashi S, Kuroyanagi H, Mori H, et al. Impaired cell cycle control of neuronal precursor cells in the neocortical primordium of presenilin-1-deficient mice. J. Neurosci. Res. 2002;70:501–513. doi: 10.1002/jnr.10430. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression pattern of a nestin-Psen1 transgene. (A,B) Horizontal sections through the telencephalon of an E13.5 nestin-Psen1 transgenic (A) or non-transgenic littermate control (B) embryo hybridized with a human-specific PSEN1 probe. There is prominent hybridization in the ventricular zone (indicated by the arrow) surrounding the lateral ventricles (V) and an absence of signal in the non-transgenic specimen. Scale bar: 200 μm.
Rescue of leptomeningeal abnormalities in Psen11–/– embryos with a nestin-Psen1 transgene. Cresyl Violet-stained horizontal sections from the lateral telencephalon of E18.5 embryos that are wild type (A), Psen11–/– (B) or have the nestin-Psen1 transgene on the Psen11–/– background (C). The leptomeninges are marked with asterisks. The Psen1–/–embryo displays the leptomeningeal thickening (B), which is normalized in the embryo with the nestin-Psen11–/– transgene on the Psen11–/– background (C). Scale bar: 50 μm.
Restoration of chondroitin sulfate proteoglycan expression in the marginal zone of Psen11–/– embryos by a nestin-Psen1 transgene. Immunohistochemical staining for CSPG (red) combined with a DAPI nuclear stain (blue) through the lateral cerebral wall of E18.5 embryos that are wild type (A) or Psen11–/– (B), or express the nestin-Psen1 transgene on the Psen11–/– background (C). The marginal zone (MZ) and cortical plate (CP) are indicated. CSPG immunoreactivity is lost in the Psen11–/– marginal zone (B) and restored when the nestin-Psen1 transgene was bred onto the Psen11–/– background. An ectopia in the marginal zone of the Psen11–/– brain is indicated by an arrow (B). Scale bar: 50 μm.
Normal appearance of Cajal-Retzius cells in Psen11–/– embryos expressing a nestin-Psen1 transgene. Reelin immunostained sections through the lateral cerebral wall of E18.5 embryos that are wild type (A,D) or Psen11–/– (B,E), or have the nestin-Psen1 transgene on the Psen11–/– background (C,F). Cajal-Retzius cells (indicated by arrows) are stained in the marginal zone of all three embryos. (D-F) Higher power views of reelin-stained cells. Cajal-Retzius cells appeared modestly depleted in the Psen11–/– embryos but normal in number when the nestin-Psen1 transgene was bred onto the Psen11–/– background. Scale bars: 50 mm in A-C; 10 μm in D-F.
Normal patterns of BrdU labeling in E12.5 embryos with or without Psen1. Pregnant females mice received one injection of BrdU 2 hours prior to sacrifice. BrdU immunostaining of horizontal sections from wild type (A,B), Psen1+/– (C,D), Psen1–/– (E,F), nestin- Psen1 transgene on Psen11–/– background (G,H) and nestin-Psen1 transgene on Psen1 background (I,J) are shown. (B,D,F,H,J) Higher power images of labeling through the lateral cerebral wall. The preplate (PP) and ventricular zone (VZ) are indicated in B. No differences were apparent between any of the genotypes. Scale bar: 80 μm for A,C,E,G,I; 10 mm for B,D,F,H,J.