

Abstract
Rpb9, a small nonessential subunit of RNA polymerase II, has been shown to have multiple transcription-related functions in Saccharomyces cerevisiae. These functions include promoting transcription elongation and mediating a subpathway of transcription-coupled repair (TCR) that is independent of Rad26, the homologue of human Cockayne syndrome complementation group B protein. Rpb9 is composed of three distinct domains: the N-terminal Zn1, the C-terminal Zn2, and the central linker. Here we show that the Zn1 and linker domains are essential, whereas the Zn2 domain is almost dispensable, for both transcription elongation and TCR functions. Impairment of transcription elongation, which does not dramatically compromise Rad26-mediated TCR, completely abolishes Rpb9-mediated TCR. Furthermore, Rpb9 appears to be dispensable for TCR if its transcription elongation function is compensated for by removing a transcription repression/elongation factor. Our data suggest that the transcription elongation function of Rpb9 is involved in TCR.
Nucleotide excision repair (NER) in eukaryotic cells is a conserved DNA repair process that removes a large variety of DNA lesions, such as UV-induced cyclobutane pyrimidine dimers (CPDs) and bulky chemical adducts (for a recent review, see reference 43). In almost all of the organisms analyzed, NER in the transcribed strand (TS) of a gene is faster than that in the nontranscribed strand (NTS) or in the nontranscribed regions of the genome (4, 33-35, 51). In human cells, so-called “transcription-coupled repair” (TCR) is dependent on Cockayne syndrome complementation group A (CSA) and B (CSB) proteins (30, 58, 63, 65). In Saccharomyces cerevisiae, it has been shown that Rad26 (62), the homologue of human CSB, and Rpb9 (26, 27), a subunit of RNA polymerase II (Pol II), mediate two subpathways of TCR. NER in the nontranscribed regions of the genome or in the NTS of an active gene is accomplished by so-called “global genomic repair” (GGR). In yeast, GGR is dependent on Rad7 and Rad16 (66).
Nucleotide-level analyses have shown that rapid NER occurs not only in the coding region of a gene, but also in regions that are upstream of the transcription start site (10, 26, 27, 41, 55, 59-61). Dissection of different NER pathways in the yeast GAL1 to -10 genes indicates that Rpb9-mediated TCR mainly operates in the coding region, whereas Rad26-mediated TCR operates equally well in the coding region and in a region upstream of the transcription start site (26, 27). It was proposed that transcription elongation may be required for Rpb9-mediated TCR, but may not be essential for Rad26-mediated TCR (26).
A functional connection between TCR and Pol II degradation has been suggested by studies with mammalian and yeast cells. Studies in human cells showed that a fraction of the largest subunit (Rpb1) of Pol II is ubiquitinated in normal but not in CSA and CSB cells following exposure to UV radiation (5). It was further shown that the ubiquitinated Pol II is degraded by the 26S proteasome (42), and the degradation occurs in normal but not in CSA and CSB cells (32). However, another study showed that despite a deficiency in ubiquitination of Pol II, the UV-induced degradation of Pol II occurs normally in CS cells (31). In yeast, the UV-induced Pol II degradation appears to be mediated by Def1, which forms a complex with the CSB homolog Rad26 (72). Interestingly, the degradation occurs much faster in cells lacking Rad26 (72). It was shown that the E3 ubiquitin ligase Rps5 catalyzes ubiquitination of Rpb1 in response to DNA damage in yeast (3). Furthermore, it was shown that yeast extracts from rsp5-1 cells incubated at nonpermissive temperature cannot ubiquitinate Pol II in vitro, even when large amounts of ubiquitin are added (44). A recent study showed that Elc1, the homologue of mammalian elongin C and an E3 ubiquitin ligase, also plays an important role in catalyzing the ubiquitination (46).
Rpb9 seems to have multiple functions that are related to transcription in yeast cells. In addition to mediating TCR, Rpb9 plays roles in transcription elongation (2, 17, 64), in selection of the correct transcription start site (13, 18, 52), and in maintaining transcriptional fidelity (37). Recently, we found that Rpb9 also plays an important role in the degradation of Pol II in response to UV radiation (R. Chen et al., unpublished results).
At present, little is known about the biochemical mechanism of Rpb9-mediated TCR. It is even unclear whether Rpb9 mediates TCR by directly recruiting NER factors or by promoting other functions of Pol II. In order to gain insight into the mechanism of this TCR subpathway, we mapped the domains of Rpb9 that are required for TCR. We found that the functions for TCR and transcription elongation are colocalized in the same Rpb9 domains, whereas the function for UV-induced degradation of Pol II is located in a different domain. Impairment of ongoing transcription, which does not significantly compromise Rad26-mediated TCR, abolishes Rpb9-mediated TCR. Furthermore, Rpb9 appears to be dispensable for TCR if its transcription elongation function is compensated for by removing a factor involved in regulation of transcription elongation. Our results suggest that the transcription elongation function of Rpb9 is involved in TCR.
MATERIALS AND METHODS
Yeast strains.
rad16 rad26 and rad16 rad26 rpb9 deletion mutants are derivatives of the wild-type strain Y452 (MATα ura3-52 his3-1 leu2-3 leu2-112 cir°) (27). The temperature-sensitive rpb1-1 mutant yRP693 (MATα rpb1-1 ura3-52 leu2-3 leu2-112) (1) was kindly provided by Carolyn J. Decker (Department of Molecular and Cellular Biology and Howard Hughes Medical Institute, University of Arizona). Deletion of RAD16 and RAD26 in the rpb1-1 background was achieved using the same procedure as described previously (27). rad7 rad26 rpb9, spt4, and rad7 rad26 rpb9 spt4 deletion mutants are derivatives of wild-type strain BJ5465 (MATa ura3-52 trp-1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1) (23). Nucleotides (with respect to the starting codon ATG) +345 to +1755, +58 to +2297, +11 to +366, +214 to +1454, and +14 to +288 were deleted for the RAD16, RAD26, RPB9, RAD7, and SPT4 genes, respectively.
Plasmids.
A series of RPB9 fragments encompassing different truncated or mutated coding sequences, the promoter (a sequence of ∼500 bp immediately upstream of the coding sequence), and 3′ mRNA processing sequence (a sequence of ∼300 bp immediately downstream of the coding sequence) were created by PCR and enzymatic ligation. These RPB9 fragments were inserted into the multiple cloning site of the centromeric vector pRS416 (50). The plasmid-borne RPB9 genes expressing different truncated or mutated Rpb9 proteins were verified by sequencing and transformed into rad16 rad26 rpb9 cells to analyze their functions.
UV and mycophenolic acid sensitivity assay.
Yeast cells were grown at 28°C in minimal media containing 2% glucose to saturation, and sequential 10-fold serial dilutions were made. For the UV sensitivity assay, the diluted samples were spotted onto YPD (2% peptone, 1% yeast extract, 2% glucose) plates. When the spots had dried, the plates were irradiated with different doses of 254-nm UV light. For the mycophenolic acicd (MPA) sensitivity assay, the diluted samples were spotted onto SD plates containing different concentrations of mycophenolic acid (Sigma). The plates were incubated at 28°C in the dark prior to being photographed.
NER analysis of UV-induced CPDs.
For experiments involving the temperature-sensitive rpb1-1 mutants, cells were cultured in minimal medium containing 2% galactose at 25°C to late log phase (A600 ≈1.0). Half of each culture was kept at the permissive temperature (25°C), and the other half was shifted to the nonpermissive temperature (36°C) and then incubated for 1 h. The cells were harvested, irradiated with 50 J/m2 UV light, and incubated in the same media at 25°C or 36°C for various times in the dark to allow DNA repair. For all other experiments that did not involve the temperature-sensitive rpb1-1 mutants, cells were grown at 28°C in minimal medium containing 2% galactose to log phase, harvested, and irradiated with 50 J/m2 UV light. The cells were incubated in repair media at 28°C for various times in the dark before being pelleted. Total genomic DNA was isolated from the pelleted cells, as described previously (27).
The gene fragments of interest were 3′-end labeled with [α-32P]dATP using a procedure described previously (28, 29). Briefly, ∼1 μg of total genomic DNA was digested with restriction enzyme(s) to release the fragments of interest and incised at CPD sites with an excess amount of purified T4 endonuclease V (Epicentre). Excess copies of biotinylated oligonucleotides, which are complementary to the 3′ end of the fragments to be labeled, were mixed with the sample. The mixture was heated at 95°C for 5 min to denature the DNA and then cooled to an annealing temperature of around 50°C. The annealed fragments were attached to streptavidin-conjugated magnetic beads (Invitrogen), and the other fragments were removed by washing the beads at the annealing temperature. The attached fragments were labeled with [α-32P]dATP (Perkin-Elmer) and resolved on sequencing gels. The gels were dried and exposed to a PhosphorImager screen (GE Healthcare or Bio-Rad).
The signal intensities at gel bands corresponding to CPD sites were quantified using Quantity One (Bio-Rad) software. The total signal intensity in a gel lane, including the band at the top of the gel (which corresponds to the undamaged or repaired full-length fragment), was obtained after the gel background signal was subtracted. The total signal intensity in a lane representing a repair time was divided by the total signal intensity in the lane of the “0-h” repair to obtain a factor of loading in the lane. The signal intensity of each band in a lane was divided by the factor of loading in that lane, to normalize loading variations. The normalized signal intensity of a band in a lane of a repair time was divided by the signal intensity of the corresponding band in the lane of the 0-h repair, to obtain the fraction of CPDs remaining at the band. The data representing the percentages of CPDs remaining presented in the plots of Fig. 3 to 7 represent at least three experiments.
FIG. 3.

TCR mediated by different Rpb9 fragments. Gels show TCR in the GAL1 gene of log-phase rad16 rad26 (A), rad16 rad26 rpb9 (B), and rad16 rad26 rpb9 cells transformed with plasmids encoding full-length (residues 1 to 122) (C) and truncated (numbers indicate residues remaining) (D to K) Rpb9. Lanes U represent unirradiated samples. Lanes 0, 1, 2, and 4 indicate different times (hours) of repair incubation following UV irradiation. An arrow to the left of the gels marks the transcription start site. Solid circles on the left of the gels mark transcribed region (+1 to +380). Open triangles mark upstream region (−1 to −180) where residual Rpb9-mediated TCR takes place. Plots underneath each of the gels show the average (mean ± standard deviation) of the percent CPDs remaining at individual sites in the transcribed (+1 to +380) (solid circles) and upstream (−1 to −180) (open triangles) regions at different times of repair incubation.
FIG. 7.
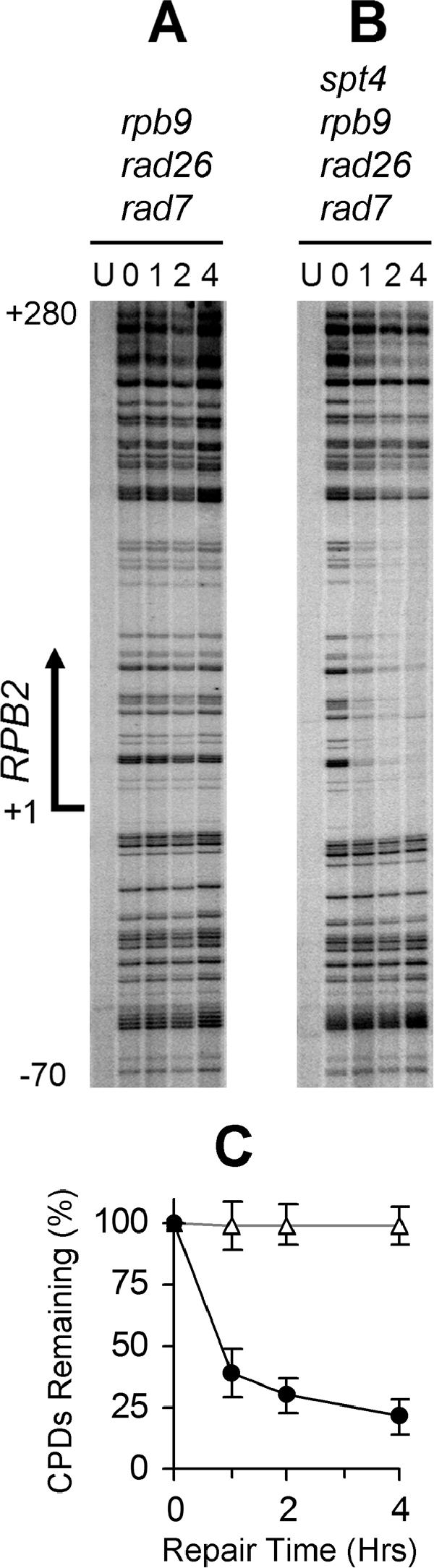
Deletion of SPT4 restores TCR in rad7 rad26 rpb9 cells. Gels show TCR in the TS of the RPB2 gene. Lanes labeled U are unirradiated samples. Other lanes are samples with different times (hours) of repair incubation. An arrow on the left of the gels marks transcription start site. Panel C shows the average (mean ± standard deviation) of the percent CPDs remaining at individual sites in the transcribed region (+1 to +280) of the RPB2 gene at different repair times in rad7 rad26 rpb9 (open triangles) and rad7 rad26 rpb9 spt4 (solid circles) cells.
RESULTS
The transcription elongation and TCR functions of Rpb9 are colocalized in the Zn1 and linker domains.
Pol II comprises 12 subunits (Rpb1 to Rpb12) in yeast (6). Rpb9 is located at the tip of the so-called “jaws” of the enzyme (Fig. 1), which is formed by portions of the Rpb1 and Rpb2 subunits (7). Rpb9 shows three distinct domains, the N-terminal Zn1 (residues 1 to 39), the C-terminal Zn2 (residues 53 to 122), and the central linker (residues 40 to 52) (Fig. 1).
FIG. 1.

Domains of Rpb9. (A) Localization of Rpb9 on the “jaw” of Pol II complex. Portions of Rpb1 and Rpb2 subunits are shown in dark gray and light gray, respectively. The Rpb9 Zn1 (residues 1 to 39), Zn2 (residues 53 to 122) and the linker (residues 40 to 52) domains are shown in red, magenta, and blue, respectively. The zinc ions bound by the Zn1 and Zn2 domains are shown as a cyan sphere. The figure was prepared from PDB file 1I50 (7) using Protein Explorer (http://www.umass.edu/microbio/chime/pe/protexpl/frntdoor.htm). (B) Sequence of Rpb9. Zn1, Zn2, and the linker are shown in red, magenta, and blue, respectively. The conserved cysteines in the Zn1 and Zn2 domains are marked with dots.
To investigate the function or functions of Rpb9 that are required for mediating TCR, we created a series of plasmids expressing Rpb9 with different truncations. These plasmids were transformed into rad16 rad26 rpb9 cells, where GGR and TCR mediated by genomic genes are eliminated and the functions of the different forms of the plasmid-encoded Rpb9 can be specifically analyzed.
Sensitivity to nucleotide depletion drugs, such as MPA and 6-azauracil, has been widely used as a landmark of transcription elongation deficiency in yeast cells (19). These drugs are inhibitors of IMP dehydrogenase, which catalyzes the rate-limiting step in the de novo synthesis of GTP (9). As shown in Fig. 2A, rad16 rad26 rpb9 cells are much more sensitive to MPA than rad16 rad26 cells, indicating Rpb9 indeed plays a role in transcription elongation (17, 64). A plasmid-expressed full-length (residues 1 to 122) Rpb9 restored the MPA resistance of rad16 rad26 rpb9 cells (Fig. 2B). A truncated Rpb9 that has a deletion of 3 amino acids from the N terminus (residues 4 to 122 remaining) can also restore the MPA resistance. However, deletions of 6 (residues 7 to 122 remaining) or 11 (residues 12 to 122 remaining) amino acids from the N terminus compromised or completely abolished the function for MPA resistance (Fig. 2; Table 1). On the other hand, deletion from the C terminus seems to have little effect. Indeed, the Rpb9 fragment of residues 1 to 49, which includes Zn1 and most of the linker (Fig. 1), retains most of its function for MPA resistance (Fig. 2 and Table 1). These results suggest that the Zn1 domain and most of the linker are essential for transcription elongation, whereas the whole Zn2 domain may play a subsidiary role.
FIG. 2.

MPA resistance conferred by different forms of Rpb9. (A) MPA resistance of rad16 rad26 and rad16 rad26 rpb9 cells. (B) MPA resistance of rad16 rad26 rpb9 cells transformed with plasmids encoding full-length (residues 1 to 122), truncated (numbers indicate residues remaining), and mutated (cysteines replaced with serines) Rpb9. Sequential 10-fold serial dilutions of yeast cells were spotted onto SD plates containing different concentrations of MPA and incubated for 6 days before being photographed.
TABLE 1.
Phenotypes of rad16 rad26 rpb9 cells transformed with plasmids encoding truncated or mutated Rpb9
| Rpb9 changea | Result forb:
|
||||
|---|---|---|---|---|---|
| Growth
|
Resistance
|
TCR activity | |||
| 25-30°C | 38°C | MPA | UV | ||
| 1-122 | ++++ | ++++ | ++++ | ++++ | ++++ |
| None | − | − | − | − | − |
| 4-122 | ++++ | ++++ | ++++ | +++ | +++ |
| 7-122 | +++ | + | ++ | ++ | + |
| 12-122 | − | − | − | − | − |
| 17-122 | − | − | − | − | − |
| 31-122 | − | − | − | − | − |
| 50-122 | − | − | − | − | − |
| 71-122 | − | − | − | − | − |
| 1-40 | − | − | − | − | − |
| 1-45 | + | + | + | − | − |
| 1-49 | +++ | +++ | +++ | +++ | +++ |
| 1-53 | +++ | +++ | +++ | +++ | +++ |
| 1-57 | +++ | +++ | +++ | +++ | +++ |
| 1-61 | +++ | +++ | +++ | +++ | +++ |
| 1-65 | +++ | +++ | +++ | +++ | +++ |
| 1-75 | +++ | +++ | +++ | +++ | +++ |
| C7S | ++++ | ++++ | ++++ | ++++ | ++++ |
| C10S | ++++ | ++++ | ++++ | ++++ | ++++ |
| C7, 10S | ++ | − | ++ | + | − |
| C29S | ++++ | ++++ | ++++ | ++++ | ++++ |
| C32S | ++++ | ++++ | ++++ | ++++ | ++++ |
| C29, 32S | ++ | − | +++ | ++ | + |
| C7, 10, 29, 32S | ++ | − | ++ | + | − |
The numbers indicate Rpb9 residues encoded by the plasmid. The format C7S indicates that a cysteine was replaced with a serine at the indicated amino acid position.
++++, +++, ++, +, and − represent different levels of function ranging from full function (++++) to no function (−).
The Zn1 domain of Rpb9 contains four conserved cysteines for zinc binding (Fig. 1) (18, 73). Mutations of the Zn1 domain, altering cysteine 7 (Fig. 1) to phenylalanine (13) or alanine (18), show a defect in selection of the correct transcription start site. To systematically examine the function(s) of the four conserved cysteines in the Zn1 domain, we replaced them with the structurally similar amino acid serine. A single cysteine replacement (C7S, C10S, C29S, and C32S) caused little detectable defect in MPA resistance (Fig. 2; Table 1). However, replacements of two (C7, 10S and C29, 32S) or all four (C7, 10, 29, and 32S) of the cysteines in the Zn1 domain greatly compromised the MPA resistance (Fig. 2; Table 1).
In agreement with a previous report (73), cells lacking Rpb9 are temperature sensitive (Table 1). Furthermore, rpb9 cells have a slower growth rate than RPB9+ cells at temperatures between 25 and 30°C. These results also suggest that the mutant cells have a deficiency in transcription elongation. Interestingly, the temperature sensitivity, growth rate at 25 to 30°C, and MPA resistance correlate quite well for cells expressing different forms of Rpb9 (Table 1).
We also examined the UV resistance of rad16 rad26 rpb9 cells transformed with the plasmids expressing the different forms of Rpb9. The UV resistance correlates almost perfectly with the temperature sensitivity, growth rate at 25 to 30°C, and MPA resistance (Table 1). This indicates that the TCR and transcription elongation functions of Rpb9 are colocalized in the same domains (i.e., the Zn1 and linker).
Next, we directly measured TCR function conferred by the different forms of plasmid-encoded Rpb9 protein. In the GAL1 gene of log-phase rad16 rad26 cells, efficient repair can be seen in the coding region (Fig. 3A). A residual repair can also be seen in the region that is within 180 nucleotides upstream of the transcription start site (Fig. 3A). However, no repair can be seen in the region that is over 180 nucleotides (nt) upstream of the transcription start site or in the entire NTS in cells lacking Rad16 (not shown), in agreement with our previous studies (26, 27). These results confirmed our previous observations that the Rpb9-mediated TCR initiates ∼180 nt upstream of the transcription start site in the GAL1 gene, but is mainly effective in the coding region of the gene (26, 27). It should be noted that the rate of Rpb9-mediated TCR is biphasic, being fast in the initial hour and very slow in the later hours of repair incubation (27) (Fig. 3A). Presumably, in log-phase cells, a gene is transcribed by a mixture of two forms of Pol II: i.e., an Rpb4-containing form and an Rpb4-free form. If it is encountered by an Rpb4-free Pol II, a lesion is quickly repaired by the Rpb9-mediated TCR, which may contribute to the early fast phase. On the other hand, if it is encountered by an Rpb4-containing Pol II, the lesion cannot be easily repaired by the Rpb9-mediated TCR, which may contribute to the later slow phase (27).
The plasmid-encoded full-length Rpb9 essentially restored TCR in the rad16 rad26 rpb9 cells (Fig. 3, compare panels A, B, and C). Deletions of 3 or 6 amino acids from the N terminus compromised the TCR activity to different extents (Fig. 3D and E; Table 1). However, a deletion of 11 amino acids from the N terminus, which includes the conserved cysteines 7 and 10, completely abolished the TCR function (Fig. 3F; Table 1). Deletions from the C terminus of the protein seemed to have a less detrimental effect on the TCR function. Indeed, a truncated Rpb9 with the whole Zn2 domain deleted (e.g., with residues 1 to 49 or 1 to 53 remaining) retains most of the TCR function (Fig. 3H to K; Table 1).
A single replacement of cysteine with serine in the Zn1 domain caused little defect in TCR (Fig. 4A, B, D, and E; Table 1). However, replacements of two or all four of the cysteines with serines in the domain greatly compromised the TCR activity (Fig. 4C and F; Table 1).
FIG. 4.

TCR mediated by mutated Rpb9. Gels show TCR in the GAL1 gene of log-phase rad16 rad26 rpb9 cells transformed with plasmids encoding Rpb9 with cysteine 7, 10, 29, and 32 replaced by serine. Lanes labeled U represent unirradiated samples. Lanes 0, 1, 2, and 4 indicate different times (hours) of repair incubation following UV irradiation. An arrow to the left of the gels marks the transcription start site. Solid circles on the left of the gels mark transcribed region (+1 to +380). Open triangles mark the upstream region (−1 to −180) where residual Rpb9-mediated TCR takes place. Plots underneath each of the gels show the average (mean ± standard deviation) of the percent CPDs remaining at individual sites in the transcribed (+1 to +380) (solid circles) and upstream (−1 to −180) (open triangles) regions at different times of repair incubation.
Taken together, our results suggest that the transcription elongation and TCR functions of Rpb9 are colocalized in the Zn1 and linker domains of Rpb9. The whole Zn2 domain may play a subsidiary role for these functions. However, the Zn2 domain appears to possess the function for promoting UV-induced degradation of Pol II (X. Chen et al., unpublished results).
Impairment of transcription elongation abolishes Rpb9-mediated TCR.
The above observation that the transcription elongation and TCR functions are possibly colocalized in the same domains of Rpb9 suggests that this Pol II subunit may mediate TCR by promoting transcription elongation. If this is the case, the Rpb9-mediated TCR will be greatly compromised or abolished if ongoing transcription elongation is impaired. To test this notion, we analyzed TCR in the rpb1-1 cells cultured at permissive and nonpermissive temperatures. rpb1-1 is a point mutation (G-to-A transition at nucleotide position 4622) in the RPB1 gene which results in a glycine-to-aspartic acid change in the C-terminal domain of the largest subunit of Pol II (38, 48). Transcription of mRNA genes occurs normally in rpb1-1 cells cultured at 25°C. However, within 5 to 15 min after the mutant cells are shifted to nonpermissive temperature (36°C), transcription is sharply reduced (38). One hour after the temperature shift, no transcription can be detected (38). Many studies have employed the rpb1-1 allele to impair ongoing transcription in yeast (14, 25, 36, 70).
RPB1+ and rpb1-1 cells were cultured in galactose media at 25°C to late log phase. Half of each culture was pulse-labeled with [32P]phosphoric acid at the permissive temperature (38). The other half was shifted to 36°C and pulse-labeled 1 h after the temperature shift. Total RNA was isolated from the labeled cells and hybridized to a membrane dot-blotted with plasmids bearing sequences of several genes of interest (e.g., GAL1 and RPB2). In agreement with a previous report (38), no transcription can be detected in the mutant cells 1 h after the temperature shift (not shown), indicating that transcription elongation is indeed greatly impaired or abolished at the nonpermissive temperature.
In wild-type and rpb1-1 cells cultured at 25°C, NER in the TS (Fig. 5A and B and 6A and B) of the GAL1-10 genes was faster than that in the upstream regions where TCR does not operate (Fig. 5A and B and 6A and B) or in the NTS (Fig. 6A and B). This indicates that TCR is operative in rpb1-1 cells at the permissive temperature. In rad16 rpb1-1, rad16 rad26, and rad16 rad26 rpb1-1 cells cultured at 25°C, apparent repair occurred in the TS of the GAL1-10 genes, but not in the upstream regions where TCR does not operate (Fig. 5C, D, and E and 6C, D, and I) or in the NTS (Fig. 6C, D, and I). These results indicate that Rpb9-mediated TCR is operative in rpb1-1 cells incubated at the permissive temperature.
FIG. 5.

NER in the TS of the GAL1 gene in cells cultured at 25°C and 36°C. Gels show repair in different strains. Lanes labeled U are unirradiated samples. Other lanes are samples of different times (hours) of repair incubation. An arrow to the left of the gels marks the transcription start site. Solid circles to the left of the gels mark transcribed region (+1 to +380). Open triangles and solid diamonds mark upstream regions where TCR is operative (−1 to −180) or inoperative (−181 to −310), respectively. Plots underneath each of the gels show the average (mean ± standard deviation) of the percent CPDs remaining at individual sites in the transcribed (+1 to +380) region (solid circles) and upstream regions where TCR is operative (−1 to −180) (open triangles) and inoperative (−181 to −310) at different times of repair incubation. WT, wild type.
FIG. 6.

NER in one strand of the GAL1-10 genes (NTS for GAL1 and TS for GAL10) in cells cultured at 25°C and 36°C. Gels show repair in different strains. Lanes labeled U are unirradiated samples. Other lanes are samples of different times (hours) of repair incubation. Arrows on the left of the gels mark transcription start sites for GAL1 and GAL10, respectively. Solid circles on the left of the gels mark transcribed region of the GAL10 gene. Open triangles indicate the upstream region of the GAL10 gene, where TCR is operative. Solid diamonds mark upstream region of the GAL1-10 genes where TCR is inoperative. Open circles denote the NTS of the GAL1 gene. Plots in panels I and J show the average (mean ± standard deviation) of the percent CPDs remaining at individual sites in the regions of the GAL1 gene where TCR is not operative (the NTS and upstream regions of the GAL1 gene). The data in panels I and J were obtained by quantification of the regions marked with open circles and solid diamonds in gel panels A to D and E to H, respectively. Wild type (WT), open circles; rpb1-1, solid circles; rad16 rpb1-1, open triangles; rad16 rpb1-1 rad26, solid triangles.
Rapid NER can be seen in the TS of the GAL1-10 genes in wild-type, rpb1-1, rad16 rpb1-1, and rad16 rad26 cells cultured at 36°C (Fig. 5F to I and 6E to G). The rapid NER in the TS appears to be accomplished by TCR, because in cells lacking the GGR protein Rad16, no repair can be seen in the upstream regions where TCR does not operate (Fig. 5H and I and 6G and J) or in the NTS (Fig. 6G and J). However, no repair can be seen in rad16 rad26 rpb1-1 cells incubated at 36°C (Fig. 5J and 6H). These results indicate that, at the nonpermissive temperature, TCR in the rpb1-1 cells was solely mediated by Rad26, and the Rpb9-mediated TCR was abolished. In other words, impairment of transcription elongation (by incubating the rpb1-1 cells at 36°C), which did not significantly compromise Rad26-mediated TCR, completely abolished Rpb9-mediated TCR.
Rpb9 is dispensable for TCR if its transcription elongation function is compensated for.
The experiments described above favor a scenario in which Rpb9 mediates TCR by promoting transcription elongation. If this is true, Rpb9 should be dispensable for TCR, provided that its transcription elongation function is compensated for. It has been shown that deletion of SPT4 restores TCR in RPB2 and URA3 genes in rad26 cells (22). We wondered if deletion of SPT4 also restores TCR in rad26 rpb9 cells. Similar to rad16 rad26 rpb9 cells (Fig. 3B), rad7 rad26 rpb9 cells showed no NER activity in RPB2 (Fig. 7A and C) and GAL1 (not shown) genes. However, apparent NER can be seen in the TS of the RPB2 (Fig. 7B and C) and GAL1 (not shown) genes, indicating deletion of SPT4 indeed restored TCR in these cells. In agreement with previous report (22), deletion of SPT4 did not significantly affect GGR (not shown).
To examine if the restoration of TCR in rad7 rad26 rpb9 cells by deletion of SPT4 was due to a restoration of transcription elongation activity, we examined the resistance of these cells to MPA. As can be seen in Fig. 8A, deletion of SPT4 dramatically increased the resistance of rad7 rad26 rpb9 cells to MPA, indicating transcription elongation activity was indeed restored. In agreement with the TCR (Fig. 7) and MPA resistance data (Fig. 8A), deletion of SPT4 also dramatically increased the UV resistance of rad7 rad26 rpb9 cells (Fig. 8B).
FIG. 8.

Deletion of SPT4 restores MPA and UV resistance of rad7 rad26 rpb9 cells. (A) Sequential 10-fold serial dilutions of yeast cells were spotted onto SD plates containing different concentrations of MPA. The plates were incubated for 6 days before being photographed. (B) Sequential 10-fold serial dilutions of yeast cells were spotted onto SD plates and irradiated with different doses of 254-nm UV light. The plates were incubated for 4 days before being photographed.
DISCUSSION
Factors that play a role in transcription elongation of Pol II can be grouped into three broad functional categories (for a review, see reference 11). The first category of factors, such as TFIIF, ELL, and elongin, suppresses transient pausing and stimulates the rate of transcript elongation. The second category stimulates elongation on chromatin templates and includes HMG14 and 17, FACT, and nucleosome remodeling complexes like SWI/SNF. The third category includes factors that are able to reactivate a Pol II molecule that has been arrested during transcription (11). TFIIS, the defining member of the third category, stimulates RNase activity that is intrinsic to Pol II, and backtracks the elongation complex to rescue it from arrest (20, 21, 45). Rpb9 may promote transcription elongation by interacting with TFIIS (17, 64, 71). Studies using purified components showed that the maximal rates of chain elongation were nearly identical for normal Pol II and Rpb9-lacking Pol II (2). However, Rpb9-lacking Pol II is defective in response to TFIIS-stimulated read-through past a block (2). The structure of the yeast TFIIS-Pol II complex shows that TFIIS binds the Rpb1/Rpb9 “jaw” of Pol II and inserts into the Pol II pore, contacting the catalytic site by its highly conserved C terminus (24).
Using an in vitro system, it was shown that mutations in the Zn1 domain of Rpb9 had little effect on transcription activity (16). In contrast, mutations in the Zn2 domain of the Pol II subunit dramatically affect transcription function (16). These in vitro studies suggest that the Zn2 domain of Rpb9 plays a critical role in transcription elongation. However, in vivo studies suggest that only the N-terminal Zn1 and the linker are essential for transcription elongation and optimal cell growth (64). Our results, which were gained from in vivo experiments, agree well with the in vivo studies (64). As noticed by Hemming et al. (17), discrepancy exists between in vitro and in vivo data regarding the functions of Rpb9 domains.
In vitro studies showed that transcription complexes arrested at a CPD (8, 56) or a cisplatin adduct (57) are subject to transcript cleavage mediated by TFIIS. The cleaved transcripts, which are up to 30 nucleotides shorter than those arrested at a lesion, can be re-elongated up to the damaged site. However, disruption of the TFIIS gene did not affect TCR in yeast cells (67). It is therefore proposed that other factors functionally homologous to TFIIS might be employed for Pol II displacement from the site of the lesion in vivo (57). The transcription elongation function of Rpb9 may be responsible for displacing Pol II arrested at a DNA lesion in vivo.
One model for TCR proposes that RNA polymerase arrested at a lesion in DNA constitutes a signal for the repair proteins to initiate repair (33). This model assumes that the polymerase must be removed from the damaged site to provide access for the repair complex to the lesion (33). In Escherichia coli, Mfd protein participates in this process (49). The Mfd protein can promote the release of the RNA polymerase and the incomplete transcript from the DNA template and target NER machinery to the site of transcription blockage (39, 49). In eukaryotic cells, it remains unclear whether Pol II is released, degraded, remodeled, or translocated away from the site of damage without dissociating from the template DNA (12). Our results argue against the degradation model and favor the “translocation” model, at least for Rpb9-mediated TCR. First, an Rpb9 fragment encompassing the Zn1 and linker domains is competent for transcription elongation and TCR, but is deficient for promoting degradation of Pol II in response to UV radiation (X. Chen et al., unpublished results). Second, by coordinating with TFIIS, the transcription elongation function of Rpb9 is very likely to play a role in backtracking the Pol II stalled at a lesion. Third, although deletion of SPT4 reactivates transcription elongation and TCR in rad7 rad26 rpb9 cells (Fig. 7 and 8), the deletion does not restore UV-induced degradation of Pol II (X. Chen et al., unpublished results).
It was shown that, in an rpb1-1 strain (wild type for NER genes), no strand bias of NER was observed in the RPB2 gene when the cells were shifted to nonpermissive temperature (54). However, we observed that the strand bias is still apparent in both the GAL1-10 (compare Fig. 5G and 6F) and RPB2 (not shown) genes in rpb1-1 cells incubated at the nonpermissive temperature. In RPB1+ cells, the strand bias is caused by both Rad26- and Rpb9-mediated TCR. In rpb1-1 cells, however, the strand bias seems to be solely caused by Rad26-mediated TCR, as the Rpb9-mediated TCR appears to be completely abolished. The reason for the discrepancy is unclear, but could be due to the difference in genetic backgrounds between our strains and those of the previous report.
The molecular basis of the dysfunction of the rpbl-1 mutant enzyme at nonpermissive temperature is not clear (38). It also remains to be elucidated as to why the mutant enzyme at nonpermissive temperature is functional for Rad26-mediated TCR, but nonfunctional for Rpb9-mediated TCR. One possibility is that, at nonpermissive temperature, the mutant Pol II may still be able to perform a very low level of “leaky” transcription, which may be enough for Rad26 to mediate TCR. However, conformational change in Rpb1 may render Rpb9 unable to stimulate the intrinsic RNase activity of Pol II and backtrack the complex stalled at a lesion, making the lesion inaccessible to the NER machinery.
It was shown that deletion of SPT4 releases Rad26-independent TCR (22). Here we show that, in the absence of Spt4, both Rad26 and Rpb9 are dispensable for TCR, indicating Spt4 is an inhibitor of TCR. Spt4 is in complex with Spt5 (15, 53). The complex was found to be homologous to the DSIF complex in human cells. Biochemical work on DSIF has provided a detailed model for the molecular action of Spt4/Spt5 (69). The complex is involved in repression of transcription elongation at the early elongation-processive elongation transition, and repression is modulated by C-terminal domain phosphorylation (68, 74). On the other hand, cells carrying mutations in SPT4 and SPT5 genes display phenotypes associated with defects in transcription elongation (53), and the gene products are thought to be involved directly in transcription elongation (15, 47). How Spt4 inhibits TCR remains to be elucidated. Spt4/5 complex is associated with Pol II throughout the elongation phase (40). It is possible that Pol II complex stalled at a lesion is stabilized by Spt4. One role of TCR factors, such as Rad26 and Rpb9, may be to destabilize the stalled complex.
Our observation that, in the absence of Spt4, both Rad26 and Rpb9 are dispensable for TCR suggests that Pol II may be intrinsically competent for recruiting NER machinery to a lesion in the TS. Alternatively, some as-yet-unidentified transcription-repair coupling factor may exist. The TCR mechanism in eukaryotic cells seems to be much more complex than was previously thought, and future work will be needed to elucidate this extremely complicated mechanism.
Acknowledgments
We thank Michael J. Smerdon for being able to initiate the work in his laboratory and for critical reading and comments on the manuscript.
This study was supported by NIH grant ES012718 from the National Institute of Environmental Health Sciences.
Footnotes
Published ahead of print on 9 October 2006.
REFERENCES
- 1.Albig, A. R., and C. J. Decker. 2001. The target of rapamycin signaling pathway regulates mRNA turnover in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 12:3428-3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Awrey, D. E., R. G. Weilbaecher, S. A. Hemming, S. M. Orlicky, C. M. Kane, and A. M. Edwards. 1997. Transcription elongation through DNA arrest sites. A multistep process involving both RNA polymerase II subunit RPB9 and TFIIS. J. Biol. Chem. 272:14747-14754. [DOI] [PubMed] [Google Scholar]
- 3.Beaudenon, S. L., M. R. Huacani, G. Wang, D. P. McDonnell, and J. M. Huibregtse. 1999. Rsp5 ubiquitin-protein ligase mediates DNA damage-induced degradation of the large subunit of RNA polymerase II in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:6972-6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohr, V. A., C. A. Smith, D. S. Okumoto, and P. C. Hanawalt. 1985. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40:359-369. [DOI] [PubMed] [Google Scholar]
- 5.Bregman, D. B., R. Halaban, A. J. van Gool, K. A. Henning, E. C. Friedberg, and S. L. Warren. 1996. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. USA 93:11586-11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cramer, P. 2002. Multisubunit RNA polymerases. Curr. Opin. Struct. Biol. 12:89-97. [DOI] [PubMed] [Google Scholar]
- 7.Cramer, P., D. A. Bushnell, and R. D. Kornberg. 2001. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 292:1863-1876.11313498 [Google Scholar]
- 8.Donahue, B. A., S. Yin, J. S. Taylor, D. Reines, and P. C. Hanawalt. 1994. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl. Acad. Sci. USA 91:8502-8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Exinger, F., and F. Lacroute. 1992. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr. Genet. 22:9-11. [DOI] [PubMed] [Google Scholar]
- 10.Ferreiro, J. A., N. G. Powell, N. Karabetsou, N. A. Kent, J. Mellor, and R. Waters. 2004. Cbf1p modulates chromatin structure, transcription and repair at the Saccharomyces cerevisiae MET16 locus. Nucleic Acids Res. 32:1617-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fish, R. N., and C. M. Kane. 2002. Promoting elongation with transcript cleavage stimulatory factors. Biochim. Biophys. Acta 1577:287-307. [DOI] [PubMed] [Google Scholar]
- 12.Friedberg, E. C., G. C. Walker, W. Siede, R. D. Wood, R. A. Schultz, and T. Ellenberger. 2006. DNA repair and mutagenesis, 2nd ed. ASM Press, Washington, D.C.
- 13.Furter-Graves, E. M., B. D. Hall, and R. Furter. 1994. Role of a small RNA pol II subunit in TATA to transcription start site spacing. Nucleic Acids Res. 22:4932-4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grigull, J., S. Mnaimneh, J. Pootoolal, M. D. Robinson, and T. R. Hughes. 2004. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol. Cell. Biol. 24:5534-5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartzog, G. A., T. Wada, H. Handa, and F. Winston. 1998. Evidence that Spt4, Spt5, and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes Dev. 12:357-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemming, S. A., and A. M. Edwards. 2000. Yeast RNA polymerase II subunit RPB9. Mapping of domains required for transcription elongation. J. Biol. Chem. 275:2288-2294. [DOI] [PubMed] [Google Scholar]
- 17.Hemming, S. A., D. B. Jansma, P. F. Macgregor, A. Goryachev, J. D. Friesen, and A. M. Edwards. 2000. RNA polymerase II subunit Rpb9 regulates transcription elongation in vivo. J. Biol. Chem. 275:35506-35511. [DOI] [PubMed] [Google Scholar]
- 18.Hull, M. W., K. McKune, and N. A. Woychik. 1995. RNA polymerase II subunit RPB9 is required for accurate start site selection. Genes Dev. 9:481-490. [DOI] [PubMed] [Google Scholar]
- 19.Hyle, J. W., R. J. Shaw, and D. Reines. 2003. Functional distinctions between IMP dehydrogenase genes in providing mycophenolate resistance and guanine prototrophy to yeast. J. Biol. Chem. 278:28470-28478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Izban, M. G., and D. S. Luse. 1993. The increment of SII-facilitated transcript cleavage varies dramatically between elongation competent and incompetent RNA polymerase II ternary complexes. J. Biol. Chem. 268:12874-12885. [PubMed] [Google Scholar]
- 21.Izban, M. G., and D. S. Luse. 1992. The RNA polymerase II ternary complex cleaves the nascent transcript in a 3′-5′ direction in the presence of elongation factor SII. Genes Dev. 6:1342-1356. [DOI] [PubMed] [Google Scholar]
- 22.Jansen, L. E., H. den Dulk, R. M. Brouns, M. de Ruijter, J. A. Brandsma, and J. Brouwer. 2000. Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J. 19:6498-6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones, E. W. 1991. Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 194:428-453. [DOI] [PubMed] [Google Scholar]
- 24.Kettenberger, H., K. J. Armache, and P. Cramer. 2003. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell 114:347-357. [DOI] [PubMed] [Google Scholar]
- 25.Li, B., C. R. Nierras, and J. R. Warner. 1999. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol. Cell. Biol. 19:5393-5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, S., and M. J. Smerdon. 2004. Dissecting transcription-coupled and global genomic repair in the chromatin of yeast GAL1-10 genes. J. Biol. Chem. 279:14418-14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, S., and M. J. Smerdon. 2002. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J. 21:5921-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, S., and R. Waters. 1996. Nucleotide level detection of cyclobutane pyrimidine dimers using oligonucleotides and magnetic beads to facilitate labelling of DNA fragments incised at the dimers and chemical sequencing reference ladders. Carcinogenesis 17:1549-1552. [DOI] [PubMed] [Google Scholar]
- 29.Li, S., R. Waters, and M. J. Smerdon. 2000. Low- and high-resolution mapping of DNA damage at specific sites. Methods 22:170-179. [DOI] [PubMed] [Google Scholar]
- 30.Lommel, L., and P. C. Hanawalt. 1991. The genetic defect in the Chinese hamster ovary cell mutant UV61 permits moderate selective repair of cyclobutane pyrimidine dimers in an expressed gene. Mutat. Res. 255:183-191. [DOI] [PubMed] [Google Scholar]
- 31.Luo, Z., J. Zheng, Y. Lu, and D. B. Bregman. 2001. Ultraviolet radiation alters the phosphorylation of RNA polymerase II large subunit and accelerates its proteasome-dependent degradation. Mutat. Res. 486:259-274. [DOI] [PubMed] [Google Scholar]
- 32.McKay, B. C., F. Chen, S. T. Clarke, H. E. Wiggin, L. M. Harley, and M. Ljungman. 2001. UV light-induced degradation of RNA polymerase II is dependent on the Cockayne's syndrome A and B proteins but not p53 or MLH1. Mutat. Res. 485:93-105. [DOI] [PubMed] [Google Scholar]
- 33.Mellon, I., V. A. Bohr, C. A. Smith, and P. C. Hanawalt. 1986. Preferential DNA repair of an active gene in human cells. Proc. Natl. Acad. Sci. USA 83:8878-8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellon, I., and P. C. Hanawalt. 1989. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature 342:95-98. [DOI] [PubMed] [Google Scholar]
- 35.Mellon, I., G. Spivak, and P. C. Hanawalt. 1987. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51:241-249. [DOI] [PubMed] [Google Scholar]
- 36.Moore, P. A., F. A. Sagliocco, R. M. C. Wood, and A. J. P. Brown. 1991. Yeast glycolytic mRNAs are differentially regulated. Mol. Cell. Biol. 11:5330-5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nesser, N. K., D. O. Peterson, and D. K. Hawley. 2006. RNA polymerase II subunit Rpb9 is important for transcriptional fidelity in vivo. Proc. Natl. Acad. Sci. USA 103:3268-3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nonet, M., C. Scafe, J. Sexton, and R. Young. 1987. Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol. Cell. Biol. 7:1602-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park, J. S., M. T. Marr, and J. W. Roberts. 2002. E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 109:757-767. [DOI] [PubMed] [Google Scholar]
- 40.Pokholok, D. K., N. M. Hannett, and R. A. Young. 2002. Exchange of RNA polymerase II initiation and elongation factors during gene expression in vivo. Mol. Cell 9:799-809. [DOI] [PubMed] [Google Scholar]
- 41.Powell, N. G., J. Ferreiro, N. Karabetsou, J. Mellor, and R. Waters. 2003. Transcription, nucleosome positioning and protein binding modulate nucleotide excision repair of the Saccharomyces cerevisiae MET17 promoter. DNA Repair 2:375-386. [DOI] [PubMed] [Google Scholar]
- 42.Ratner, J. N., B. Balasubramanian, J. Corden, S. L. Warren, and D. B. Bregman. 1998. Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II. Implications for transcription-coupled DNA repair. J. Biol. Chem. 273:5184-5189. [DOI] [PubMed] [Google Scholar]
- 43.Reardon, J. T., and A. Sancar. 2005. Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79:183-235. [DOI] [PubMed] [Google Scholar]
- 44.Reid, J., and J. Q. Svejstrup. 2004. DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro. J. Biol. Chem. 279:29875-29878. [DOI] [PubMed] [Google Scholar]
- 45.Reines, D., P. Ghanouni, Q. Q. Li, and J. Mote, Jr. 1992. The RNA polymerase II elongation complex. Factor-dependent transcription elongation involves nascent RNA cleavage. J. Biol. Chem. 267:15516-15522. [PMC free article] [PubMed] [Google Scholar]
- 46.Ribar, B., L. Prakash, and S. Prakash. 2006. Requirement of ELC1 for RNA polymerase II polyubiquitylation and degradation in response to DNA damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 26:3999-4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rondon, A. G., M. Garcia-Rubio, S. Gonzalez-Barrera, and A. Aguilera. 2003. Molecular evidence for a positive role of Spt4 in transcription elongation. EMBO J. 22:612-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scafe, C., C. Martin, M. Nonet, S. Podos, S. Okamura, and R. A. Young. 1990. Conditional mutations occur predominantly in highly conserved residues of RNA polymerase II subunits. Mol. Cell. Biol. 10:1270-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selby, C. P., and A. Sancar. 1993. Molecular mechanism of transcription-repair coupling. Science 260:53-58. [DOI] [PubMed] [Google Scholar]
- 50.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smerdon, M. J., and F. Thoma. 1990. Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell 61:675-684. [DOI] [PubMed] [Google Scholar]
- 52.Sun, Z. W., A. Tessmer, and M. Hampsey. 1996. Functional interaction between TFIIB and the Rpb9 (Ssu73) subunit of RNA polymerase II in Saccharomyces cerevisiae. Nucleic Acids Res. 24:2560-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swanson, M. S., and F. Winston. 1992. SPT4, SPT5 and SPT6 interactions: effects on transcription and viability in Saccharomyces cerevisiae. Genetics 132:325-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sweder, K. S., and P. C. Hanawalt. 1992. Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription. Proc. Natl. Acad. Sci. USA 89:10696-10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teng, Y., S. Li, R. Waters, and S. H. Reed. 1997. Excision repair at the level of the nucleotide in the Saccharomyces cerevisiae MFA2 gene: mapping of where enhanced repair in the transcribed strand begins or ends and identification of only a partial rad16 requisite for repairing upstream control sequences. J. Mol. Biol. 267:324-337. [DOI] [PubMed] [Google Scholar]
- 56.Tornaletti, S., B. A. Donahue, D. Reines, and P. C. Hanawalt. 1997. Nucleotide sequence context effect of a cyclobutane pyrimidine dimer upon RNA polymerase II transcription. J. Biol. Chem. 272:31719-31724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tornaletti, S., S. M. Patrick, J. J. Turchi, and P. C. Hanawalt. 2003. Behavior of T7 RNA polymerase and mammalian RNA polymerase II at site-specific cisplatin adducts in the template DNA. J. Biol. Chem. 278:35791-35797. [DOI] [PubMed] [Google Scholar]
- 58.Troelstra, C., A. van Gool, J. de Wit, W. Vermeulen, D. Bootsma, and J. H. Hoeijmakers. 1992. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71:939-953. [DOI] [PubMed] [Google Scholar]
- 59.Tu, Y., S. Bates, and G. P. Pfeifer. 1997. Sequence-specific and domain-specific DNA repair in xeroderma pigmentosum and Cockayne syndrome cells. J. Biol. Chem. 272:20747-20755. [DOI] [PubMed] [Google Scholar]
- 60.Tu, Y., S. Bates, and G. P. Pfeifer. 1998. The transcription-repair coupling factor CSA is required for efficient repair only during the elongation stages of RNA polymerase II transcription. Mutat. Res. 400:143-151. [DOI] [PubMed] [Google Scholar]
- 61.Tu, Y., S. Tornaletti, and G. P. Pfeifer. 1996. DNA repair domains within a human gene: selective repair of sequences near the transcription initiation site. EMBO J. 15:675-683. [PMC free article] [PubMed] [Google Scholar]
- 62.van Gool, A. J., R. Verhage, S. M. Swagemakers, P. van de Putte, J. Brouwer, C. Troelstra, D. Bootsma, and J. H. Hoeijmakers. 1994. RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J. 13:5361-5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Hoffen, A., A. T. Natarajan, L. V. Mayne, A. A. van Zeeland, L. H. Mullenders, and J. Venema. 1993. Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells. Nucleic Acids Res. 21:5890-5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Mullem, V., M. Wery, M. Werner, J. Vandenhaute, and P. Thuriaux. 2002. The Rpb9 subunit of RNA polymerase II binds transcription factor TFIIE and interferes with the SAGA and elongator histone acetyltransferases. J. Biol. Chem. 277:10220-10225. [DOI] [PubMed] [Google Scholar]
- 65.Venema, J., L. H. Mullenders, A. T. Natarajan, A. A. van Zeeland, and L. V. Mayne. 1990. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. USA 87:4707-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verhage, R., A.-M. Zeeman, N. de Groot, F. Gleig, D. D. Bang, P. van de Putte, and J. Brouwer. 1994. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol. Cell. Biol. 14:6135-6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verhage, R. A., J. Heyn, P. van de Putte, and J. Brouwer. 1997. Transcription elongation factor S-II is not required for transcription-coupled repair in yeast. Mol. Gen. Genet. 254:284-290. [DOI] [PubMed] [Google Scholar]
- 68.Wada, T., G. Orphanides, J. Hasegawa, D. K. Kim, D. Shima, Y. Yamaguchi, A. Fukuda, K. Hisatake, S. Oh, D. Reinberg, and H. Handa. 2000. FACT relieves DSIF/NELF-mediated inhibition of transcriptional elongation and reveals functional differences between P-TEFb and TFIIH. Mol. Cell 5:1067-1072. [DOI] [PubMed] [Google Scholar]
- 69.Wada, T., T. Takagi, Y. Yamaguchi, A. Ferdous, T. Imai, S. Hirose, S. Sugimoto, K. Yano, G. A. Hartzog, F. Winston, S. Buratowski, and H. Handa. 1998. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 12:343-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang, Y., C. L. Liu, J. D. Storey, R. J. Tibshirani, D. Herschlag, and P. O. Brown. 2002. Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. USA 99:5860-5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wery, M., E. Shematorova, B. Van Driessche, J. Vandenhaute, P. Thuriaux, and V. Van Mullem. 2004. Members of the SAGA and Mediator complexes are partners of the transcription elongation factor TFIIS. EMBO J. 23:4232-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woudstra, E. C., C. Gilbert, J. Fellows, L. Jansen, J. Brouwer, H. Erdjument-Bromage, P. Tempst, and J. Q. Svejstrup. 2002. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 415:929-933. [DOI] [PubMed] [Google Scholar]
- 73.Woychik, N. A., W. S. Lane, and R. A. Young. 1991. Yeast RNA polymerase II subunit RPB9 is essential for growth at temperature extremes. J. Biol. Chem. 266:19053-19055. [PubMed] [Google Scholar]
- 74.Yamaguchi, Y., T. Takagi, T. Wada, K. Yano, A. Furuya, S. Sugimoto, J. Hasegawa, and H. Handa. 1999. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97:41-51. [DOI] [PubMed] [Google Scholar]