

Abstract
In eukaryotes, three pairs of structural-maintenance-of-chromosome (SMC) proteins are found in conserved multisubunit protein complexes required for chromosomal organization. Cohesin, the Smc1/3 complex, mediates sister chromatid cohesion while two condensin complexes containing Smc2/4 facilitate chromosome condensation. Smc5/6 scaffolds an essential complex required for homologous recombination repair. We have examined the response of smc6 mutants to the inhibition of DNA replication. We define homologous recombination-dependent and -independent functions for Smc6 during replication inhibition and provide evidence for a Rad60-independent function within S phase, in addition to a Rad60-dependent function following S phase. Both genetic and physical data show that when forks collapse (i.e., are not stabilized by the Cds1Chk2 checkpoint), Smc6 is required for the effective repair of resulting lesions but not for the recruitment of recombination proteins. We further demonstrate that when the Rad60-dependent, post-S-phase Smc6 function is compromised, the resulting recombination-dependent DNA intermediates that accumulate following release from replication arrest are not recognized by the G2/M checkpoint.
The eukaryotic structural-maintenance-of-chromosome (SMC) proteins form heterodimers that are the cores of several evolutionarily conserved multisubunit protein complexes. An Smc1/3 complex mediates sister chromatid cohesion during both mitosis and meiosis and is known as cohesin. Chromosome condensation requires two related Smc2/4-based complexes known as condensins (17). Smc5 and Smc6 form a complex along with six non-Smc components (Nse1 to Nse6). An additional protein, Rad60, has been shown to associate with the Smc5/6-Nse1-6 core complex nonstoichiometrically. In Schizosaccharomyces pombe, Smc5, Smc6, and Nse1-4 complex components are essential and loss of function results in chromosomal fragmentation that requires passage through S phase. Hypomorphic smc6, nse1, nse2, nse3, and nse4 mutants are all defective in homologous recombination repair (HRR) (5, 14, 16, 27, 33, 37, 39, 45, 54). Similar observations have been made for Saccharomyces cerevisiae, where compromising the Smc5/6 complex function interferes with ribosomal DNA (rDNA) segregation and sister chromatid exchange (11, 19, 53). In contrast, S. pombe Nse5 and Nse6 are nonessential. nse5 and nse6 null mutants grow slowly and exhibit repair defects similar to those of the smc5/6 complex hypomorphic mutants (46).
Of the non-SMC components, Nse1 contains a RING domain typical of ubiquitin E3 ligases, although its substrates and activity remain unidentified. Nse2 is a SUMO E3 ligase and directs the sumoylation of multiple substrates, including S. pombe Smc6 and S. cerevisiae and human Smc5. Nse3 is a member of the MAGE (melanoma antigen-encoding gene) superfamily and, like Nse4, is a conserved protein of an unknown function (2, 33, 45, 47, 50, 56). The core subunit components localize to the nucleus together with the noncore component Rad60, which has a C-terminal ubiquitin-like domain. Upon S-phase checkpoint activation by the ribonucleotide reductase inhibitor hydroxyurea (HU), Cds1CHK2 kinase phosphorylates Rad60, which is then excluded from the nucleus (5).
The Smc5/6 complex function was first characterized by genetic analysis of S. pombe smc6, formerly known as rad18 (14, 27, 54). smc6-X and smc6-74 are two separation-of-function mutants that are DNA damage sensitive and are defective in HRR pathways but are proficient in the essential function at all temperatures. Conditional mutants (e.g., smc6-T2) exhibit similar but temperature-dependent sensitivities to DNA damage and lose viability over several generations at 35°C. All the S. pombe conditional mutations map to the hinge region (50) (Fig. 1A). The smc6-X mutation (R706C) also maps close to the hinge. In contrast, the smc6-74 mutation (A151T) maps to the highly conserved arginine finger (25) close to the ATP binding site in the N-terminal globular domain, suggesting that it might affect DNA-dependent ATP binding/hydrolysis. (ATP binding site mutations are lethal [14, 54].) smc6-74 mutant cells have previously been shown to be proficient in DNA damage checkpoint initiation following UV-C irradiation but do not maintain checkpoint arrest in the presence of unrepaired damage (16, 54). Intriguingly, smc6-74 damage sensitivity, but not smc6-X sensitivity, is suppressed by overexpression of Brc1, a 6-BRCT (BRCA1 C-terminal) domain protein (54). Brc1 has homology to S. cerevisiae Esc4/Rtt107 and similarities with human Pax2 transactivation domain-interacting protein (PTIP), both of which function in DNA repair during S phase (20, 48, 49). Brc1-dependent smc6-74 suppression is dependent on structure-specific nucleases (Slx1/4 and Mus81/Eme1) and homologous recombination (HR), and we have interpreted this to suggest that smc6-74 cells are defective in a step of recombination that can be bypassed or resolved by alternative mechanisms (51).
FIG. 1.

smc6 mutants are sensitive to HU. (A) Diagram of the Smc5/6 complex, showing the relative positions of the smc6-T2 (ts), smc6-X, and smc6-74 mutations. (B) smc6 mutants are sensitive to HU, and the sensitivity of smc6-74 but not smc6-X can be rescued by overexpression of brc1. Overexpression of brc1 slightly sensitizes smc6+ cells to HU. Row 1, smc6+ plus pREP41 (vector control); row 2, smc6+ plus pREP41brc1; row 3, smc6-74 plus pREP41; row 4, smc6-74 plus pREP41brc1; row 5, smc6-X plus pREP41; row 6, smc6-X plus pREP41brc1. (C) Epistasis analysis of smc6 mutants with cds1CHK2 and recombination null mutants in response to HU. Representative double mutants are shown. cds1CHK2 with recombination null double mutants are slightly more sensitive than cds1CHK2 null mutants (compare the samples in the third and sixth rows at 1 mM HU in each panel). smc6 cds1-d CHK2 double mutants are more sensitive than the parental strains. Middle panel, smc6-X cds1-d; top and bottom panels, smc6-74 cds1-d. rad22-dRAD52 smc6 double mutants are more sensitive than the parental strains. Top panel, rad22-d smc6-74. rhp51RAD51 or rhp55RAD55 rescues the sensitivities of cells carrying smc6 single and smc6 cds1CHK2 double mutations. Middle panel, rhp51-d smc6-X; bottom panel, rhp55-d smc6-74. (D) Epistasis analysis with swi5 shows that swi5 mutants are not sensitive at 5 mM HU and show no genetic interaction with smc6. wt, wild type; OP, overproduction.
Previous work characterizing the smc6-X mutant HRR response showed that at low UV doses, survival is rescued by concomitant rhp51RAD51 deletion (27). A similar phenomenon was observed for the S. pombe RecQ helicase mutant rqh1-d (26). During HRR in G2, Rqh1 and Top3 act in a manner consistent with double Holliday junction (HJ) dissolution (7, 8). In vitro, the human RecQ helicase BLM and Top3 converge the two HJs and decatenate the resulting hemicatene (55). Because RecQ helicases have been implicated in the regulation of recombination at stalled or collapsed replication forks and since functions of the Smc5/6 complex have been indirectly ascribed to S phase, we examined the role of Smc6 during replication arrest due to nucleotide (deoxynucleoside triphosphate [dNTP]) depletion promoted by HU.
We show that replication intermediates are normal in smc6 mutants both in the presence and in the absence of replication arrest in checkpoint-proficient cells. During replication arrest in the absence of Cds1Chk2 (when replication forks rapidly collapse in the presence of HU [29]), a phenomenon associated with dissociation of the replicative polymerases (Pol) (10, 21, 32), replication intermediates are stable in the smc6 mutant cells compared to those in smc6+ cells. Despite the fact that a similar phenomenon is observed in recombination mutants (34), smc6 mutants are proficient in recruiting HR proteins to the chromatin when forks collapse, suggesting that the Smc5/6 complex is involved in later stages of HR under these circumstances. Consistent with this, we show that Smc5/6-dependent functions are required for effective HR-dependent repair of lesions resulting from HU-induced replication arrest and that the HR-dependent DNA structures that accumulate in smc6 mutants are not recognized by the G2 DNA damage checkpoint. We also show that, unlike Rad60, which is subjected to nuclear exclusion during S-phase arrest, Smc6 functions during, as well as upon release from, HU arrest and is chromatin bound in HU-treated cells.
MATERIALS AND METHODS
Genetics and cell biology techniques.
Strains were constructed by standard genetic techniques (36) and are listed in Table 1. The smc6-MYC strain, constructed by one-step integration (3), has a wild-type generation time at all temperatures and no sensitivity to DNA-damaging agents, such as ionizing radiation (IR) and UV, but is slightly sensitive to higher doses of HU (7.5 mM as opposed to 10 mM for the wild type). Replication was inhibited by the addition of 10 mM HU, and cells were grown at 30°C unless otherwise indicated. The protocols for checkpoint measurements, cell scoring, centrifugal elutriation, and fluorescence-activated cell sorter (FACS) analysis are all described in reference 13.
TABLE 1.
Strains
| Strain | Genotype | Source or reference |
|---|---|---|
| Wild type | leu1-32 ade6-704 ura4-d18 h− | |
| smc6-X | smc6-X leu1-32 ade6-704 ura4-d18 h− | 27 |
| smc6-74 | smc6-74 leu1-32 ade6-704 ura4-d18 h− | 54 |
| smc6-13MYC | smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 h− | This study |
| smc6-T2 | smc6-ts leu1-32 ade6-704 ura4-d18 rad22-GFP::G418 | 50 |
| rad60-1 | rad60-1 leu1-32 ade6-704 ura4-d18 rad22-GFP::G418 | 39 |
| rhp51-d | rhp51-d::ura4 leu1-32 ade6-704 ura4-d18 | 40 |
| rhp55-d | rhp55-d::ura4 leu1-32 ade6-704 ura4-d18 | 41 |
| rhp51-d smc6-X | rhp51-d::ura4 smc6-X leu1-32 ade6-704 ura4-d18 | 27 |
| rhp51-d smc6-74 | rhp51-d::ura4 smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| rhp55-d smc6-X | rhp55-d::ura4 smc6-X leu1-32 ade6-704 ura4-d18 | This study |
| rhp55-d smc6-74 | rhp55-d::ura4 smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d | cds1-d::ura4 leu1-32 ade6-704 ura4-d18 h− | 29 |
| cds1-d rhp51-d | cds1-d:: ura4 rhp51-d::ura4 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d rhp55-d | cds1-d:: ura4 rhp55-d::ura4 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d smc6-X | cds1-d::ura4 smc6-X leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d smc6-74 | cds1-d::ura4 smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d rhp51-d smc6-X | cds1-d:: ura4 rhp51-d::ura4 smc6-X leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d rhp51-d smc6-74 | cds1-d:: ura4 rhp51-d::ura4 smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d rhp55-d smc6-X | cds1-d:: ura4 rhp55-d::ura4 smc6-X leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d rhp55-d smc6-74 | cds1-d:: ura4 rhp55-d::ura4 smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| rad22-d | rad22-d::LEU leu1-32 ade6-M216 his3-d1 ura4-d18 | P. Werler |
| rad22-d smc6-X | rad22-LEU smc6-X leu1-32 ade6-704 ura4-d18 | This study |
| rad22-d smc6-74 | rad22-d::LEU smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d rad22 | cds1-d:: ura4 rad22-d::LEU leu1-32 ade6-704 ura4-d18 | This study |
| swi5-d | swi5-d::ura4 leu1-32 ura4-d18 | 1 |
| swi5-d smc6-74 | swi5-d::ura4 smc6-74 leu1-32 ura4-d18 | This study |
| rad22-GFP | rad22-GFP::G418 leu1-32 ade6-704 ura4-d18 h− | J. Cooper |
| smc6-X rad22-GFP | rad22-GFP::G418 smc6-X leu1-32 ade6-704 ura4-d18 h− | This study |
| smc6-74 rad22-GFP | rad22-GFP::G418 smc6-74 leu1-32 ade6-704 ura4-d18 h− | This study |
| cds1-d rad22-GFP | rad22-GFP::G418 cds1-d::ura4 leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d smc6-X rad22-GFP | rad22-GFP::G418 cds1-d::ura4 smc6-X leu1-32 ade6-704 ura4-d18 | This study |
| cds1-d smc6-74 rad22-GFP | rad22-GFP::G418 cds1-d::ura4 smc6-74 leu1-32 ade6-704 ura4-d18 | This study |
| taz1-GFP | taz-GFP::G418 leu1-32 ura4-d18 | J. Cooper |
| gar2-GFP | gar2-GFP::G418 leu1 ura4 h− | M. Yamamoto |
| swi6-GFP | nmt81-swi6-GFP leu1-32 ade6-M210 ura4-d18 h− | R. Allshire |
| cdc1-P7 | cdc1-p7 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc6-23 | cdc6-23 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc17-K42 | cdc17-k42 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc20-M10 | cdc20-M10 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc21-M68 | cdc21-M68 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc22-M45 | cdc22-M45 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc23-M36 | cdc23-M36 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
| cdc24-M38 | cdc24-M38 smc6-13MYC::G418 leu1-32 ade6-704 ura4-d18 | This study |
Microscopy.
For Smc6 visualization, an in vivo chromatin binding assay (22) was employed. Anti-Smc6 (50) was used at 1:60 and anti-MYC at 1:200. To visualize asynchronous and HU-blocked cells under the same coverslip, Triton X-100-extracted cells were washed twice in 1 mM CaCl2-Tris-buffered saline (TBS) and HU-blocked cells incubated in 1 mM CaCl2-TBS containing 2 μg/ml Texas Red GS-1 lectin. Cells were washed in CaCl2-TBS and fixed in methanol-acetone before mixing HU-treated/untreated cells and subjecting them to antibody staining. Live-cell imaging of Rad22-GFP foci was carried out in minimal media (EMM2) at room temperature. To determine the percentages of cells with Rad22 foci, >300 nuclei were scored for each sample.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed using a protocol modified from that described in reference 52. Anti-Smc6 was used at 1:15, and anti-MYC was used at 1:100. Antibodies were captured with G-protein Dynabeads. Efficiency of immunoprecipitation (IP) was monitored by Western blot analysis of input and IP samples. The relative amounts of PCR products were quantified by real-time quantitative PCR (qPCR) on 5 μl of each sample, using QuantiTect SYBR green PCR master mix (QIAGEN). Enrichments (n-fold) were calculated as previously described (9). Briefly, since the number of molecules doubles in each cycle, enrichment (n-fold) was calculated according to the formula 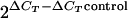 , in which ΔCT is the difference between the number of cycles required to go above background in input and IP samples and ΔCTcontrol is calculated from parallel input and IP samples from untagged strains or beads only or from nonspecific antibody controls. Since enrichment is calculated from the difference in the cycles, it is independent of the copy number of the genomic locus. Two or three sets of primer pairs were tested for the three positions within the rDNA repeat (the replication fork barrier [RFB], the autonomously replicating sequence ars3001, and the 17S gene), and a representative pair was used. Primers corresponding to the ade6 locus, ars3005 and ars2004 and control regions, 10 kb and 20 kb telomere proximal, respectively, were used to amplify unique loci. Primer sequences are available upon request. qPCR reactions were carried out in duplicate and ChIP data averaged over two or three independent experiments.
, in which ΔCT is the difference between the number of cycles required to go above background in input and IP samples and ΔCTcontrol is calculated from parallel input and IP samples from untagged strains or beads only or from nonspecific antibody controls. Since enrichment is calculated from the difference in the cycles, it is independent of the copy number of the genomic locus. Two or three sets of primer pairs were tested for the three positions within the rDNA repeat (the replication fork barrier [RFB], the autonomously replicating sequence ars3001, and the 17S gene), and a representative pair was used. Primers corresponding to the ade6 locus, ars3005 and ars2004 and control regions, 10 kb and 20 kb telomere proximal, respectively, were used to amplify unique loci. Primer sequences are available upon request. qPCR reactions were carried out in duplicate and ChIP data averaged over two or three independent experiments.
2-D gel electrophoresis.
Two-dimensional (2-D) gel electrophoresis was performed as described elsewhere (6). Genomic DNA was prepared by CsCl purification (23) from asynchronous cultures (−HU) or HU-treated cells (2.5 h, 10 mM HU) (+HU) and digested with XhoI and KpnI.
RESULTS
smc6 mutants are HR defective in response to replication inhibition.
smc6-X and smc6-74 are sensitive to the inhibition of DNA replication by HU (Fig. 1B). smc6-74 was consistently less sensitive than smc6-X, and HU sensitivity was suppressed in smc6-74, but not in smc6-X, by overexpression of brc1. This is consistent with the previously reported suppression of methyl methanesulfonate (MMS) and UV sensitivity (51, 54). Overexpression of brc1 slightly sensitizes smc6+ cells to HU. Thus, survival is dependent on a balance between Smc6- and Brc1-dependent pathways. The difference between the two hypomorphic smc6 mutants suggests that they have distinct defects in responding to HU, as we have previously observed for MMS resistance (51). We therefore further characterized both smc6 mutants in parallel.
Since Smc6 has been shown to function in HRR and, at low UV doses, sensitivity is rescued by concomitant rhp51RAD51 deletion (27), we examined the roles of Smc6 and HRR during replication inhibition by HU. Double mutants with rhp51rad51 and rhp55rad55 null alleles are not more sensitive than the single mutants, consistent with a function for Smc6 in the HR pathway. Figure 1C shows epistasis between smc6-X and rhp51 (the most sensitive pair) (Fig. 1C, middle panel) and smc6-74 and rhp55-d (the least sensitive pair) (Fig. 1C, bottom panel). Similar results were obtained with smc6-X rhp55-d and smc6-74 rhp51-d (data not shown). smc6 mutants are more sensitive than the recombination mutants but are partially rescued by concomitant deletion of recombination genes. This suggests either that stalled replication forks in the smc6 mutants are the target of illegitimate lethal recombination events or that Smc6 is required at a late stage of legitimate HR in response to HU. In the second eventuality, the HR-dependent structures that accumulate in smc6 mutants would be lethal but, in the absence of HR, an alternative pathway could repair the initial structure.
In S. pombe, Rhp55Rad55 acts upstream of Rhp51Rad51 in parallel to a second recombination mediator, Swi5 (1). Like loss of rhp51RAD51, loss of rhp55RAD55 rescues smc6 mutant sensitivity to HU. Loss of swi5 does not (Fig. 1D). This is reminiscent of the requirement for the Rhp55/57 complex for Brc1-mediated suppression of the MMS sensitivity of smc6-74, which is also independent of Swi5 (16). A similar pattern of suppression is observed for the S. pombe RecQ helicase mutant rqh1-d (18, 35; also unpublished data) and for rad60-1 (35). In contrast, the sensitivities of nse5 and nse6 mutants (nonessential components of the Smc5/6 complex) to UV are suppressed by rhp51-d and also weakly by both swi5-d and rhp55-d (46).
S. pombe Rad22Rad52 is required for all recombination. We have consistently observed that smc6 mutants are more sensitive in rad22rad52 mutant backgrounds than the single mutants (Fig. 1C, top panel). However, because rad22rad52 mutant cells are slow growing and rapidly acquire suppressor mutations in fbh1 (38, 44), further epistasis analysis is not feasible.
Replication forks arrested in HU are stabilized by activation of the Cds1CHK2-dependent intra-S-phase checkpoint. In the absence of Cds1CHK2, forks “collapse” (29), a process thought to involve the dissociation of the replicative apparatus from nascent DNA ends (10, 21, 32). Collapsed forks recruit recombination proteins (30, 34), and this is suggested to help restart replication. This requirement for recombination during replication resumption would predict that loss of recombination functions would increase cds1CHK2 mutant sensitivity to low doses (1 to 2 mM) of HU. A slight increase in sensitivity is indeed seen in recombination cds1CHK2 double mutants (Fig. 1C). In contrast, smc6 cds1CHK2 double mutants are much more sensitive than either of the single mutants (Fig. 1C). Loss of recombination genes rhp51RAD51 and rhp55RAD55 partially rescues smc6 cds1CHK2 double-mutant sensitivity. This again suggests either that smc6 mutants are defective at a late stage of HR or that in smc6 mutants, some collapsed replication forks are illegitimately processed by HR. The latter scenario would result in lethal HR-dependent intermediates. The rescue of viability is not at the level seen in rhp51-d cds1-d cells, suggesting that the Smc5/6 complex also has functions independent of HR in tolerating the HU arrest of replication forks.
smc6 mutants are defective in processing collapsed forks.
To establish how smc6 mutants affected recombination at stalled and collapsed forks, we used 2-D gel electrophoresis to visualize replication intermediates (31) from the intergenic region of the rDNA (Fig. 2). This region contains ars3001 and RFBs that ensure unidirectional rDNA replication. In both asynchronous cultures and HU-arrested cells, the replication intermediates observed in smc6 mutants were similar to those from smc6+ cells and replication forks were stably maintained at the RFBs, as judged by the occurrence of pause spots on the large Y arc. The X spike (recombination intermediates) was slightly increased in smc6-X cells both with and without treatment. Notably, no aberrant structures are seen in smc6-74. Consistent with previous analysis (24), we also observed the loss of Swi1/Swi3-dependent pause sites in the presence of HU (in the asynchronous cultures, two pause sites are visible on the Y arc, and only one is visible in HU-blocked cells). These data indicate that in the smc6 mutants, replication forks were stably maintained when stalled both specifically at the replication fork barriers and randomly due to dNTP depletion.
FIG. 2.

smc6 mutants are defective in processing collapsed forks. rDNA replication intermediates were analyzed using 2-D gels. (A) The structure of the rDNA region. (B) The expected position of each intermediate. (C) Genomic DNA was prepared from smc6+ (wt), smc6-X (X), smc6-74 (74), the cds1CHK2 null mutant (cds1), and the indicated double mutants, digested with XhoI and KpnI, run in both dimensions, and probed with the XhoI and KpnI fragment (XK) spanning ars3001 and the RFB. Asynchronous cultures (−HU) or cultures treated with 10 mM HU for 2.5 h (+HU) were analyzed. The bubble arc is visible in the HU-treated cells. Asynchronous cultures have two pause sites on the large Y arc. Only one is seen in HU-arrested cells. In smc6 mutants, replication intermediates are similar to those in smc6+ with or without HU treatment. The X spike (X-shaped molecules, such as recombination intermediates) is slightly increased in smc6-X. No aberrant structures are seen in smc-74. In the HU-arrested cds1CHK2 null cells, no large Y or bubble arc structures are visible, consistent with fork collapse. In the cds1CHK2 smc6 double mutants, these replication intermediates are preserved and the X spike increases, consistent with an increase in recombination intermediates.
We next examined a role for Smc6 at collapsed replication forks. In cds1CHK2 mutants treated with HU, replication forks collapse (29). This leads to the loss of replication intermediates (bubble and large Y arcs) and the accumulation of small Y molecules and a cone-shaped signal resulting from molecules migrating similarly to double-Y- and/or X-shaped structures (31, 34, 43). Consistently, we see neither the bubble arc nor large Y structures and no pause sites in HU-treated cds1CHK2 cells. (No cone-shaped signal was detected using this probe, but this signal was visible when a shorter HindIII-KpnI fragment lacking the RFBs was used as a probe, suggesting that the pause sites are particularly unstable [data not shown].) Surprisingly, bubble arcs, large Y, and pause structures are clearly present in DNA from HU-treated smc6-X cds1CHK2 and smc6-74 cds1CHK2 double-mutant cells. A similar stabilization of replication intermediates was recently reported for rhp51-d cds1CHK2 double mutants (34). Our results are thus consistent with Smc6 acting with HRR. However, neither smc6 nor rhp51rad51 mutants rescue the sensitivity of cds1-d (Fig. 1C), suggesting that illegitimate HR is not the cause of cds1CHK2 mutant sensitivity. Thus, the processing of collapsed replication forks is defective in smc6 mutants and there is an accumulation of potential recombination intermediates since smc6 cds1CHK2 mutants show a significant accumulation of X structures.
smc6 mutants lose viability upon entry into mitosis.
In HU-treated cells, replication forks are stably maintained due to the activation of the intra-S-phase checkpoint. Checkpoint-defective cds1CHK2 null cells rapidly lose viability during an HU-induced S-phase arrest but do not enter mitosis because the DNA damage checkpoint is activated by the structures that arise as forks collapse, and the cells elongate due to the checkpoint delay (29). In contrast, recombination mutants, such as rhp51-d, also elongate in response to HU but are not specifically sensitive to a transient inhibition of replication. This would be consistent with a requirement for HRR at only the small proportion of forks that collapse, the majority of forks being stabilized by the intra-S checkpoint. We assayed the viability of smc6 and smc6 cds1CHK2 double mutants during a transient HU exposure. smc6-X and smc6-74 cells start to die 4 to 6 h after exposure (Fig. 3A). At 6 h, wild-type and smc6 mutant cells overcome the replication block, have completed bulk DNA synthesis to obtain a 2N DNA content (Fig. 3B), and start entering mitosis. Thus, unlike Cds1CHK2, Smc6 is required when cells resume/complete replication rather than to maintain stalled replication forks. In smc6 cds1CHK2 double mutants, where the intra-S-phase checkpoint defect results in the collapse of the majority of forks, viability is lost with the same kinetics as that seen for cds1CHK2 null cells. These data suggest that in checkpoint-proficient cells, Smc6 is required at a specific subset of stalled forks which are not stable and collapse.
FIG. 3.

smc6-X and smc6-74 cells start to lose viability upon entry into mitosis. (A) Upon transient HU exposure, smc6 mutants start to lose viability after about 4 to 6 h. At 6 h, cells are overcoming the S-phase block and entering mitosis. smc6 cds1CHK2 double mutants lose viability with kinetics similar to that of cds1CHK2 null cells, dying in S phase. wt, wild type. (B) FACS analysis of smc6+, smc6-X, and smc6-74 cells in 10 mM HU. S. pombe is a haploid organism that spends ∼70% of its time in G2. In asynchronous cultures (time zero), there is a strong 2C peak. In an unperturbed cell cycle, G1 is very short and cytokinesis coincident with S phase, giving rise to the 2 to 2x2C shoulder. When cells are blocked in HU, cytokinesis proceeds as normal but S phase is slowed, leading to uncoupling of septation and replication and the appearance of a 1C peak. Thus, the FACS profile of cells blocked in HU shows a movement from a 2C peak to a 1C peak with time. By 2.5 h (the generation time in rich media at 30°C), all the cells are arrested with a 1C DNA content and stay arrested for approximately 3 h. However, cells gradually overcome the block such that by 6 h, most cells have completed replication (2C peak), and by 8 h, they have divided and are into the next S phase (2x2C shoulder). smc6 mutants progress similarly to wild-type cells but start to lose viability at about 6 h, coincident with entry into mitosis.
Rad22Rad52 focus formation is normal in smc6 mutants.
Previous work (30, 34) has shown a temporal separation of replication and recombination. HU-arrested cells have low levels of recombination foci. Upon release from arrest, approximately 20% of cells transiently acquire one or two foci. In order to further investigate the role of Smc6 in the resumption of replication, we monitored recombination protein foci both during HU arrest and upon release from HU. We assayed the occurrence of Rad22Rad52 foci in HU-treated smc6+ and smc6 mutant cells. After 4 h in HU, only ∼2% of smc6+ cells displayed Rad22Rad52 (Fig. 4A and B) and Rhp51Rad51 (data not shown) foci. smc6-X and smc6-74 cells showed similar levels (4% and 2%, respectively) (Fig. 4B). Thus, in the smc6 mutants, stalled replication forks do not lead to an increase in recombination foci.
FIG. 4.

Recombination focus formation is normal in smc6 mutants. (A) Rad22Rad52 foci (live-cell imaging) in HU and upon release. rad22-GFP cells were arrested for 4 h (10 mM HU) at 30°C and released at 18°C. In smc6+, smc6-74, and smc6-X, the Rad22Rad52 signal was diffuse in the nucleus in HU, but cells with one or two nuclear foci became visible upon release. (B) Quantification of Rad22Rad52 foci and mitotic cells upon HU release. Foci formed with similar kinetics in smc6+, smc6-X, and smc6-74 cultures, peaking at ∼90 min after release, and disappeared prior to mitosis. (C and D) In contrast, smc6 cds1CHK2 double mutants, like cds1CHK2 null cells, showed multiple nuclear Rad22Rad52 foci during HU treatment (examples are shown in panel C) and these remained for the duration of the experiment after release (D).
Ninety minutes after release from HU arrest at room temperature, ∼20% of smc6+ cells have one or two Rad22Rad52 foci. Similar percentages of smc6-X and smc6-74 cells acquired foci upon release from HU arrest (Fig. 4A and B). Focus numbers peaked at 90 min and had mostly disappeared by 120 min, before cells entered mitosis at ∼150 min (Fig. 4B). Focus loss and mitotic entry were both slightly delayed in smc6-X cells. Thus, the functions defective in smc6 mutants are not required for recombination foci upon release from HU.
When replication forks collapse in checkpoint-defective cds1CHK2 null cells, the majority of nuclei contain multiple recombination foci (34). We found that cds1CHK2 null cells and cds1CHK2 smc6 double mutants exposed to HU acquired multiple Rad22Rad52 foci that remained after release (Fig. 4C). The cells remain checkpoint arrested and do not progress into mitosis during the course of the experiment (Fig. 4D). This shows that the functions defective in smc6 mutants are not required to form recombination foci in response to replication fork collapse.
smc6 mutants have a checkpoint maintenance defect.
The kinetics of mitotic entry in smc6 mutants following release from an HU arrest is similar to that seen for smc6+ (Fig. 5A). This is consistent with previous work showing that Smc5/6 complex mutants are proficient in the activation of the G2 DNA damage checkpoint (11, 16, 19, 46, 53). However, in contrast to smc6+ cells, smc6 mutant cells exhibit high levels of morphologically aberrant mitosis; the chromosomes missegregate or fail to separate before septation, leading to a cut phenotype (Fig. 5A and B). This indicates a failure to maintain the Chk1-dependent G2 DNA damage checkpoint even though DNA repair is not complete. Consistent with a dependence on HR for this incomplete repair, the appearance of cut cells is suppressed by deletion of recombination genes (rhp55RAD55 [Fig. 5A] and rhp51RAD51 [not shown]). A similar suppression of the cut phenotype is seen when brc1 is overexpressed in smc6-74 cells (Fig. 5C). This is consistent with the suppression of the checkpoint maintenance defect after UV treatment (54).
FIG. 5.

smc6 mutants have a checkpoint maintenance defect. Cells were arrested for 4 h (10 mM HU) at 30°C, and cell cycle progression was monitored (diamonds, mitotic index; open squares, septation index) following release at 30°C. (A) smc6+, smc6-X, and smc6-74 cultures all delayed mitosis for ∼90 min, indicating that the G2/M checkpoint is intact but smc6-X and smc6-74 cultures accumulate aberrant mitotic figures and “cut” cells (closed squares) (examples are shown in panel B). Under the same conditions, few “cut” cells were seen in rhp55-d, rhp55-d smc6-74, or rhp55-d smc6-X cultures. Thus, in smc6 mutants, recombination-dependent structures cause mitotic aberrations. These structures are not recognized by the G2/M checkpoint. (C) Overexpression of brc1 suppresses the accumulation of “cut” cells in smc6-74. Left, smc6-74 cells with an empty vector control (pREP41); right, smc6-74 cells with pREP41brc1. Symbols are as defined in panel A.
Smc6 is required for viability when replication is inhibited.
Recent work has demonstrated that rad60-1 hypomorphic mutant cells accumulate Rhp51Rad51-dependent recombination intermediates upon release from HU and that, like those that accumulated in smc6 mutants, such structures do not generate a checkpoint arrest and thus result in aberrant mitoses (35). Rad60 associates with the Smc5/6 complex but unlike the core subunits is excluded from the nucleus in response to HU following its phosphorylation by Cds1CHK2 (5). Rad60 is thus likely to be required only after release from HU but not during arrest in HU. Consistent with this, Miyabe et al. used a temperature-sensitive rad60 mutant (rad60-1) to demonstrate that loss of the Rad60 function during HU arrest did not result in viability loss (35).
Because Smc6 remains in the nucleus during an HU arrest, we wished to ascertain whether Smc6 is required for chromosome integrity during this arrest in addition to its common requirement with Rad60 upon resumption of cell cycle progression following HU withdrawal. We thus arrested smc6+, rad60-1, and a temperature-sensitive smc6 mutant (smc6-T2) (50) in HU at 36°C (inactive Rad60 or Smc6), followed by release at 22°C (permissive for rad60-1 and smc6-T2). We compared the results to those for a regimen in which the same mutants were arrested in HU at 22°C and released at 36°C (Fig. 6). Inactivating Smc6 during HU blocking led to the appearance of aberrant mitotic cells, which were not seen when Rad60 was inactivated during the HU block, and approximately 55% viability compared to that of smc6+ (rad60-1 was 88% viable). As expected, inactivation of either Smc6 or Rad60 immediately after HU release caused the appearance of aberrant mitotic cells in both cultures (Fig. 6) and loss of viability (rad60-1 was 48% viable and smc6-T2 was 51% viable). Consistent with smc6-T2 taking at least two cell cycles to start to lose viability (50), incubation at the nonpermissive temperature for 5 h in the absence of HU did not lead to significant loss of viability (rad60-1 was 87.7% viable and smc6-T2 was 89.8% viable). Nor did a regime where cells were blocked and released at the permissive temperature (rad60-1 was 89.7% viable and smc6-T2 was 93% viable). Together, these data suggest that Smc6 is required for chromosome integrity both during the HU block and upon release. This contrasts with Rad60, which is cytoplasmic during arrest and is required only for mitotic fidelity during recovery from the HU-induced blockade. However, a significant caveat for this interpretation is that we have no measure for how quickly Smc6 becomes active following a downshift to the permissive temperature (Smc6-T2 protein levels remain unchanged).
FIG. 6.

Smc6 is required in HU and upon release. smc6+, rad60-1, and smc6-T2 cells were incubated in HU (10 mM, 2.5 h) at the nonpermissive temperature for smc6-T2 and rad60-1 (36°C) and released into fresh media at the permissive temperature (22°C) (A), incubated in HU (10 mM, 4.5 h) at the permissive temperature (22°C) and released into fresh media at the nonpermissive temperature for smc6-T2 and rad60-1 (36°C) (B), or incubated in HU (10 mM, 4.5 h) at the permissive temperature and released at the same temperature (22°C) (C). Cell cycle progression following release (time zero) was monitored by septation index, and the percentages of aberrant mitotic or “cut” cells were scored. smc6+, rad60-1, and smc6-T2 entered mitosis with similar kinetics. rad60-1 accumulated aberrant mitotic cells only in regimen B (in which cells were released into the nonpermissive temperature) but not in regimen A (in which cells were HU blocked at the nonpermissive temperature and released into the permissive temperature), indicating that the Rad60 function is required only when the cells resume replication. In contrast, smc6-T2 accumulated aberrant cells in both regimen A and regimen B, suggesting that the Smc6 function is required both when replication is inhibited (Rad60 is not required) and when replication resumes upon release (a common requirement with Rad60). wt, wild type. (D) Viabilities observed after treatment with regimens A, B, and C or after incubation for 5 h at 36°C expressed as percentages of the smc6+ value.
Smc6 localizes to the nucleolus and accumulates after HU.
Smc5/6 complex components localize to the nucleus (5, 33, 53, 54, 56). We used an in situ chromatin binding assay to establish the subnuclear localization of Smc6 in untreated, S-phase-arrested, and DNA-damaged cells (22). In asynchronous cultures, all cells showed a background diffuse nuclear staining and Triton X-100-insoluble nuclear foci that were not visible after DNase I treatment. The foci did not colocalize with Swi6 in heterochromatin (centromeres, MAT loci, and telomeres) or with Taz1 at telomeres but did colocalize with the nucleolar component Gar2 (Fig. 7A). Smc6 had a more compact distribution than Gar2, which could be indicative of a distinct localization within the nucleolus. HU treatment significantly increased focus intensity but did not change the pattern of localization (Fig. 7B). In order to confirm the increases in Smc6 focus intensity, asynchronous and HU-blocked cells (cell wall-stained with Texas Red-conjugated lectin) were observed under the same coverslip (Fig. 7C). The intensity of the Smc6 nuclear signal was increased in HU-blocked cells compared to that in asynchronous cells. However, incubation in the lectin staining buffer led to the distribution of Smc6 throughout the nucleus rather than restricted to nucleolar foci, suggesting that the subcellular localization of Smc6 is sensitive to the method of sample preparation. The increase in intensity was independent of cell wall staining, as a parallel experiment with the asynchronous cells marked with Texas Red-conjugated lectin also showed the HU-blocked cells to have a more intense nuclear staining (data not shown).
FIG. 7.

Smc6 localizes to the nucleolus and increases in intensity upon exposure to replication stress. (A). Indirect immunofluorescence with anti-Smc6 identifies foci that colocalize with Gar2-green fluorescent protein (GFP) (nucleolus) but not Swi6-GFP (heterochromatin) or Taz1-GFP (telomeres). Smc6 (red), GFP (green), and DNA visualized with DAPI (4′,6′-diamidino-2-phenylindole) (blue) are shown. Bar, 5 μm. (B). Smc6 foci are more intense in HU-blocked cells. Smc6 (red) and DNA visualized with DAPI (blue) are shown. (C) Asynchronous and HU-blocked cells under the same coverslip. HU-blocked cells, identified by Texas Red-conjugated lectin staining cell walls, have increased nuclear Smc6 (green). DNA visualized with DAPI (blue) is shown. (D) Increased nuclear Smc6 in the replication mutant cdc23-M36 (MCM10) at the nonpermissive temperature. Top panel, smc6-MYC cells at 35°C; bottom panel, smc6-MYC cdc23-M36 cells at 35°C. Smc6-MYC (green) and DNA visualized with DAPI (blue) are shown. (E) Total Smc6 protein levels do not change upon inhibition of replication. Column 1, smc6-MYC cells at 25°C; column 2, smc6-MYC cells at 35°C; column 3, smc6-MYC cdc23-M36 cells at 25°C; column 4, smc6-MYC cdc23-M36 cells at 35°C. Top, anti-MYC; bottom, anti-Cdc2 loading control.
Similar increases in intensity were seen in replication mutants cdc23-M36 (MCM10), cdc20-M10 (Pol ɛ), cdc1-P7 (Pol δ), cdc6-23 (Pol δ), cdc22-M45 (RNR), cdc17-K42 (DNA ligase 1), and cdc24-M38 (RFC and PCNA interacting factor) at the restrictive temperature (Fig. 7D and data not shown). Notably, no increase was seen in cdc21-M68 (MCM4) at the restrictive temperature (data not shown), suggesting that either replication initiation is required or loss of the MCM replicative helicase does not lead to an increase in nuclear Smc6. By Western blot analysis, this increase in Smc6 is due to protein relocalization, as total protein levels do not change (Fig. 7E). Since these replication mutants arrest with a 2C DNA content (42), we used IR to activate the G2 DNA damage checkpoint. No change was observed after IR (not shown), demonstrating that the increased focus intensity correlated with replicative stress. Localization of Smc6 to the nucleolus could indicate a compartmentalization of Smc6 away from Rad60 (cytoplasmic in HU) but is also consistent with a requirement for Smc6 at the ∼150 rDNA repeats, as has been previously described for S. cerevisiae (53).
Smc6 is associated with chromatin.
Our data suggest that Smc6 is required both during HU arrest and upon release from arrest. To ascertain whether Smc6 associated with chromatin within the rDNA or other loci, we performed ChIP assays. These assays employed qPCR with primers at three positions within the rDNA repeat (RFB, ars3001, and the 17S gene) and five unique loci (ars3005, 10 kb telomere proximal to ars3005, ars2004, 20 kb centromere proximal to ars2004, and the ade6 ORF) (Fig. 8). In contrast to S. cerevisiae Smc6, which was reported to be enriched at repeat sequences, including rDNA and telomeres (53), Smc6 was modestly enriched at all loci, both rDNA and unique, compared to negative controls (beads only or nonspecific antibody). Consistent with the microscopy results, HU treatment modestly but reproducibly increased enrichment. Similar data were obtained using anti-MYC ChIP and smc6-13MYC cells normalized to anti-MYC ChIP from an untagged strain. Thus, Smc6 is chromatin associated, and this increased in response to inhibition of replication.
FIG. 8.

Smc6 is chromatin associated. Results for qPCR analysis of Smc6 chromatin IP are shown. Using either anti-Smc6 (smc6+ cells) or anti-MYC (smc6-MYC cells), Smc6 precipitates are enriched at all loci tested (see schematic). HU treatment modestly increased enrichment. Data shown are mean values for two or three independent experiments. Enrichment (n-fold) is calculated compared to beads only or untagged controls. Error bars indicate standard errors.
DISCUSSION
Function for Smc5/6 at collapsed replication forks.
We have examined the role of Smc6 during replication arrest due to dNTP depletion promoted by HU. While smc6 mutants are sensitive to HU, we show by using 2-D gels that HU-arrested replication intermediates are normal in smc6 mutants when checkpoints are intact. Upon transient HU exposure, smc6 mutant cells lose viability when the cells are overcoming the block, have a 2C DNA content, and are starting to enter mitosis. A significant proportion of smc6 mutant cells, but not smc6+ cells, exhibit cut phenotypes, suggesting the persistence of unrepaired DNA structures during mitosis that impede chromosome segregation. These data suggest that, rather than being required for stabilization of stalled replication forks, the Smc5/6 complex is required either for the resumption of replication at or for the repair of a subset of forks that are unstable and have collapsed.
We examined the requirement for Smc6 at collapsed forks by arresting the checkpoint-defective mutant cds1-d in HU. One important function of the Cds1CHK2-dependent inter-S-phase checkpoint in yeast is to stabilize stalled replication forks and prevent them from collapsing (29). Fork collapse is thought to involve the movement of replicative helicases ahead of the site of DNA incorporation (21) and the dissociation of DNA polymerases from nascent strands (10, 32). A stable fork is capable of rapid resumption when dNTP synthesis resumes. A collapsed fork, however, requires processing to restore the ability to replicate. It is not known precisely how this occurs, but a role for HR proteins is proposed in most models. We observed that when forks collapse in HU-treated cds1CHK2 cells, replication intermediates remain stable in smc6 mutants, while they decay in smc6+ cells. This phenomenon has recently been noted for rhp51rad51 mutants. However, this common phenotype was not a consequence of an inability to associate recombination proteins with chromatin when replication forks collapse, because we observed that focus formation was normal. Together, these data indicate that Smc6 functions to regulate aspects of HR-dependent DNA processing in response to fork collapse.
Regulation of recombination at HU-arrested forks.
A role for the Smc5/6 complex in HRR in response to a variety of DNA damaging agents has been well documented. We show here that the HU sensitivities of smc6 mutants can be suppressed by deletion of recombination gene rhp51 or rhp55 but not by deletion of rad22 or swi5. This is similar to the suppression of smc6-X at low UV doses by concomitant rhp51RAD51 deletion (27). Such suppression can be interpreted in two ways: Smc6 could have an antirecombinogenic role, with stalled forks in the smc6 mutants becoming targets for illegitimate and lethal recombination events, or alternatively, the smc6 mutants may be defective in a late stage of HR and accumulate lethal recombination intermediates.
To distinguish between these possibilities, we assayed the formation of recombination foci. If the Smc5/6 complex acts to suppress recombination at stalled replication forks, then smc6 mutants arrested in HU are predicted to have increased levels of Rad22 foci and recombination intermediates. While for smc6-X cells, cells with recombination foci (4%, compared to 2% in smc6+ cells [Fig. 4]) and X-shaped DNA structures increased slightly (Fig. 2), the effect is not dramatic and no increase was observed in smc6-74 cells, suggesting that this is not the major defect in these cells.
It is not clear why HRR would be required at replication forks stalled by dNTP depletion, as there is no apparent lesion to be repaired, but HRR mutants are sensitive to HU, and this has led to the idea that HRR is required for a small proportion of stalled forks that become unstable and collapse. Collapsed forks are known to recruit recombination proteins (30, 34). While few recombination foci are seen in cells arrested in HU, cells with recombination foci transiently become visible upon release, suggesting a separation of replication and recombination (30, 34) and consistent with the time when smc6 mutant cells lose viability. This leads us to propose that the Smc5/6 complex is required to regulate a late stage of recombination for the subset of stalled forks that become unstable and collapse.
Smc6 mutants accumulate aberrant DNA structures not recognized by the damage checkpoint.
We examined a role for Smc6 during release of checkpoint-proficient cells from HU. Following release from HU arrest, smc6 mutants complete bulk DNA synthesis and enter mitosis with kinetics similar to that of smc6+ cells. However, mitosis is highly aberrant in a significant proportion of mutant cells. The aberrant mitoses were suppressed by deletion of rhp51rad51 (35) or rhp55rad55 (Fig. 5), indicating that HR-dependent DNA structures that are not recognized by the G2 DNA damage checkpoint accumulate, and this is again consistent with a requirement for Smc6 during a late stage in recombination at the subset of stalled forks, which become unstable and collapse. A similar phenomenon was recently reported for rad60 mutants (35). Rad60 is an Smc5/6 complex-interacting protein that is excluded from the nucleus in HU-arrested cells. Our data predict that Rad60 reenters the nucleus upon HU release to participate with the Smc5/6 complex in a late repair event. In the absence of this function, DNA structures that are not visible to the checkpoint accumulate and result in mitotic catastrophe. Thus, Smc5/6 and Rad60 are required for the proper resolution of HR intermediates.
A similar role has been proposed for RecQ helicases (7, 8), and in vitro, the human BLM-Top3 complex is able to converge the two HJs and decatenate the resulting hemicatene (55). Miyabe et al. (35) showed that the S. pombe RecQ helicase mutant rqh1-d accumulated HR-dependent DNA structures in response to HU arrest (35). Similarly, in S. cerevisiae sgs1-d cells, Rad51-dependent DNA structures have been shown to accumulate at damaged replication forks and checkpoint activation is reduced. Intriguingly, proper activation of Rad53 is restored by concomitant deletion of RAD51 (28). Thus, the accumulation of late recombination structures in RecQ helicase mutants in response to replication problems appears to be a common theme.
The accumulation of “checkpoint-silent” late recombination structures in rqh1-defective cells is consistent with the observation that expression of the bacterial resolvase RusA partially complemented the UV and HU sensitivities and the associated aberrant mitoses of rqh1-d (12). Recently, a similar suppression of the UV sensitivity of nse6-d (null for a nonessential Smc5/6 complex component) was reported (46). However, while deletion of rhp51 efficiently suppressed the UV sensitivity of nse6-d, rhp55-d did not, and overexpression of Brc1 (see below) sensitized nse6-d to UV. Since wild-type cells are similarly sensitized to HU (Fig. 1) by Brc1 overexpression, this suggests that there are both separate and overlapping requirements for the essential and nonessential Smc5/6 components in repair processes.
The DNA damage sensitivity of smc6-74, but not smc6-X, is efficiently suppressed by overexpression of the multiple-BRCT-domain protein Brc1 (54). We show here that overexpression of Brc1 also suppresses the sensitivity of smc6-74 to inhibition of replication by HU, and this correlates with a decrease in cut cells upon entry into mitosis (Fig. 1B and 5). Overexpression of Brc1 had no effect on the HU sensitivity of smc6-X. Thus, either the more severe defect in smc6-X cannot be efficiently suppressed or smc6-74 and smc6-X cause the accumulation of distinct structures late in the recombination process. Both of these hypomorphic smc6 alleles are synthetically lethal with brc1 loss, which itself also results in an S-phase-specific DNA repair defect (51, 54). Taken together, these data suggest that the Smc5/6 and Brc1 pathways overlap in processing at least some aberrant DNA structures that can arise as a result of replication stress. The balance between these functions is important, as overexpression of brc1 sensitized smc6+ cells to HU.
Genome-wide function for Smc5/6 following replication fork collapse.
The accumulation of Smc6 at foci within the nucleolus (Fig. 7) and, in S. cerevisiae, efficient rDNA segregation (53) may reflect a specific function for Smc5/6 confined to the rDNA. However, genome-wide DNA fragmentation in conditional alleles of Smc5/6 complex members and the lack of complementation by episomal rDNA suggest that while the rDNA provides a specific readout of Smc5/6 defects (probably because of the intimate link between replication and recombination at this repetitive locus), Smc5/6 functions are globally important as opposed to rDNA specific (39, 53). Such a notion is also consistent with the observation that, while the rDNA is restricted to one chromosome in S. pombe, the HR-mediated repair of IR-induced strand breaks is equally defective for all chromosomes in the smc6 mutants and that defects in the segregation of all chromosomes are observed in the aberrant mitoses following irradiation or treatment with HU.
We found that, without treatment, Smc6 was equally enriched at the rDNA and unique loci. The association of Smc6 with all loci was modest, but similar profiles were seen with two different antibodies for ChIP (Fig. 8), including anti-Myc against an epitope-tagged Smc6. This is in contrast to observations made by Torres-Rosell et al. (53), who found Smc6 to be enriched at repetitive sequences, including rDNA and telomeres in S. cerevisiae (53), though the use of different methodologies to quantify the ChIP signal may bias their study toward repetitive sequences. Indeed, a recent genome-wide analysis using ChIP-on-Chip concluded that Smc5/6 is associated at other specific loci, including centromeres (4). Our data do not exclude such a distribution for Smc6 in S. pombe, as we have examined only a limited number of loci and we may not have covered more extensively enriched regions. Alternatively, we cannot rule out the possibility that there are real differences in Smc5/6 localization between the two yeasts. A genome-wide analysis of S. pombe and other species would be required to resolve this.
After HU treatment, the levels of Smc6 were modestly but reproducibly increased at all loci. This was most noticeable at the 17S region of the rDNA. Thus, in contrast to Rad60, which is excluded from the nucleus in response to HU, Smc6 is chromatin bound in response to inhibition of replication, and this may reflect a separate requirement for Smc5/6 to act at stalled replication forks.
Model.
We and others have provided evidence that Smc5/6 functions genome wide when forks collapse. HR proteins associate with collapsed forks independently of the Smc6 functions defective in the hypomorphic mutants (Fig. 4), but these Smc6 functions are necessary for the processing of collapsed replication forks (Fig. 2), for normal mitosis, and for optimal cell survival (Fig. 1 and 5). Smc6 is assigned to the HRR pathway by epistasis (27) and presumably participates in DNA repair and/or the HR-dependent restoration of DNA replication. However, the two smc6 hypomorphic mutants analyzed are more sensitive to HU exposure than recombination mutants and are also partially rescued by loss of recombination genes (Fig. 1). This suggests that the function of Smc5/6 in the repair/resumption of collapsed replication forks has several aspects, only some of which are HR dependent. This is consistent with the fact that complete loss of the Smc5/6 complex function is lethal because of global genome fragmentation associated with DNA replication (39, 53). In contrast, HR-deficient cells remain viable, demonstrating that Smc5/6 coordinates DNA-processing functions during replication in addition to HR.
We propose that Smc5/6 with Rad60 plays a late role in resolving as-yet-unidentified DNA structures associated with DNA repair by HR of both canonical DNA damage and collapsed replication forks (Fig. 9). Our results suggest that these structures can arise at unstable replication forks from both HR-dependent events (partial suppression of smc6 mutant sensitivity by concomitant deletion of HR genes) and HR-independent events (loss of HR is not lethal). In addition to the Rad60-dependent late role in the resolution of recombination intermediates after fork collapse, we also propose that Smc5/6 functions independently of Rad60, possibly in resetting unstable forks.
FIG. 9.

Model for the Smc5/6 complex function. Cds1Chk2 activity stabilizes stalled replication forks, and replication complexes remain associated. Forks collapse (replication complexes are disassociated), which can lead to the generation of a polar double-strand break. Restoration of the fork then occurs through break-induced replication (BIR) initiated by HR-dependent strand invasion of the intact template and subsequent HJ resolution (15). Rad22Rad52 and other recombination proteins (not shown) associate with the damaged chromatin at collapsed forks independently of Smc5/6, but the complex is required with Rad60 at a later stage for HR repair and/or restoration of replication (bottom right). Both smc6-74 and smc6-X are defective in this process (indicated by bars). Fork resetting without Rhp51Rad51-dependent HR occurs by multiple mechanisms, some of which require Smc5/6 (top right, shown in gray) or Brc1. In smc6-74 mutants, Brc1-dependent resetting of unstable forks can still occur inefficiently, whereas in smc6-X mutants, this process is nonfunctional (indicated by bar and ?X). Overexpression of brc1 could thus bypass the requirement for smc6-74 but not for smc6-X.
It remains unclear whether the two hypomorphic alleles analyzed here define distinct roles for Smc6 or whether the ability of brc1 overexpression to suppress the sensitivity of smc6-74 but not of smc6-X is a result of differential penetrances in the same process. For example, it is possible that in smc6-74 mutants, Brc1-dependent resetting of unstable forks can still occur inefficiently, whereas in smc6-X mutants, this process is nonfunctional. Overexpression of brc1 could thus bypass the requirement for smc6-74 but not for smc6-X. Overexpression of Brc1 cannot suppress null mutations in Smc5/6 complex genes (our unpublished data).
Conclusions.
The genetic analysis of Smc5/6 complex components demonstrates that the complex has multiple functions in DNA metabolism. We provide evidence that one “late” function of Smc5/6 complex, in association with Rad60 (35), is to resolve structures established by the processing of collapsed replication forks by HR proteins. The Brc1/nuclease-dependent pathway(s) can alternatively resolve a subset of these structures. These data predict that the highly conserved human Smc5/6 complex participates in pathways that prevent diseases related to genome instability by coordinating DNA repair responses with DNA replication and cell cycle progression.
Acknowledgments
We thank J. Cooper for taz1-GFP and rad22-GFP, M. Yamamoto for gar2-GFP, and R. Allshire for Swi6-GFP strains; E. Taylor for Smc6 antibodies and the smc6-T2 strain; A. Watson for help with ChIP; K. Furuya for help with 2-D gels; and colleagues at the GDSC for advice and discussions.
This work was supported by Cancer Research United Kingdom, the BBSRC (J.M.), and the NIH/NCI (CA100076) (M.O.).
Footnotes
Published ahead of print on 9 October 2006.
REFERENCES
- 1.Akamatsu, Y., D. Dziadkowiec, M. Ikeguchi, H. Shinagawa, and H. Iwasaki. 2003. Two different Swi5-containing protein complexes are involved in mating-type switching and recombination repair in fission yeast. Proc. Natl. Acad. Sci. USA 100:15770-15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews, E. A., J. Palecek, J. Sergeant, E. Taylor, A. R. Lehmann, and F. Z. Watts. 2005. Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol. Cell. Biol. 25:185-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahler, J., J. Q. Wu, M. S. Longtine, N. G. Shah, A. McKenzie III, A. B. Steever, A. Wach, P. Philippsen, and J. R. Pringle. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14:943-951. [DOI] [PubMed] [Google Scholar]
- 4.Betts Lindroos, H., L. Strom, T. Itoh, Y. Katou, K. Shirahige, and C. Sjogren. 2006. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 22:755-767. [DOI] [PubMed] [Google Scholar]
- 5.Boddy, M. N., P. Shanahan, W. H. McDonald, A. Lopez-Girona, E. Noguchi, I. J. Yates, and P. Russell. 2003. Replication checkpoint kinase Cds1 regulates recombinational repair protein Rad60. Mol. Cell. Biol. 23:5939-5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brewer, B. J., and W. L. Fangman. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51:463-471. [DOI] [PubMed] [Google Scholar]
- 7.Carr, A. M. 2002. DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair 1:983-994. [DOI] [PubMed] [Google Scholar]
- 8.Caspari, T., J. M. Murray, and A. M. Carr. 2002. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 16:1195-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakrabarti, S. K., J. C. James, and R. G. Mirmira. 2002. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J. Biol. Chem. 277:13286-13293. [DOI] [PubMed] [Google Scholar]
- 10.Cobb, J. A., L. Bjergbaek, K. Shimada, C. Frei, and S. M. Gasser. 2003. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22:4325-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cost, G. J., and N. R. Cozzarelli. 2006. Smc5p promotes faithful chromosome transmission and DNA repair in Saccharomyces cerevisiae. Genetics 172:2185-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doe, C. L., J. Dixon, F. Osman, and M. C. Whitby. 2000. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 19:2751-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards, R. J., and A. M. Carr. 1997. Analysis of radiation-sensitive mutants of fission yeast. Methods Enzymol. 283:471-494. [DOI] [PubMed] [Google Scholar]
- 14.Fousteri, M. I., and A. R. Lehmann. 2000. A novel SMC protein complex in Schizosaccharomyces pombe contains the Rad18 DNA repair protein. EMBO J. 19:1691-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haber, J. E., and W. D. Heyer. 2001. The fuss about Mus81. Cell 107:551-554. [DOI] [PubMed] [Google Scholar]
- 16.Harvey, S. H., D. M. Sheedy, A. R. Cuddihy, and M. J. O'Connell. 2004. Coordination of DNA damage responses via the Smc5/Smc6 complex. Mol. Cell. Biol. 24:662-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano, T. 2002. The ABCs of SMC proteins: two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev. 16:399-414. [DOI] [PubMed] [Google Scholar]
- 18.Hope, J. C., M. Maftahi, and G. A. Freyer. 2005. A postsynaptic role for Rhp55/57 that is responsible for cell death in Δrqh1 mutants following replication arrest in Schizosaccharomyces pombe. Genetics 170:519-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu, B., C. Liao, S. H. Millson, M. Mollapour, C. Prodromou, L. H. Pearl, P. W. Piper, and B. Panaretou. 2005. Qri2/Nse4, a component of the essential Smc5/6 DNA repair complex. Mol. Microbiol. 55:1735-1750. [DOI] [PubMed] [Google Scholar]
- 20.Jowsey, P. A., A. J. Doherty, and J. Rouse. 2004. Human PTIP facilitates ATM-mediated activation of p53 and promotes cellular resistance to ionizing radiation. J. Biol. Chem. 279:55562-55569. [DOI] [PubMed] [Google Scholar]
- 21.Katou, Y., Y. Kanoh, M. Bando, H. Noguchi, H. Tanaka, T. Ashikari, K. Sugimoto, and K. Shirahige. 2003. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078-1083. [DOI] [PubMed] [Google Scholar]
- 22.Kearsey, S. E., S. Montgomery, K. Labib, and K. Lindner. 2000. Chromatin binding of the fission yeast replication factor mcm4 occurs during anaphase and requires ORC and cdc18. EMBO J. 19:1681-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, S. M., and J. A. Huberman. 2001. Regulation of replication timing in fission yeast. EMBO J. 20:6115-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krings, G., and D. Bastia. 2004. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 101:14085-14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lammens, A., A. Schele, and K. P. Hopfner. 2004. Structural biochemistry of ATP-driven dimerization and DNA-stimulated activation of SMC ATPases. Curr. Biol. 14:1778-1782. [DOI] [PubMed] [Google Scholar]
- 26.Laursen, L. V., E. Ampatzidou, A. H. Andersen, and J. M. Murray. 2003. Role for the fission yeast RecQ helicase in DNA repair in G2. Mol. Cell. Biol. 23:3692-3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehmann, A. R., M. Walicka, D. J. Griffiths, J. M. Murray, F. Z. Watts, S. McCready, and A. M. Carr. 1995. The rad18 gene of Schizosaccharomyces pombe defines a new subgroup of the SMC superfamily involved in DNA repair. Mol. Cell. Biol. 15:7067-7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberi, G., G. Maffioletti, C. Lucca, I. Chiolo, A. Baryshnikova, C. Cotta-Ramusino, M. Lopes, A. Pellicioli, J. E. Haber, and M. Foiani. 2005. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 19:339-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindsay, H. D., D. J. Griffiths, R. J. Edwards, P. U. Christensen, J. M. Murray, F. Osman, N. Walworth, and A. M. Carr. 1998. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 12:382-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lisby, M., J. H. Barlow, R. C. Burgess, and R. Rothstein. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118:699-713. [DOI] [PubMed] [Google Scholar]
- 31.Lopes, M., C. Cotta-Ramusino, A. Pellicioli, G. Liberi, P. Plevani, M. Muzi-Falconi, C. S. Newlon, and M. Foiani. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412:557-561. [DOI] [PubMed] [Google Scholar]
- 32.Lucca, C., F. Vanoli, C. Cotta-Ramusino, A. Pellicioli, G. Liberi, J. Haber, and M. Foiani. 2004. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 23:1206-1213. [DOI] [PubMed] [Google Scholar]
- 33.McDonald, W. H., Y. Pavlova, J. R. Yates III, and M. N. Boddy. 2003. Novel essential DNA repair proteins Nse1 and Nse2 are subunits of the fission yeast Smc5-Smc6 complex. J. Biol. Chem. 278:45460-45467. [DOI] [PubMed] [Google Scholar]
- 34.Meister, P., A. Taddei, L. Vernis, M. Poidevin, S. M. Gasser, and G. Baldacci. 2005. Temporal separation of replication and recombination requires the intra-S checkpoint. J. Cell Biol. 168:537-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyabe, I., T. Morishita, T. Hishida, S. Yonei, and H. Shinagawa. 2006. Rhp51-dependent recombination intermediates that do not generate checkpoint signal are accumulated in Schizosaccharomyces pombe rad60 and smc5/6 mutants after release from replication arrest. Mol. Cell. Biol. 26:343-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreno, S., A. Klar, and P. Nurse. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194:795-823. [DOI] [PubMed] [Google Scholar]
- 37.Morikawa, H., T. Morishita, S. Kawane, H. Iwasaki, A. M. Carr, and H. Shinagawa. 2004. Rad62 protein functionally and physically associates with the smc5/smc6 protein complex and is required for chromosome integrity and recombination repair in fission yeast. Mol. Cell. Biol. 24:9401-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morishita, T., F. Furukawa, C. Sakaguchi, T. Toda, A. M. Carr, H. Iwasaki, and H. Shinagawa. 2005. Role of the Schizosaccharomyces pombe F-box DNA helicase in processing recombination intermediates. Mol. Cell. Biol. 25:8074-8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morishita, T., Y. Tsutsui, H. Iwasaki, and H. Shinagawa. 2002. The Schizosaccharomyces pombe rad60 gene is essential for repairing double-strand DNA breaks spontaneously occurring during replication and induced by DNA-damaging agents. Mol. Cell. Biol. 22:3537-3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muris, D. F., K. Vreeken, A. M. Carr, B. C. Broughton, A. R. Lehmann, P. H. Lohman, and A. Pastink. 1993. Cloning the RAD51 homologue of Schizosaccharomyces pombe. Nucleic Acids Res. 21:4586-4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muris, D. F., K. Vreeken, A. M. Carr, J. M. Murray, C. Smit, P. H. Lohman, and A. Pastink. 1996. Isolation of the Schizosaccharomyces pombe RAD54 homologue, rhp54+, a gene involved in the repair of radiation damage and replication fidelity. J. Cell Sci. 109:73-81. [DOI] [PubMed] [Google Scholar]
- 42.Nasmyth, K., and P. Nurse. 1981. Cell division cycle mutants altered in DNA replication and mitosis in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 182:119-124. [DOI] [PubMed] [Google Scholar]
- 43.Noguchi, E., C. Noguchi, L. L. Du, and P. Russell. 2003. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 23:7861-7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osman, F., J. Dixon, A. R. Barr, and M. C. Whitby. 2005. The F-box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell. Biol. 25:8084-8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pebernard, S., W. H. McDonald, Y. Pavlova, J. R. Yates III, and M. N. Boddy. 2004. Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol. Biol. Cell 15:4866-4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pebernard, S., J. Wohlschlegel, W. H. McDonald, J. R. Yates III, and M. N. Boddy. 2006. The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol. Cell. Biol. 26:1617-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potts, P. R., and H. Yu. 2005. Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol. Cell. Biol. 25:7021-7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts, T. M., M. S. Kobor, S. A. Bastin-Shanower, M. Ii, S. A. Horte, J. W. Gin, A. Emili, J. Rine, S. J. Brill, and G. W. Brown. 2006. Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol. Biol. Cell 17:539-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rouse, J. 2004. Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J. 23:1188-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sergeant, J., E. Taylor, J. Palecek, M. Fousteri, E. A. Andrews, S. Sweeney, H. Shinagawa, F. Z. Watts, and A. R. Lehmann. 2005. Composition and architecture of the Schizosaccharomyces pombe Rad18 (Smc5-6) complex. Mol. Cell. Biol. 25:172-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheedy, D. M., D. Dimitrova, J. K. Rankin, K. L. Bass, K. M. Lee, C. Tapia-Alveal, S. H. Harvey, J. M. Murray, and M. J. O'Connell. 2005. Brc1-mediated DNA repair and damage tolerance. Genetics 171:457-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strahl-Bolsinger, S., A. Hecht, K. Luo, and M. Grunstein. 1997. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 11:83-93. [DOI] [PubMed] [Google Scholar]
- 53.Torres-Rosell, J., F. Machin, S. Farmer, A. Jarmuz, T. Eydmann, J. Z. Dalgaard, and L. Aragon. 2005. SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat. Cell Biol. 7:412-419. [DOI] [PubMed] [Google Scholar]
- 54.Verkade, H. M., S. J. Bugg, H. D. Lindsay, A. M. Carr, and M. J. O'Connell. 1999. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell 10:2905-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu, L., and I. D. Hickson. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426:870-874. [DOI] [PubMed] [Google Scholar]
- 56.Zhao, X., and G. Blobel. 2005. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 102:4777-4782. [DOI] [PMC free article] [PubMed] [Google Scholar]