

Abstract
Large-scale association studies are being undertaken with the hope of uncovering the genetic determinants of complex disease. We describe a computationally efficient method for inferring genealogies from population genotype data and show how these genealogies can be used to fine map disease loci and interpret association signals. These genealogies take the form of the ancestral recombination graph (ARG). The ARG defines a genealogical tree for each locus, and, as one moves along the chromosome, the topologies of consecutive trees shift according to the impact of historical recombination events. There are two stages to our analysis. First, we infer plausible ARGs, using a heuristic algorithm, which can handle unphased and missing data and is fast enough to be applied to large-scale studies. Second, we test the genealogical tree at each locus for a clustering of the disease cases beneath a branch, suggesting that a causative mutation occurred on that branch. Since the true ARG is unknown, we average this analysis over an ensemble of inferred ARGs. We have characterized the performance of our method across a wide range of simulated disease models. Compared with simpler tests, our method gives increased accuracy in positioning untyped causative loci and can also be used to estimate the frequencies of untyped causative alleles. We have applied our method to Ueda et al.’s association study of CTLA4 and Graves disease, showing how it can be used to dissect the association signal, giving potentially interesting results of allelic heterogeneity and interaction. Similar approaches analyzing an ensemble of ARGs inferred using our method may be applicable to many other problems of inference from population genotype data.
Unraveling the genetic basis of complex disease is one of the main goals of human genetics. In the case-control association study design,1,2 nonfamilial individuals are genotyped for a panel of SNPs that capture most but not all of the genetic variation in a population. Each individual is labeled as either a “case” (affected by the disease) or as a “control” (unaffected), and, by analyzing the segregation of SNP alleles between cases and controls, it is possible to identify loci with statistical association with the disease.
One of the simplest analyses for case-control data is Pearson’s χ2 test applied to each marker. This tests for nonindependence between genotype and phenotype, and, in certain circumstances, it will successfully identify disease associations—such as when causative polymorphisms are typed or are in strong linkage disequilibrium (LD) with typed markers.3,4 But, by the testing of each marker independently, information about the population history is discarded (in particular, information about the coinheritance of markers) that, if exploited, can yield a substantial increase in power.
A potentially more powerful approach is to interpret the pattern of variation by considering the evolutionary processes that produced it.5,6 In this article, we present an algorithm for reconstructing the genealogical history of a population sample and show how these genealogies can be used to fine map disease loci. Additionally, we use the genealogies to dissect the association signal—estimating the frequencies of untyped polymorphisms and searching for allelic heterogeneity and epistasis.
The formalism we use for representing these genealogies is the ancestral recombination graph (ARG).7 For a population of chromosome sequences, the ARG describes how they are related to each other—through mutation, recombination, and coalescence—back to a common ancestor (fig. 1A). Note that we are using the term “ARG” to mean the data structure for representing genealogical histories. The distribution of these under the Wright-Fisher model with recombination is described by a stochastic process called the “coalescent-with-recombination” model.7–10
Figure 1. .

The ARG. A, Example ARG for four chromosome sequences. The sequences label the leaves of the ARG and are written as strings of 0s and 1s (coding SNP alleles). Moving backward in time (up the ARG), one first encounters a mutation. A mutation is denoted by a black dot and a number specifying its marker position. The second event is a recombination between markers 2 and 3. As one works backward in time, this corresponds to splitting a lineage into two, with the alleles at positions 1 and 2 following the left lineage and the allele at position 3 following the right lineage. After this is a coalescence, merging two lineages into one, and so on, to the grand common ancestor. B, Marginal tree for the SNPs at positions 1 and 2. C, Marginal tree for the SNP at position 3. To test a marginal tree for disease association, mutations are dropped onto each of the branches in turn, defining hypothetical allelic states of the leaves, which can then be tested for statistical association with the phenotype. The black dot labeled “2” best segregates the cases (D) from the controls (U) and would be identified as the most likely causative mutation event. D–G, Logic behind the ARG inference algorithm. D, The two sequences have a shared tract over the region 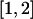 . E, To coalesce over the tract region, we must add a recombination breakpoint to the right of it—that is, between positions 2 and 3. This results in two parent sequences. F, We let undefined material (denoted by ·) coalesce with anything. We can now coalesce the left recombination parent and the other sequence. G, We can add a mutation.
. E, To coalesce over the tract region, we must add a recombination breakpoint to the right of it—that is, between positions 2 and 3. This results in two parent sequences. F, We let undefined material (denoted by ·) coalesce with anything. We can now coalesce the left recombination parent and the other sequence. G, We can add a mutation.
For each position on the chromosome, there is a genealogical tree, called a “marginal tree,” embedded in the ARG. As one moves along the chromosome, the topologies of consecutive marginal trees shift according to the impact of historical recombination events (fig. 1B and 1C). In this way, historical recombination events define the chromosomal region that each marginal tree spans, and, since many recombination events have occurred in population history, the resolution is very fine.
If there is a disease-predisposing mutation at a particular chromosomal location, it would have occurred on some internal branch of the marginal tree at that location. So, one way to find disease associations is to scan across the marginal trees, looking for those with branches that discriminate well between cases and controls—that is, that have a large number of cases beneath them and significantly fewer controls. Such a clustering of the cases underneath a branch suggests that a causative mutation arose on that branch.
If the true ARG were known, it would provide the optimal amount of information for mapping—no extra information would be available from the genotypes. Not only would disease-associated regions be identified, but the ARG would give the ages of the causative mutations, would specify the haplotypic background of those mutations, and so forth. It would also be possible to optimally impute missing data. But, unfortunately, the true ARG is unknowable, and inference under the coalescent-with-recombination model has proven computationally prohibitive. This is in part because there are infinitely many ARGs compatible with any set of genotype data, and very many of these are of comparable likelihood.11,12
The difficulties involved in coalescent-based inference have partly motivated the development of faster haplotype-clustering methods.13–17 These cluster the haplotype sequences (for small nonrecombining regions) and perform statistical tests on these clusters. The clustering hierarchy is often organized as a cladogram, which is assumed to approximate the marginal tree for that region. However, compared with the ARG, cladograms are a coarse approximation of population evolution, and there is often difficulty in modeling the relationships between similar haplotypes and in handling rare haplotypes. Additionally, it is often assumed that haplotypes are observed directly and that one can define nonrecombining haplotype blocks, which, in general, is not the case.
We have developed an ARG-based mapping method that has computational efficiency nearing that of haplotype-clustering methods. We achieve this by using a heuristic approach for ARG inference and are thereby able to construct ARGs for thousands of individuals typed for hundreds of SNPs; this is sufficiently fast that the analysis may be windowed over the whole genome, fitting the scale of proposed large-scale case-control studies. However, in this article, we focus our attention on fine mapping and interpretation of a signal at a potentially associated locus, in part because there are currently no publicly available genomewide-association-study data sets, experimental or simulated. Because the algorithm is heuristic, we do not claim to sample ARGs from the coalescent-with-recombination model; instead, we suggest that we infer plausible ARGs, a claim that can be tested by seeing how well these ARGs infer properties of causative polymorphisms. In this way, our method fills the gap between methods that are based on more-sophisticated coalescent models18–20 but require prohibitive computation and haplotype-based methods that model less precisely the structure and evolution of a disease locus.
Methods
A related problem to constructing plausible ARGs is that of constructing minimal ARGs21–23—that is, those with the smallest number of recombination events required to derive a sample of sequences. An algorithm that is similar to our ARG inference method has been developed independently for this problem.24 Our emphasis is, however, on inference of plausible ARGs rather than minimal ones.
To develop an intuition for how our ARG inference algorithm works, we will give an informal description in two stages—first, by describing a way to construct genealogical trees for nonrecombining chromosome sequences and then by extending this to include recombination, so that ARGs can be constructed for any set of sequences.
When the sequences are nonrecombining, we only need to use coalescences and mutations to describe their genealogy, and there are efficient algorithms for this.25,26 Working backward in time, two haplotype sequences can coalesce into a parent sequence (that is, their lineages merge into one) only if they are identical. Since the goal of our algorithm is to coalesce back to a single common ancestor, we perform coalescences whenever possible. Unless all the sequences are identical, we will also need to infer mutation events and remove mutant alleles from ancestral sequences. We will assume the infinite-sites model throughout, which stipulates that there are no back or recurrent mutations. Consequently, a mutant allele can be removed from a set of ancestral sequences only if it occurs on exactly one of those sequences. By performance of mutations and coalescences as described, ancestral populations are defined, and, if there were no recombinations, it will be possible to coalesce back to a single common ancestor.
If recombination did occur, it may not be possible to construct a tree for the sequenced region, and, instead, an ARG must be inferred. To infer recombination events, our algorithm looks for pairs of sequences that are identical over a contiguous region (fig. 1D–1G). We assume that such a shared tract is inherited intact from an ancestor and that the sequence mismatches at either end of the tract were caused by historical mutation or recombination events. If recombination events are added at both ends of a shared tract, the tract becomes decoupled from the genetic material to the left and right of it and is then free to coalesce.
To understand this, consider working backward in time, putting a recombination event on a sequence. This results in two parental sequences, a left parent and a right parent, that are only defined to the left and right of the recombination breakpoint, respectively—they failed to pass on the rest of their genetic material to the next generation. Since undefined regions have no constraint on what they can coalesce with, the number of mismatching alleles preventing a coalescence is reduced, possibly to zero. By incorporating recombination into genealogy construction, it is always possible to construct an ARG that coalesces back to a single common ancestor.
What follows is a more detailed description of the algorithm. It can infer ARGs from population data with missing genotypes and unknown haplotype phase, although, for ease of exposition, we initially describe the simpler case of perfect phase-known data.
ARG Inference Algorithm
The algorithm works backward in time from the contemporary, typed population of chromosome sequences to a single ancestor sequence. Each step back in time, accomplished with a recombination, mutation, or coalescence, defines an ancestral population of sequences. We denote the set of sequences at time T as ST, and the sequences are, in the phase-known case, strings of length m from the alphabet {0,1,·}, where m is the number of markers, 0 is one of the SNP alleles, 1 is another allele, and “·” denotes an undefined allele—undefined because it was not inherited by any sequences in the contemporary, typed population. The allelic state of a SNP on sequence C is denoted C[i], where i is the marker position, numbered from 1, so 1⩽i⩽m. We define C1[i]∼C2[i] if and only if C1[i]=C2[i], or C1[i]=·, or C2[i]=·. We define a complement operator, ¬, such that, if C[i]=0, then ¬C[i]=1 and vice versa, and · is its own complement.
There is a shared tract between sequences C1 and C2, over the contiguous set of markers a,…,b, (1) if C1[i]∼C2[i] for all a⩽i⩽b; (2) if there is at least one i for which C1[i]=C2[i]≠·; (3) if a>1, then C1[a-1]≠C2[a-1] and neither is · ; and (4) if b<m, then C1[b+1]≠C2[b+1] and neither is · . Item (1) requires that the two sequences have the same allelic state over the shared tract; item (2) requires that, for at least one position in the tract, both sequences are defined; and items (3) and (4) require that the shared tract is maximal. We denote such a shared tract as {C1,C2}[a,b].
The algorithm is initialized at time T=1 (T is incremented as we move back in time) by setting S1 to be the set of contemporary, typed sequences. The algorithm proceeds by finding which coalescences, mutations, and recombinations can be performed, determining this according to the rules below. Applying one of these operations defines an ancestral population ST+1, which is constructed from ST by use of the transitions also described below. The algorithm continues in this way until it arrives at a population with only one sequence.
- Coalescence.
- Rule. If there exist two sequences, C1 and C2, in ST such that, for all i, C1[i]∼C2[i], then C1 and C2 can be coalesced into an ancestor.
- Transition.
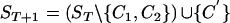 where C′[i]=C1[i] when C1[i]≠· and C′[i]=C2[i] otherwise. (By
where C′[i]=C1[i] when C1[i]≠· and C′[i]=C2[i] otherwise. (By 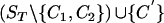 , we mean ST with the sequences C1 and C2 removed and the sequence C′ added in.)
, we mean ST with the sequences C1 and C2 removed and the sequence C′ added in.)
- Mutation.
- Rule. If there exists a sequence C1 in ST and a marker i, where, for all C2 in ST∖{C1}, we have C2[i]=¬C1[i] or · , then we can remove the derived allele (C1[i]) from the population.
- Transition.
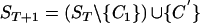 , where C′[i]=¬C1[i] and C′[j]=C1[j] for all j≠i.
, where C′[i]=¬C1[i] and C′[j]=C1[j] for all j≠i.
- Recombination.
- Rule. When the rules for coalescence and mutation are not satisfied, we must perform a recombination (or a pair of recombinations) instead. We denote a recombination breakpoint as
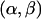 , meaning that it occurs between markers α and β. Picking a shared tract {C1,C2}[a,b] from those available in ST, we aim to put recombinations on the lineages of C1 and C2 such that one recombination parent of C1 and one recombination parent of C2 satisfy the rule for coalescence. To do this, we must put a breakpoint at
, meaning that it occurs between markers α and β. Picking a shared tract {C1,C2}[a,b] from those available in ST, we aim to put recombinations on the lineages of C1 and C2 such that one recombination parent of C1 and one recombination parent of C2 satisfy the rule for coalescence. To do this, we must put a breakpoint at 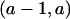 if a≠1 and put a breakpoint at
if a≠1 and put a breakpoint at 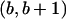 if b≠m.
if b≠m. - Transition. From the tract {C1,C2}[a,b], pick (1) a valid breakpoint
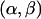 , where either
, where either 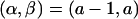 or
or 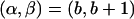 , and (2) a recombinant sequence CR, where either CR=C1 or CR=C2. Then,
, and (2) a recombinant sequence CR, where either CR=C1 or CR=C2. Then, 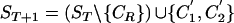 , where C′1[i]=CR[i] for i⩽α and C′1[i]=· otherwise, and C′2[i]=CR[i] for all i⩾β and C′2[i]=· otherwise. If both
, where C′1[i]=CR[i] for i⩽α and C′1[i]=· otherwise, and C′2[i]=CR[i] for all i⩾β and C′2[i]=· otherwise. If both  and
and  are valid breakpoints (i.e., a≠1 and b≠m), we must put the second recombination (taking us to state ST+2) on an appropriate ancestor of C1 or C2. See figure 1D–1G for an example.
are valid breakpoints (i.e., a≠1 and b≠m), we must put the second recombination (taking us to state ST+2) on an appropriate ancestor of C1 or C2. See figure 1D–1G for an example.
These rules define the constraints on the algorithm that must be enforced if it is to produce legal ARGs. However, at any stage of the algorithm, there may be several different coalescences, mutations, or recombinations that satisfy the rules. We choose between these, using the heuristics below, and the stochastic elements mean that different ARGs are generated each time the algorithm is run.
Heuristics. (1) Perform a recombination only if no mutations or coalescences are possible. (2) If it is possible to add multiple mutations and/or multiple coalescences at the same time, the order in which these are done is chosen arbitrarily. (3) Coalesce sequences only if they have an overlapping region of defined material—that is, the two sequences must match for at least one position that is not · . This restriction reflects ideas in the sequentially Markovian coalescent-with-recombination model.11 (4) Recombinations are added at the ends of longer shared tracts first. During the recombination step, we choose a shared tract {C1,C2}[a,b] such that the base-pair distance between markers a and b is maximized, reflecting that longer shared tracts tend to arise from more-recent recombination events. However, because this is only a tendency, not absolute, we break this heuristic with a certain probability (which, throughout this article, is 0.1), and, in these cases, a randomly selected tract is used to position recombination breakpoint(s). (5) The first coalescence after a recombination is based on the shared segment that was used to decide the location of that recombination.
Handling Unphased and Missing Data
By extending our algorithm, it is possible to resolve haplotype phase and impute missing data while constructing an ARG. Handling the missing data is the simpler of the two cases. A missing character is allowed to coalesce with any other character (0,1,·, or another missing character), and, when it coalesces with a state-known character (0 or 1), the missing character becomes fixed to that state and this assignment is propagated down the ARG to the leaves.
Phasing the data is similar, except that a record of the diploid pairings of chromosomes is kept. A phase-unknown character may not coalesce with the corresponding phase-unknown character on its sister chromosome (because the individual is heterozygous at that position). When a phase-unknown character coalesces with a state-known character, its phase becomes fixed, as does the character on its sister chromosome, although to the complement state. When phase-unknown characters from two chromosomes coalesce, these chromosomes and their sisters become dependent on each other; neither of those chromosomes may coalesce with the sister of the other one, and, when one of the chromosomes has a character phase resolved, that character is also resolved on the other chromosome and to the complement state on the two sister chromosomes. Of course, many more than four chromosomes can become involved in such interdependencies.
Fine-Scale Mapping Using ARGs
An ARG generated as described above defines a marginal tree for each chromosome position (fig. 1A–1C). For a given position, the marginal tree can be extracted from the ARG by tracing the genealogy of that position back in time from the leaves. When a recombination is encountered, the genealogy follows the path of the left recombination parent if the breakpoint is to the right of the position in question; otherwise, it follows the right recombination parent.
We can test a position for association by seeing whether its marginal tree has a branch on which we can place a hypothetical causative mutation that suitably explains the observed disease states of the genotyped individuals—such as mutation 2 in figure 1. (Note that, although such a branch extends over an interval of markers in the ARG, localization is refined by recombination events lower down the ARG; these change the number of case and control chromosomes under the branch at each position. Therefore, our method gives a different score at each marker.)
Our test is as follows: since the true ARG is unknown, we infer an ensemble of 100 plausible ARGs. These are generated by running the ARG inference algorithm 100 times, and stochastic choices made during ARG construction (such as which pairs of sequences to coalesce first) mean that these ARGs are all different. For each marker, the 100 marginal trees are extracted from the ARGs. For each marginal tree, hypothetical disease-predisposing mutations are put on each branch in turn. These cause the case-control individuals (the leaves of the tree) to be bipartitioned into those with the mutant allele and those with the ancestral allele. A χ2 test can then be used to detect nonindependence between inferred allelic state and disease state. If there are n leaves, then there are n-3 nonequivalent, nonunary bipartitions of a tree, and, hence, n-3 χ2 test statistics for a tree. Under the assumption that the region spanned by one tree harbors, at most, one causative mutation, we take the maximum of these n-3 test statistics, calling this the “best-cut score.” After finding the best-cut score for each of the 100 trees, we take the mean, giving an association score for the marker (this assumes that all the inferred ARGs are equally likely).
Although we test for nonindependence between alleles and disease, the test could easily be modified to test for association between genotype and disease. Similarly, a regression could be performed, rather than a χ2 test, allowing our method to be applied to quantitative phenotype data. Or we could calculate the likelihood of the data given the tree, although this would require an explicit disease and mutation model. Also, we need not assume that there is only one causative mutation on a tree.20
We calculate the statistical significance of the mapping score at each marker—the markerwise P value—by permuting the assignments of case and control labels of the individuals and repeating the test above. By performance of multiple permutations, an empirical null distribution is generated from which the P value can be calculated.27 For P values exceeding the precision of the permutations, we fit an extreme value distribution to the empirical distribution.28
Since multiple markers are being tested for association, there is a multiple-testing issue, which we can correct for by calculating, for each marker, an experimentwise P value: the probability that any of the typed markers show such a strong association signal by chance. Again, this is done by permutation; after shuffling the case and control labels, the maximum association score of all the markers is recorded, thus defining an empirical experimentwise null distribution. Once again, an extreme value distribution can be fitted, to estimate small P values.
Simulation of Case-Control Studies
To evaluate the performance of our method under a variety of disease models, we simulated suites of case-control studies. Each suite contains 50 studies simulated under the same model, which was parameterized according to (1) the recombination model of the population from which the cases and controls were sampled, (2) the tagging SNP (tSNP) ascertainment scheme, (3) whether the sequences are phased or unphased and the amount of missing data, and (4) the disease model parameters: genotype relative risk, disease-allele frequency, and size of study.
The case-control studies were sampled from one of two populations, which we call “constant” and “hot.” Both populations contain 20,000 1-Mb chromosome sequences, which were simulated using the FREGENE forward simulator29 (BARGEN Web site) and are available from the Margarita Web site. The constant population was simulated using the simple (i.e., no population expansion or complex demography) Wright-Fisher model with a constant recombination rate. The mutation rate was 1.1×10-8 per generation per nucletide, and the recombination crossover rate was 2.2×10-8 per generation per nucleotide. In contrast, the hot population was simulated with recombination hotspots. These were 2 kb in length and accounted for 1% of the length of the region but 60% of all recombinations. The average recombination crossover rate was the same as before, resulting in recombination crossover rates within and between hotspots of 6.56×10-7 and 4.44×10-9 per bp per generation, respectively. Gene conversions were also included, with a constant tract length of 50 bp and average rate across the genome of 1.1×10-7. Gene conversions were assigned the same hotspots as crossovers, and their rates within and between hotspots were 6.56×10-6 and 4.44×10-8 per bp per generation, respectively. For both populations, all SNPs with minor-allele frequency (MAF) ⩾0.005 were recorded (yielding 4,621 SNPs in the constant population and 4,825 SNPs in the hot population).
For each population, tSNPs were selected using three schemes, as follows.
“Full” ascertainment. A total of 120 chromosomes were sampled without replacement from the population and were presented to the tagging program TAGGER.30 (For the constant population, 4,235 of the 4,621 SNPs were polymorphic in this sample and thus were considered for tagging; for the hot population, 4,389 of the 4,825 SNPs were polymorphic). We set TAGGER to use a maximum tagging distance of 100 kb (the distance between a tag and the SNPs it tags) and that the tags be designed for single-marker tests.
5% Ascertainment. This ascertainment is like the full ascertainment, but only SNPs with MAF⩾0.05 in the population were considered in the tagging process.
Random. tSNPs were evenly spaced but otherwise were selected at random from the SNPs with MAF⩾0.05 in the population.
In all three cases, 300 tSNPs were chosen. For full and 5% ascertainments, these were the best 300 tSNPs according to TAGGER.
The disease model for each suite of 50 case-control studies was specified by parameters q, GRR(Aa), GRR(AA), and ncc, where q is the frequency of the disease-predisposing allele, GRR(Aa) is the genotype relative risk of the heterozygote, GRR(AA) is the genotype relative risk of the mutant homozygote, and ncc is the number of case chromosome sequences (which, in our simulations, is the same as the number of control sequences). GRR(Aa) was varied between 1.4 and 2.4, GRR(AA) was set to 2×GRR(Aa)-1 (an additive effect), q was varied between 0.02 and 0.20, and ncc was varied between 500 and 3,000. To calculate the penetrances of each genotype at a disease locus, it was also necessary to specify the population prevalence of the disease; this was set to 1% for all simulated studies.
To simulate a case-control study, the following process was used:
-
Step 1.
From one of the FREGENE populations (all SNPs with MAF⩾0.005), a SNP with MAF between q-0.005 and q+0.005 was picked at random to be causative.
-
Step 2.
Two sequences (a diploid individual) were picked at random (with replacement) from the population.
-
Step 3.
The individual was assigned to the case set or control set according to the probability of having the disease, given his or her genotype at the causative SNP.
-
Step 4.
Steps 2 and 3 were repeated until ncc case sequences and ncc control sequences were sampled.
-
Step 5.
Only the 300 tSNPs were output.
Resampling from the population is not ideal, but we are limited by the size of population, which it is computationally feasible to simulate. The resampling may be thought of as performing an additional round of Wright-Fisher evolution with a sudden increase in population size or as there being unidentified consanguinity in the study. This approach has been used elsewhere.30
Results
We have implemented the algorithm as a program called “Margarita” and have assessed it with both simulated and real data sets involving thousands of individuals typed for hundreds of markers across megabase-scale regions. The performance of a mapping method may be measured according to three criteria: (1) power—the probability of obtaining a significant association signal in a region around a causative polymorphism, (2) localization—how accurately one can estimate the position of an untyped causative polymorphism, and (3) interpretation—the ability to estimate properties of an untyped causative polymorphism (in addition to its position), such as its frequency, which can then guide further investigation.
The power and localization of Margarita across a range of disease models were compared with those of two other methods: the single-marker χ2 test and the CLADH haplotype-clustering method.15 Single-marker and haplotype-based tests are those most commonly used in practice; coalescent methods such as LATAG20 are not computationally feasible for the scale of data we consider here. The single-marker χ2 test is often used in practice, and, for our simulations, we have selected tSNPs that capture much of the population variation, meaning that this test is not as “naive” as it may be when markers are chosen at random. From the many available haplotype-based methods, we chose to compare our method with CLADH because it is designed to be applied to megabase-scale regions, is computationally feasible, and has been shown to perform well against similar methods.31
To illustrate how our method can be used to interpret and dissect an association signal, we analyzed data from Ueda et al.’s study32 of association between CTLA4 (MIM 123890) and Graves disease (MIM 275000).
Results for a Simulated Suite of Case-Control Studies
We simulated case-control studies typed for 300 markers across a 1-Mb region, as described in the “Methods” section. These correspond to fine-mapping studies where one has detected or suspects a causative polymorphism in the region and wishes to finely localize and interpret that signal.
We first compared Margarita, CLADH, and the χ2 test, using a suite of 50 case-control studies with parameters GRR(Aa)=2, GRR(AA)=3, q=0.04, and ncc=2,000, sampled from the constant population with the full ascertainment tSNP set, and with the use of the true phased haplotype sequences with no missing data. The association structure for one of those studies is shown in figure 2A.
Figure 2. .

Analysis of a suite of case-control studies with disease parameters GRR(Aa)=2, GRR(AA)=3, q=0.04, and ncc=2,000, sampled from the constant population with the full ascertainment tSNP set. A, Association structure for a simulated case-control study. ▵ denotes the position of the (untyped) causative SNP. B,Probability of there being a significant association within an interval around the causative SNP. C and D, Markerwise P values at the marker closest to the causative SNP for each of the 50 studies. E, Cumulative distribution of distances between the association peak and the causative SNP. F, Distribution of estimated allele frequency.
All Margarita P values for the simulated studies were calculated by performing 10,000 permutations, and P values <.0001 were estimated by fitting extreme value distributions. Analysis of one case-control study (4,000 haplotype sequences of 300 SNPs) by use of Margarita on a 2.8-GHz Pentium IV processor requires 3–4 min to construct 1 ARG and 6 h to perform the mapping test with 10,000 permutations on 100 ARGs. The mapping test for Margarita is for marginal trees, which potentially change at each marker; therefore, we took the location of the typed marker to be the point location of the test. However, the branch that best segregates the cases and controls will be linked to that marker and may not correspond to it.
When using CLADH, the user is required to specify the number of SNPs in each haplotype window. We tried the range of window widths used in the CLADH article,15 and we report the best results obtained (using windows of size 5). All CLADH P values were calculated with 10,000 permutations, and we took the location of the typed SNP closest to the center of the window as the point location of the test. We now consider the mapping performance measures of power, localization, and interpretation, in turn.
Power
To determine power, we defined a window around the causative SNP and calculated the proportion of case-control studies with a significant signal (P⩽.05) within that window. Figure 2B shows the probability of detecting a markerwise and experimentwise significant association within a window around the untyped causative SNP. We are unable to report the experimentwise significances for CLADH, because it does not calculate these. When one considers markerwise significance (top three lines in fig. 2B), the χ2 test and CLADH have greater power than that of Margarita for windows >25 kb around the causative SNP. However, when one corrects for multiple testing, Margarita has greater power than that of the χ2 test (lower two lines). This difference arises because Margarita’s tests at adjacent SNPs are more strongly correlated through shared ancestry than are those of the χ2 test (see fig. 3), reducing the effective number of independent tests across the region.
Figure 3. .

Marginal tree correlation versus r2 LD for part of ENCODE region 7p15.2 from the phase I HapMap.38 Tree correlation is measured as the proportion of the n-3 nonequivalent, nonunary bipartitions of the leaves of each tree (defined by cutting branches) that are shared between trees at different positions.
In figure 2C and 2D, we report the markerwise P values for the test that is closest (according to its point location) to the untyped causative SNP in each of the 50 case-control studies. The P values attained by Margarita are typically stronger than those obtained by the other methods.
We have compared the false-positive rates of the three methods by counting the number of associations with markerwise P⩽.05 at a distance >250 kb from the untyped causative SNP. An association is counted when the signal breaks below the 0.05 cutoff and then returns above it. The mean number of such false-positive results for a case-control study from this suite is 0.70 for Margarita, 6.16 for CLADH, and 10.48 for the χ2 test. This may explain, in part, the apparent difference in markerwise power at longer distances (fig. 2B).
Localization
By localization, we mean how accurately we can estimate the position of the causative SNP. For each of the methods, we take the point location of the test with the strongest markerwise P value as the estimate of causative SNP location. Figure 2E shows that our method gives better localization than do CLADH and the χ2 test for this suite of studies.
Interpretation
In studies where the causative SNPs are untyped, it is useful to estimate properties of those SNPs, thus guiding the design of subsequent studies. For example, an estimate of causative-allele frequency (which one can also obtain with haplotype-clustering methods17) can be used to calculate the sample size required to achieve significance. To estimate this, we take the ensemble of marginal trees at the marker closest to the causative SNP and record the branch (bipartition) of each tree that shows the strongest disease association—we call this the “best cut.” For each tree, an estimate of causative-allele frequency can be obtained by calculating the frequency of chromosomes in the general population that fall under the best-cut branch.
Figure 2F shows the distribution of causative-allele frequencies as estimated by the ARGs constructed for this suite (causative-allele frequency 0.04). The median estimate is 0.036. Note that we report only frequency estimates from studies with a significant association signal. Additionally, we obtain a sample of estimated ancestral haplotypes from which the causative allele may have arisen (data not shown).
Results across a Range of Simulated Disease Models
So far, we have reported only the performances of the three methods for one suite of case-control studies—that is, under one disease model. In this section, we explore a range of models by varying each parameter (either GRR(Aa), the causative-allele frequency q, or the study size ncc) in turn while fixing the others at “default” values of GRR(Aa)=2, GRR(AA)=2×GRR(Aa)-1, q=0.04, and ncc=2,000. In all these simulations, we used the constant population with the full tSNP ascertainment scheme.
Figure 4A compares the power of Margarita and the power of the χ2 test to detect an experimentwise significant (P⩽.05) association within 100 kb of the untyped causative SNP. CLADH is excluded from this comparison because it does not calculate experimentwise P values. In a comparison of experimentwise P values, Margarita outperforms the χ2 test.
Figure 4. .

Power, localization, and interpretation for a range of disease models. Each point on the X-axis corresponds to a suite of 50 studies. Each of the disease parameters is varied between suites, whereas the other parameters are held at “default” values of GRR(Aa)=2, GRR(AA)=2×GRR(Aa)-1, q=0.04, and ncc=2,000. All studies are sampled from the constant population with the full ascertainment tSNP set. A, Probability of an experimentwise significant signal within 100 kb of the causative SNP (calculated as the proportion of studies in each suite that meet this criterion). B, Probability that the association peak is within 100 kb of the causative SNP. C, Estimated causative-allele frequency versus true frequency q.
Figure 4B shows the localization performance of the three methods. For the majority of disease models, Margarita outperforms both the χ2 test and CLADH.
Finally, figure 4C shows the median estimated causative-allele frequency for a range of suites with varying causative-allele frequency (we report only estimates from studies with a significant association signal). We compared the performance of Margarita with that of a simple haplotype approach. For this, we considered all windows of length up to 10 SNPs around the causative polymorphism. We tested each haplotype allele for association with the disease and used the frequency of the most strongly associated haplotype allele to estimate the frequency of the causative polymorphism. Margarita has a slight downward bias in its estimate, but, nevertheless, it is reasonable and outperforms the simple haplotype approach just described, which has a significant upward bias and a higher variance.
Results across a Range of Simulated Population Models and Ascertainment Schemes
For our final set of simulations, the disease model was fixed to GRR(Aa)=2, GRR(AA)=3, q=0.04, and ncc=2,000, whereas the data quality, population model, and SNP ascertainment scheme were varied.
In figure 5A, we examine the effect of missing and unphased data on the performance of our method. For this figure, the same suite of case-control studies (sampled from the constant population and with the full tSNP ascertainment scheme) were used, but with the sample output either as phased haplotype sequences, unphased genotype sequences, or phased sequences with 10% missing data. These results show that Margarita is robust against both these complications. We did not compare Margarita with CLADH because the latter requires phased haplotypes with no missing data.
Figure 5. .

Localization for different data, populations, and tSNP models. A, Performance on a suite of case-control studies with GRR(Aa)=2, GRR(AA)=3, q=0.04, and ncc=2,000, sampled from the constant population and with the full ascertainment tSNP set. Margarita is applied to this suite under three scenarios: when the data are phased, when they are unphased, and when they are phased but have 10% missing data. B, Performance on a suite of case-control studies sampled from the hot population (and with GRR(Aa)=2, GRR(AA)=3, q=0.04, and ncc=2,000). Performance is compared using three different tSNP ascertainment schemes (described in the “Methods” section).
In figure 5B, we evaluate the performance of Margarita for case-control studies sampled from a population simulated using a recombination hotspot model (the hot population described in the “Methods” section). Under this scenario, we see a performance increase for the χ2 test, compared with under the scenario of using the constant population (compare fig. 5B with 5A). However, it still performs less well than Margarita. The χ2 test has increased performance because recombination hotspots give rise to blocks of strong LD, resulting in tSNPs that capture more of the population variation.
In figure 5B, we also compare the effect of the tSNP ascertainment scheme on mapping performance. The same suite of case-control studies was used, but the samples were “typed” using each of the three tSNP selection schemes described in the “Methods” section. tSNP selection based on less complete data (specifically, when the causative polymorphism is not included in the data used to select tSNPs) results in significantly reduced performance of the χ2 test but has less of an effect on Margarita's performance. Furthermore, the SNP ascertainment scheme that is best for the χ2 test (full ascertainment) is not necessarily the best for Margarita (which seems to prefer markers with frequency ⩾0.05). For clarity, we did not plot the results from CLADH in figure 5; although consistent with the previous studies, the performance of CLADH tends to fall between Margarita and the χ2 test.
Results for Real Data
In this section, we analyze data from a fine-mapping study of association between polymorphisms in the region of the CTLA4 gene and an autoimmune disorder,32 showing how our method can be used to dissect an association signal. In the study by Ueda et al.,32 a 300-kb region (CD28-CTLA4-ICOS) was genotyped for 108 SNPs in 384 individuals with Graves disease and 652 controls. In their analysis, three association peaks were identified; moving from left to right in figure 6A, these peaks are at SNPs MH30, CT60, and CTBC217_1. By performing a regression analysis, they concluded that the causative variant is more likely to be around the CT60 peak than around the others. Furthermore, CT60 was found to be correlated to the expression of CTLA4 mRNA isoforms.
Figure 6. .

Analysis of the CTLA4 data. A, Association structure of the region. B, Distribution of estimated causative-allele frequency, by use of marginal trees at CT60. C, Test for allelic heterogeneity, by calculation of the proportion of inferred marginal trees at each position for which a chromosome appears under the branch that best segregates the cases and controls. D, Association structure for a subset of the CTLA4 data—only those chromosomes with the protective CT60 allele.
Since the data are unphased and there are missing genotype data, we used the ability of Margarita to infer these (in inferring 100 ARGs, we obtain 100 different phase resolutions, thus marginalizing over phase uncertainty when performing the mapping test). However, the data being unphased will present a hurdle for mapping methods that require phased haplotype sequences. One way to overcome this problem is to run a phasing algorithm33 on the data and then pass the result to the mapping method, as though it is the true phase resolution.34 To examine the effect of this, we also ran Margarita on the “best” phase resolution of the data after applying one run of the program PHASE.35
Figure 6A shows that CT60 has the strongest disease association in our analysis (when using both the PHASEd and the unphased data), which agrees with the analysis by Ueda et al.32 All Margarita P values in this section were calculated by performing up to 1 million permutations.
Margarita used on the unphased data gives a stronger association signal at CT60 (P≈2×10-6) than does Margarita used on the PHASEd sequences (P≈2×10-5). This result agrees with that of Morris et al.,34 who similarly show that a two-stage approach results in a loss of power compared with the handling of genotypes directly and marginalizing over unknown phase. Both Margarita analyses have lower significance than that of the χ2 test (P≈1.6×10-6), which would be expected if CT60 is indeed the causative polymorphism, a hypothesis that can be explored further by using the ARGs to dissect the association signal.
Figure 6B gives the distribution of the estimated susceptibility-allele frequency in the general population (calculated using the observation that Graves disease has population prevalence of 0.5%). The mean estimate for the causative allele in the cases and controls is 65% and 54%, respectively, corresponding to the G allele of CT60 (63% and 52% in cases and controls, respectively). This suggests that the bulk of the association signal at CT60 is due to susceptibility caused by CT60, or something extremely tightly linked to it.
However, in 43% of the inferred ARGs for the unphased data, Margarita is able to find internal branches of the marginal tree at CT60 that segregate the cases and controls with χ2 test P values ⩽∼10−7, with the strongest being ∼10−9; this suggests a second causative polymorphism. We therefore used the inferred ARGs to test explicitly for allelic heterogeneity. We took the 100 marginal trees for each marker and counted the number of times each chromosome appeared under the branch corresponding to the best partitioning of cases and controls—the best-cut branch. When a chromosome is under the best-cut branch, it means that, if there is a disease-causing allele at that position, then it is likely that the chromosome possesses it. Figure 6C shows this analysis for an illustrative sample of 167 case chromosomes (with phase inferred on the ARGs). For each marker and chromosome, the intensity of the plot represents the proportion of trees for which the chromosome is under the best cut. Case chromosomes 131–167 show a different pattern than do the others. They occur less frequently under the best cut at CT60 and more frequently under the best cut at CT53 and CTBC358, whereas case chromosomes 1–130 appear frequently under the best cut at CT60 but infrequently under the best cut at CT53 and CTBC358. There are other case chromosomes not associated with any of these loci (not shown). To test whether CT53 or CTBC358 are also susceptibility loci (or linked to susceptibility loci), we stratified the case-control population in three ways, as follows.
Only those chromosomes with the protective allele at CT60
We took the PHASEd chromosomes and removed all those with the CT60 susceptibility allele, running our analysis on the remaining 282 case chromosomes and 620 controls with the protective allele (fig. 6D). When the population is stratified in this way, the association signals at MH30 and CTBC217_1 collapse into the background, suggesting that the association signals at those locations are due to LD with CT60. Furthermore, there is an association peak at CT53 (markerwise χ2 test P≈5×10-5; Margarita P≈2×10-4). By use of Margarita, the estimated frequency of the causative allele (92% in the cases and 82% in the controls) matches that of the A allele at CT53 in this subpopulation (93% in the cases and 83% in the controls), suggesting that the A allele confers susceptibility on this CT60 background.
Only those chromosomes with the susceptibility allele at CT60
When we condition on the CT60 susceptibility allele, we obtain 486 case chromosomes and 684 control chromosomes. In this subpopulation, CT53 has a weak signal of association with the disease (markerwise χ2 test P≈.023; Margarita P≈.016). In contrast to the previous stratification, the A allele at CT53 is less frequent in the cases (2%) than in the controls (5%), suggesting that A may be protective on this haplotypic background. This reversal of the effect of CT53 dependent on CT60 status may explain why CT53 is not detected in simple analyses using the full data.
Only those individuals who are homozygous for the CT60 protective allele
To check that the CT53 association is not due to some spurious signal resulting from the selection of chromosomes on the basis of their inferred haplotype phase, we took the genotype sequences homozygous for the CT60 protective allele and ran Margarita on these unphased sequences. There are 102 case chromosomes and 300 controls, which give a weaker but still significant signal of association (markerwise χ2 test P≈.012; Margarita P≈.013). As expected, on this background the A allele of CT53 is the susceptibility allele.
These results suggest epistasis between CT60 and CT53, with the A allele at CT53 conferring susceptibility on a CT60 protective background but being protective on a CT60 susceptibility background. To test explicitly for epistasis between CT60 and CT53, we performed a logistic regression test for interaction36,37 and obtained P≈.004 for interaction effects over and above single-marker effects.
Given the small samples sizes of these subpopulations, further genotyping in more samples is required to determine whether the observed signal at CT53 is a true positive or an artifact of the data. Nevertheless, we have shown that it is possible to interrogate real data by use of inferred ARGs.
Discussion
Advantages of the Method
Association studies rely on LD to summarize the recombination history. However, LD statistics (such the r2 measure of LD, which is strongly related4 to the χ2 test) are not pure measures of recombination distance; they are affected by other factors, such as the relative timing of mutation events and nonrandom mating patterns. This confounds our ability to map disease loci.
Shifts in marginal tree topology, however, are entirely dependent on the positions of observable recombinations, so the degree of correlation between marginal trees is a better measure of (observable) recombination distance. Figure 3 illustrates this and helps explain the enhanced performance of our method over the χ2 test. Compared with r2, the correlation between adjacent trees is (1) tighter into the diagonal, giving finer localization and fewer false-positive results caused by distant markers being stochastically correlated; (2) stronger, because, when the causative SNP is untyped, there is a stronger association signal from adjacent markers, giving greater power; and (3) smoother in its decay—because tests are more correlated, we observe an improvement in experimentwise significance.
Furthermore, the inferred ARGs can be used to make useful inferences about causative polymorphisms in addition to their position. Taking the branches of the marginal trees that show the strongest clustering of cases beneath them, we can estimate the frequencies of causative alleles, the ancestral haplotypes on which they arose, and which cases are caused by each allele, giving a clustering of the cases and possibly identifying allelic, locus, or phenotypic heterogeneity. These three estimates are particularly useful for designing follow-up studies and provide important advantages of our approach.
Compared with haplotype-clustering methods, our approach naturally handles rare haplotypes and unphased data and does not require the specification of nonrecombining haplotype blocks. It also gives more-precise fine mapping. Using ARGs is not the same as using haplotype-based methods; recombinations subsequent to the formation of the haplotype interval can give additional mapping information, which our method uses. Compared with other ARG-based methods, it is faster. We are able to build ARGs for large data sets, involving thousands of individuals typed for hundreds of SNPs, and it remains computationally feasible when the data are unphased. By use of a cluster of processors, it is feasible to window the analysis over the whole genome and thus apply this approach to whole-genome case-control studies.
We now consider some other potential features of our approach that have not been tested in this article. Our method could be adapted to handle allelic heterogeneity by allowing multiple causative mutations on each marginal tree when the mapping test is performed, as in LATAG20.
Along with various types of heterogeneity, cryptic population substructure is another confounder of many population genetic analyses. One may, however, expect the subpopulations to tend to cluster in the ARG, which makes identification of substructure and analyses that allow for this possible.
Because our method can handle missing and unphased data by resolving both on the ARG, it may be possible to combine samples from different association studies that do not necessarily have overlapping markers. The missing SNPs in each study could be imputed, thus merging the studies in a principled way—according to population history. In particular, it should be possible to include additional densely typed samples, such as the HapMap samples.38 This would allow imputed estimation in the cases and controls of the values of SNPs that had been typed in the additional samples and allow explicit testing of direct association with those SNPs as well as the tree-based analysis described here.
Similarly, it may be possible to combine and analyze data from multiple family-based linkage studies. Known relationships between individuals could be enforced as rules in the ARG inference algorithm, whereas the founders of pedigrees could be allowed to coalesce and recombine according our standard ARG inference rules. In this way, it may be possible to extract more information from previous studies.
Limitations of the Method
By avoiding inference under the coalescent-with-recombination model, we have discarded an understood probabilistic framework that lends itself naturally to a Bayesian formulation. Consequently, we do not attach probabilities to the inferred ARGs but consider them all to be equally likely. Nor have we provided a strong definition of what “plausible” ARG inference means. The heuristics described in the “Methods” section suggest some aspects of why our inferred ARGs are plausible, but providing justification is that the inferred ARGs work for disease mapping.
A second limitation, which is probably true of all the more sophisticated mapping methods, is that, when the causative mutations are typed, our method is unlikely to provide a significant mapping advantage (in terms of localization and power) over a single-marker test. We might expect this to be typically the case in the future, if it becomes routine to fully resequence individuals.39 Nevertheless, resequencing would not render ARG inference redundant, since we suggest that there are potential applications to other population genetic analyses, such as the identification of selection, population substructure, and estimation of the ages of alleles.
Acknowledgments
We are grateful to Clive Hoggart and David Balding, for providing the FREGENE simulated population, to Neil Walker and John Todd, for providing the CTLA4 data, and to Chris Lowe and Joanna Howson, for commenting on our analysis of epistasis. We also thank Simon Tavaré, Ralph McGinnis, and Lachlan Coin, for helpful discussions, and two anonymous reviewers whose comments helped us improve the manuscript. This work was supported by the Wellcome Trust.
Web Resources
The URLs for data presented herein are as follows:
- BARGEN, http://www.ebi.ac.uk/projects/BARGEN (for downloading the FREGENE simulator)
- Margarita, http://www.sanger.ac.uk/Software/analysis/margarita (for downloading the Java program Margarita, plus high-resolution versions of the figures from this article)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CTLA4 and Graves disease)
References
- 1.Cordell HJ, Clayton DG (2005) Genetic association studies. Lancet 366:1121–1131 10.1016/S0140-6736(05)67424-7 [DOI] [PubMed] [Google Scholar]
- 2.Palmer LJ, Cardon LR (2005) Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet 366:1223–1234 10.1016/S0140-6736(05)67485-5 [DOI] [PubMed] [Google Scholar]
- 3.Devlin B, Risch N (1995) A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311–322 10.1006/geno.1995.9003 [DOI] [PubMed] [Google Scholar]
- 4.Pritchard JK, Przeworski M (2001) Linkage disequilibrium in humans. Am J Hum Genet 69:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordborg M, Tavaré S (2002) Linkage disequilibrium: what history has to tell us. Trends Genet 18:83–90 10.1016/S0168-9525(02)02557-X [DOI] [PubMed] [Google Scholar]
- 6.McVean GA (2002) A genealogical interpretation of linkage disequilibrium. Genetics 162:987–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffiths RC, Marjoram P (1997) An ancestral recombination graph. In: Donnelly P, Tavaré S (eds) Progress in population genetics and human evolution. Springer Verlag, New York, pp 257–270 [Google Scholar]
- 8.Hudson RR (1983) Properties of a neutral allele model with intragenic recombination. Theor Popul Biol 23:183–201 10.1016/0040-5809(83)90013-8 [DOI] [PubMed] [Google Scholar]
- 9.Nordbord M (2001) Coalescent theory. In: Balding DJ, Bishop MJ, Cannings C (eds) Handbook of statistical genetics. John Wiley & Sons, Chichester [Google Scholar]
- 10.Stephens M (2001) Inference under the coalescent. In: Balding DJ, Bishop MJ, Cannings C (eds) Handbook of statistical genetics. John Wiley & Sons, Chichester [Google Scholar]
- 11.McVean GA, Cardin NJ (2005) Approximating the coalescent with recombination. Philos Trans R Soc Lond B Biol Sci 360:1387–1393 10.1098/rstb.2005.1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song YS, Lyngsø R, Hein J (2006) Counting all possible ancestral configurations of sample sequences in population genetics. IEEE/ACM Trans Comput Biol Bioinform 3:239–251 [DOI] [PubMed] [Google Scholar]
- 13.Templeton AR, Boerwinkle E, Sing CF (1987) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in Drosophila. Genetics 117:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molitor J, Marjoram P, Thomas D (2003) Fine-scale mapping of disease genes with multiple mutations via spatial clustering techniques. Am J Hum Genet 73:1368–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durrant C, Zondervan KT, Cardon LR, Hunt S, Deloukas P, Morris AP (2004) Linkage disequilibrium mapping via cladistic analysis of single-nucleotide polymorphism haplotypes. Am J Hum Genet 75:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Templeton AR, Maxwell T, Posada D, Stengard JH, Boerwinkle E, Sing CF (2005) Tree scanning: a method for using haplotype trees in phenotype/genotype association studies. Genetics 169:441–453 10.1534/genetics.104.030080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waldron ER, Whittaker JC, Balding DJ (2006) Fine mapping of disease genes via haplotype clustering. Genet Epidemiol 30:170–179 10.1002/gepi.20134 [DOI] [PubMed] [Google Scholar]
- 18.Larribe F, Lessard S, Schork NJ (2002) Gene mapping via the ancestral recombination graph. Theor Popul Biol 62:215–229 10.1006/tpbi.2002.1601 [DOI] [PubMed] [Google Scholar]
- 19.Morris AP, Whittaker JC, Balding DJ (2002) Fine-scale mapping of disease loci via shattered coalescent modeling of genealogies. Am J Hum Genet 70:686–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zollner S, Pritchard JK (2005) Coalescent-based association mapping and fine mapping of complex trait loci. Genetics 169:1071–1092 10.1534/genetics.104.031799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers SR, Griffiths RC (2003) Bounds on the minimum number of recombination events in a sample history. Genetics 163:375–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gusfield D, Eddhu S, Langley C (2004) Optimal, efficient reconstruction of phylogenetic networks with constrained recombination. J Bioinform Comput Biol 2:173–213 10.1142/S0219720004000521 [DOI] [PubMed] [Google Scholar]
- 23.Song YS, Hein J (2005) Constructing minimal ancestral recombination graphs. J Comput Biol 12:147–169 10.1089/cmb.2005.12.147 [DOI] [PubMed] [Google Scholar]
- 24.Lyngsø R, Song YS, Hein J (2005) Minimum recombination histories by branch and bound. Proceedings of Workshop on Algorithms in Bioinformatics 2005, Lecture Notes in Computer Science 3692:239–250 [Google Scholar]
- 25.Gusfield D (1991) Efficient algorithms for inferring evolutionary trees. Networks 21:19–28 [Google Scholar]
- 26.Griffiths RC, Tavaré S (1995) Unrooted genealogical tree probabilities in the infinitely-many-sites model. Math Biosci 127:77–98 10.1016/0025-5564(94)00044-Z [DOI] [PubMed] [Google Scholar]
- 27.Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dudbridge F, Koeleman BP (2004) Efficient computation of significance levels for multiple associations in large studies of correlated data, including genomewide association studies. Am J Hum Genet 75:424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoggart CJ, Clark T, Lampariello R, De Iorio M, Whittaker J, Balding D (2005) FREGENE: software for simulating large genomic regions. Technical Report, Department of Epidemiology and Public Health, Imperial College, University of London, London [Google Scholar]
- 30.de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D (2005) Efficiency and power in genetic association studies. Nat Genet 37:1217–1223 10.1038/ng1669 [DOI] [PubMed] [Google Scholar]
- 31.Bardel C, Danjean V, Hugot JP, Darlu P, Genin E (2005) On the use of haplotype phylogeny to detect disease susceptibility loci. BMC Genet 6:24 10.1186/1471-2156-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, et al (2003) Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423:506–511 10.1038/nature01621 [DOI] [PubMed] [Google Scholar]
- 33.Marchini J, Cutler D, Patterson N, Stephens M, Eskin E, Halperin E, Lin S, Qin ZS, Munro HM, Abecasis GR, Donnelly P (2006) A comparison of phasing algorithms for trios and unrelated individuals. Am J Hum Genet 78:437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris AP, Whittaker JC, Balding DJ (2004) Little loss of information due to unknown phase for fine-scale linkage-disequilibrium mapping with single-nucleotide-polymorphism genotype data. Am J Hum Genet 74:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cordell HJ (2002) Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet 11:2463–2468 10.1093/hmg/11.20.2463 [DOI] [PubMed] [Google Scholar]
- 37.Macgregor S, Khan IA (2006) GAIA: an easy-to-use Web-based application for interaction analysis of case-control data. BMC Med Genet 7:34 10.1186/1471-2350-7-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balding D (2005) The impact of low-cost, genome-wide resequencing on association studies. Hum Genomics 2:79–80 [DOI] [PMC free article] [PubMed] [Google Scholar]