

Abstract
One of the most striking features of several X-ray structures of CoA-independent ALDHs (aldehyde dehydrogenases) in complex with NAD(P) is the conformational flexibility of the NMN moiety. However, the fact that the rate of the acylation step is high in GAPN (non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase) from Streptococcus mutans implies an optimal positioning of the nicotinamide ring relative to the hemithioacetal intermediate within the ternary GAPN complex to allow an efficient and stereospecific hydride transfer. Substitutions of serine for invariant Thr244 and alanine for Lys178 result in a drastic decrease of the efficiency of hydride transfer which becomes rate-limiting. The crystal structure of the binary complex T244S GAPN–NADP shows that the absence of the β-methyl group leads to a well-defined conformation of the NMN part, including the nicotinamide ring, clearly different from that depicted to be suitable for an efficient hydride transfer in the wild-type. The ∼0.6-unit increase in pKapp of the catalytic Cys302 observed in the ternary complex for both mutated GAPNs is likely to be due to a slight difference in positioning of the nicotinamide ring relative to Cys302 with respect to the wild-type ternary complex. Taken together, the data support a critical role of the Thr244 β-methyl group, held in position through a hydrogen-bond interaction between the Thr244 β-hydroxy group and the ϵ-amino group of Lys178, in permitting the nicotinamide ring to adopt a conformation suitable for an efficient hydride transfer during the acylation step for all the members of the CoA-independent ALDH family.
Keywords: acylation step, aldehyde dehydrogenase (ALDH), hydride transfer, nicotinamide conformation, Streptococcus mutans, X-ray structure
Abbreviations: ALDH, aldehyde dehydrogenase; GAPN, non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase; G3P, glyceraldehyde 3-phosphate; 2PDS, 2,2′-dipyridyl disulfide; r.m.s.d., root mean square deviation
INTRODUCTION
Non-phosphorylating ALDHs (aldehyde dehydrogenases) belong to a family of phylogenetically and structurally related enzymes that catalyse the NAD(P)-dependent oxidation of a wide variety of aldehydes to their corresponding non-activated or CoA-activated acids. ALDHs are known to participate in many biological functions, such as intermediary metabolism, detoxification, osmotic protection and cellular differentiation. They share a similar chemical mechanism involving two major steps: first, an acylation step common to both families and, second, a deacylation step that differs by the nature of the acyl acceptor. The acylation step involves the formation of a thiohemiacetal intermediate via nucleophilic attack of the catalytic Cys302 on the aldehydic function followed by an oxidoreduction process leading to the formation of a thioacylenzyme intermediate and NAD(P)H. (The amino acid numbering used for the biochemical and structural data is that defined by Wang and Weiner [1], and this nomenclature has been used for all biochemical studies performed on members of the ALDH family.) The deacylation step includes the nucleophilic attack of a water or CoA molecule on the thioacylenzyme intermediate and release of the non-activated or CoA-activated acids. Moreover, previous studies have highlighted major differences in the kinetic mechanism of ALDHs depending upon the nature of the deacylation step. In CoA-independent ALDHs, kinetic data support an ordered sequential mechanism in which NAD(P)H dissociates last [2,3], whereas CoA-dependent ALDHs exhibit a Ping Pong mechanism in which the release of the reduced cofactor occurs before the trans-thioesterification step [4,5].
Mechanistic aspects were studied extensively for CoA-independent ALDHs, and several invariant residues were shown to be critical for the chemical mechanism [1,6–8]. In addition, evidence has been provided for the chemical activation of catalytic Cys302 upon cofactor binding to GAPN (non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase) from Streptococcus mutans through a local conformational rearrangement which has been shown to be strongly kinetically favoured by substrate binding to the binary complex GAPN–NADP [9]. Moreover, enzymatic and structural studies on GAPN and other ALDHs have highlighted the essential roles of: (i) an oxyanion hole composed of the side chain of invariant Asn169 and the main-chain nitrogen of Cys302 that allows an efficient hydride transfer without base catalyst assistance [10–13], and (ii) invariant Glu268 in the rate-limiting hydrolysis step through activation and orientation of the attacking water molecule [2]. Additionally, numerous crystal structures of CoA-independent ALDHs have already been solved in the presence of NAD(P). One of the most striking features of many ALDH structures is the conformational flexibility of the NMN moiety of the cofactor, which is seen as having missing or weak electron density (see, e.g., [11,14–15]). However, two discrete conformations, compatible with the two-step catalytic mechanism, were described for class 1 and class 2 ALDHs [10,16]. In the first conformation, the NMN moiety, in particular that of the nicotinamide ring, occupies a position suitable for hydride transfer, but not for deacylation because it would sterically exclude catalytic Glu268 from playing its role in the hydrolytic process. The fact that the cofactor remains bound to the enzyme along the two-step catalytic mechanism, strongly suggested that movement of the reduced NMN (NMNH) is a prerequisite for completion of the second half of the reaction. The second conformation in which the NMN moiety adopts a contracted position was hypothesized to be suitable for hydrolysis. This view was supported by comparison of human ALDH2 structures in complex with either NAD or NADH, which provided arguments in favour of a conformational preference for the oxidized and reduced cofactors [17]. Recently, the first structural evidence for a conformational isomerization of the cofactor during the catalytic cycle of CoA-independent ALDH was put forward by the crystal structure of a GAPN thioacylenzyme intermediate–NADPH complex [18]. This structure revealed that the reduced cofactor adopts a new conformation with a flip of the NMNH moiety which has been proposed to be suitable for the deacylation step for all members of the CoA-independent ALDH family. In contrast, the key question about the positioning of the nicotinamide ring during the acylation step and the structural factors involved remains to be addressed. In the case of GAPN from S. mutans, inspection of the three-dimensional structure of the binary complex GAPN–NADP revealed a poorly defined electron density around the NMN portion. The fact that the rate of the acylation step is high (>500 s−1) is, however, indicative of a defined positioning of the nicotinamide ring relative to the transient hemithioacetal intermediate competent for an efficient hydride transfer [2]. In the conformation depicted to be suitable for an efficient hydride transfer in CoA-independent ALDHs, including GAPN, the nicotinamide ring is positioned between Cys302 and the β-methyl group of the invariant Thr244 residue, each at a distance of ∼3.5 Å (1 Å=0.1 nm) from the ring [10,11,17]. This suggested a major role of the β-methyl group of Thr244 in the positioning of the nicotinamide ring within the covalent ternary complex. Such an assumption is also supported by the fact that the Thr244 side chain could be held in position through hydrogen-bond interaction between its β-hydroxy group and the ϵ-amino group of Lys178, a residue highly conserved in all known primary structures of CoA-independent ALDHs.
In the present study, the catalytic properties of the T244S, T244V and K178A mutated GAPNs were determined. These substitutions result in a drastic effect on the acylation step, which becomes rate-limiting for the overall reaction in contrast with the wild-type for which the deacylation step is rate-limiting. Moreover, the crystal structure of a binary complex T244S GAPN–NADP shows that the NMN moiety of the NADP molecule is now well defined in the electron-density maps. The nicotinamide ring adopts a unique conformation clearly different from that hypothesized to be operative in the ALDH–GAPN acylating ternary complex [10,15–17]. Taken together, these results support a critical role of Thr244, in particular of its β-methyl group, in permitting the nicotinamide ring to adopt a conformation suitable for efficient hydride transfer. All the results are discussed in relation to available data on the CoA-independent ALDH family and for their relevance to the two-step catalytic mechanism.
EXPERIMENTAL
Production and purification of mutated GAPNs from S. mutans, and materials
T244S, T244S/C382S, T244V and K178A mutated GAPNs were produced as described previously [9]. For purification, cells were harvested by centrifugation at 3000 g for 20 min, resuspended in 50 mM potassium phosphate buffer, pH 8.2 (buffer A) and disrupted by sonication for 5 min using a Branson Sonifier 250. The extract was clarified by centrifugation at 15000 g for 90 min, and solid ammonium sulfate was added to either 40% saturation for T244S and T244S/C382S GAPNs or 30% saturation for T244V and K178A. The precipitate was removed by centrifugation at 15000 g for 90 min, and the supernatant was applied to a high-performance phenyl-Sepharose column using a FPLC system (Amersham Biosciences) equilibrated previously with buffer A containing 1 M ammonium sulfate. Following equilibration, mutated GAPNs were eluted using a descending gradient (1–0 M) of ammonium sulfate at 5 ml·min−1. Fractions containing mutated GAPNs were pooled, concentrated and dialysed exhaustively against buffer A and applied to a Q-Sepharose column equilibrated with buffer A. After washing with buffer A, mutated GAPNs were eluted using a 0–1 M KCl gradient at 5 ml·min−1. At this stage, mutated GAPNs were considered to be pure as judged by SDS/7.5% PAGE and by electrospray-MS analysis. T244V and K178A GAPNs were isolated as apo forms, whereas 0.8 mol of NADP remained bound per tetramer of T244S and T244S/C382S GAPNs. Enzyme concentrations were determined spectrophotometrically as the apo form by using a molar absorption coefficient of 2.04×105 M−1·cm−1 at 280 nm [9] and are expressed in molarity. All other materials were reagent-grade or better and were used without further purification. NADP (Roche) was dissolved in water, and the stock concentration was determined spectrophotometrically by using a molar absorption coefficient of 18000 M−1·cm−1 at 259 nm. D,L-G3P (G3P is glyceraldehyde 3-phosphate) or D-G3P (Sigma) was obtained by hydrolysing D,L-G3P diethyl acetal or D-G3P diethyl acetal respectively, according to the manufacturer's instructions, and its concentration was assessed enzymatically using wild-type GAPN.
Quantification of NADP content
The NADP content of purified GAPNs was quantified enzymatically after addition of 0.2 mM D,L-G3P to 1.25 μM GAPN by following the appearance of NADPH on a SAFAS flx spectrofluorometer. Fluorescence measurements were carried out in 50 mM potassium phosphate buffer at pH 8.2. The excitation wavelength was set at 358 nm, and spectra were recorded in the range 380–540 nm. Maximum emission intensity was observed at 460 nm.
Steady-state kinetic analyses
Initial rate measurements were carried out on a Kontron Uvikon 933 spectrophotometer by following the appearance of NADPH at 340 nm. Determination of the kinetic parameters was carried out at the optimum pH of 8.2 in 50 mM Tes buffer and 5 mM 2-mercaptoethanol. The temperature of the solutions was maintained at 25 °C by thermostatically controlled sample holders using a circulating water bath for all measurements. Catalytic constants kcat and Km were determined by measuring steady-state velocity at various D,L-G3P and NADP concentrations under conditions where no inhibitory effect was observed either with NADP or D,L-G3P. Data were plotted according to the Michaelis–Menten equation using least-squares regression analysis.
Deuterium isotope effects
D-[1-2H]G3P was prepared as described previously [19] and its concentration was determined enzymatically. Isotopic effects on T244S and K178A GAPNs were measured in 50 mM Tes buffer, pH 8.2, and 5 mM 2-mercaptoethanol by direct comparison of the initial velocities measured with D-G3P and D-[1-2H]G3P at concentrations of 0.1 and 0.2 mM for both mutated GAPNs in the presence of NADP at 25 °C.
pH-dependence of the steady-state rate constant of K178A and T244S GAPNs
The pH-dependence of the steady-state rate constant, kcat, which was shown to be associated with the rate-limiting acylation step (see the Results section), was studied over the pH range 5.25–8.75 or 5.25–9.00 in a mixed reaction buffer consisting of 120 mM Tris, 30 mM imidazole and 30 mM ethanoic (acetic) acid (pH-adjusted with NaOH or HCl), adjusted with 3 M NaCl to a constant ionic strength of 0.15 M. The apparent pKa parameters governing the pH-dependence of kcat were extracted from kobs against pH profiles and analysed as described by Marchal and Branlant [9].
Kinetic reactions with 2PDS (2,2′-dipyridyl disulfide)
Owing to the high reactivity of cysteine residues in all mutated GAPNs, kinetic measurements were carried out on a SX18MV-R Applied Photophysics stopped-flow apparatus (Leatherhead) at 25 °C, under pseudo-first-order conditions in 30 mM ethanoic acid, 30 mM imidazole and 120 mM Tris buffer at a constant ionic strength of 0.15 M over the pH range 5.50–8.75 for T244S and T244/C382S GAPNs and 5.75–9.25 for K178A GAPN. The pH range was used to study the pH stability of the mutated GAPNs. One syringe contained the apo or apo-like form of GAPN, and the other contained 2PDS. (In contrast with the term ‘holo form’, the term ‘apo-like form’ indicates here an apo form saturated by NADP but in which the cofactor has not yet induced the local conformational rearrangement of the active site, including the decrease of the pKa of Cys302 and the loss of the salt-bridge between Glu268 and Arg459 [9,15].) The reaction was monitored at 343 nm with 2.5 μM GAPN and 200 μM 2PDS and pyridine-2-thione was quantified using an molar absorption coefficient of 8080 M−1·cm−1. The pseudo-first-order kobs values were determined at each pH by fitting the absorbance A at 343 nm against time t to a monophasic expression (eqn 1):
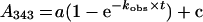 |
(1) |
where kobs is the observed rate constant, a is the burst magnitude and c represents the value of the ordinate intercept, or to a biphasic expression (eqn 2):
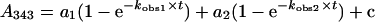 |
(2) |
where kobs1 and kobs2 are the observed rate constants for the fast and the slower phase respectively, a1 and a2 are the relative amplitude values for the two phases, and c represents the value of the ordinate intercept.
The second-order rate constants, k2, were determined at each pH value and data were analysed as described by Marchal and Branlant [9].
Crystallization, data collection and processing
Crystals of the T244S GAPN with NADP were obtained using the hanging-drop vapour-diffusion method at 293 K. A 2–6 μl volume of a 5 mg·ml−1 protein solution in 50 mM imidazole buffer at pH 6.8 was mixed with 1–2 μl of reservoir solution composed of 1.9–2.0 M ammonium sulfate, 0.1 M Hepes buffer, pH 7.5, and 2% PEG [poly(ethylene glycol)] 400 [12,15]. The crystals grew within a few days. A 2.50 Å resolution data set was collected at 100 K at the synchrotron source in Trieste. The crystals belong to the space group P21212, with unit cell dimensions a=143.7 Å, b=154.4 Å and c=115.0 Å. Diffracted data were processed using the HKL crystallographic data reduction package [20]. Statistics of the data set are summarized in Table 1.
Table 1. Data collection and refinement statistics.
Values in parentheses refer to the outermost resolution shell.
| (a) Data collection | |
|---|---|
| Parameter | Value |
| Space group | P21212 |
| a (Å) | 143.7 |
| b (Å) | 154.4 |
| c (Å) | 115.0 |
| Z | 4 |
| Resolution (Å) | 2.5 (2.59–2.50) |
| Temperature (K) | 100 |
| Total reflections | 323307 |
| Independent reflections | 78424 |
| Completeness (%) | 88.2 (90.1) |
| R-sym | 10.4 (37) |
| I/σ(I) | 11.7 (4.5) |
| (b) Refinement | |
| Parameter | Value |
| R-factor (%) | 17.6 |
| R-free (%) | 22.7 |
| R.m.s.d. from ideal geometry: | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.26 |
| Average B-factor (Å2) | |
| Main chain | 24.3 |
| Side chains | 27.9 |
| Water molecules | 26.4 |
| NADP (AMP moiety) | 28.98 |
| NADP (NMN) | 32.87 |
Phasing, refinement and quality of the model
The structure of T244S GAPN was solved by difference Fourier techniques, using the wild-type apo-like structure as a starting model (PDB code 2EUH) [15].
Several cycles of rigid-body refinement carried out with CNS [21] led to a decrease in the crystallographic R-factor and R-free values to 0.23 and 0.28 respectively. The analysis of the (2Fo−Fc) electron-density map at this stage of refinement was very well defined for all the structure, including the mutated residue. The ordered water molecules were added automatically and checked individually. Moreover, seven sulfate ions were added (one in each of the A and C monomers, two in the B monomer and three in the D monomer) and refined with occupancy factors ranging from 0.5 to 1. After refinement (CNS) and manual rebuilding (TurboFrodo; Silicon Graphics) the final crystallographic R-factor and R-free values calculated for the 78424 independent reflections (in the 20.00–2.50 Å resolution range) were 0.176 and 0.227 respectively. The stereochemical quality of the model was assessed using PROCHECK [23]. The most favoured and additionally allowed regions of the Ramachandran plot contained 99.6% of the nonglycine residues. The final model consisted of 14362 non-hydrogen atoms, 1109 water molecules and seven sulfate ions coming from the crystallization solution. The refined structure presented a good geometry with r.m.s.d. (root mean square deviation) from ideal bond lengths and angles of 0.005 Å and 1.26° respectively. The average temperature factor (B) for all atoms was 24.6 Å2. The statistics for refinement and model geometry are summarized in Table 1. Figure 1(A) was drawn using MOLMOL [24] and Figure 1(B) with BOBSCRIPT [25]
Figure 1. Active site of T244S GAPN.

(A) Superimposition of the active-site regions of the apo-like wild-type enzyme (in grey) and of the T244S GAPN (in black). The conformation of the NMN moiety in the wild-type structure is the one described to be suitable for efficient hydride transfer [9,17]. Single letter amino acid codes are used. (B) Stereo view of the cofactor molecule in the (2Fo−Fc) electron-density map (contour level: 1σ).
RESULTS
Rationale for the mutations
As indicated in the Introduction, we have postulated that Thr244 and its potential interaction with Lys178 could be critical in the correct positioning of the NMN moiety of the cofactor during acylation, so that hydride transfer can occur with an optimum efficiency. In order to validate this assumption, the roles of Thr244 and Lys178 have been probed by site-directed mutagenesis. Introduction of a residue such as serine at position 244 would provide experimental support for the role of the van der Waals interaction between the β-methyl group of Thr244 and the nicotinamide ring, whereas T244V and K178A substitutions would assess the role of the Lys178/Thr244 hydrogen-bond interaction in the correct positioning of the β-methyl group of Thr244 relative to the nicotinamide ring of the cofactor. In the apo and apo-like forms of GAPN, Cys302 and Cys382 were shown previously to be reactive towards 2PDS [9]. Therefore, to assign pKapp and k2 values to Cys302 in the apo-like form of T244S GAPN, Cys382 has been replaced by a serine residue.
Determination of the pKapp values and reactivities of the cysteine residues in the apo form of K178A GAPN and apo-like forms of K178A, T244S and T244S/C382S GAPNs with 2PDS
For all mutated GAPNs, the reaction with 2PDS under native conditions obeyed pseudo-first-order kinetics with formation of 2 mol of pyridine-2-thione per subunit as determined from the A343 value, except that only 1 mol of 2PDS reacted per subunit of T244S/C382S GAPN. This is in agreement with what was described for the wild-type in which two residues, i.e. Cys302 and Cys382 were shown to be reactive per monomer [9]. For the K178A GAPN apo form, the two cysteine residues exhibited distinct k2 constants. The pH–k2 profile for each thiolate showed sigmoidal behaviour with pKapp values of 8.80 and 9.15 and k2 values of 3.3×105 M−1·s−1 and 3.05×104 M−1·s−1 respectively (Figure 2A and Table 2). The high k2 values indicated that both cysteine residues are accessible within the active site of K178A GAPN, in contrast with what was observed for the wild-type apo form [9]. The high accessibility of Cys302 is probably not catalytically relevant, but rather is another example of the great conformational flexibility exhibited by this residue within the active site [9,26]. The apo form of K178A GAPN incubated at 4 °C in the presence of saturating concentrations of NADP for 5 h at pH 5.5 under conditions where the conformational rearrangement of the active site was shown to occur in the apo wild-type, displayed a similar pH–k2 profile to that described for the apo K178A GAPN with pKapp values of 8.85 and 9.25 and k2 values of 3.0×105 and 2.9×104 M−1·s−1 respectively (curves not shown, see Table 2). Since the apo form of T244S GAPN could not be obtained irrespective of the experimental procedure used (results not shown), titration by 2PDS was only performed on the apo-like form of T244S GAPN after incubation for 5 h at 4 °C and pH 5.5 in the presence of saturating concentrations of NADP. A pH–k2 profile similar to that of apo and apo-like forms of K178A GAPN was obtained with pKapp values of 8.5 and 9.0 and k2 values of 3×103 and 9.5×104 M−1·s−1 respectively (Figure 2B, inset, and Table 2). The pH–k2 curve of the apo-like form of T244S/C382S GAPN fitted only to a monosigmoidal profile with a pKapp value of 9.0 and a k2 value of 2×105 M−1·s−1 (Figure 2B and Table 2). Such a result strongly suggests that the pKapp of 9.0 determined in the T244S GAPN represents that of Cys302.
Figure 2. pH-dependence of the second-order rate constant, k2, for the reaction of 2PDS with apo K178A and apo-like T244S and T244S/C382S GAPNs.

Kinetics were determined at 25 °C as indicated in the Experimental section. The GAPN and 2PDS concentrations were 2.5 μM and 200 μM respectively. Rate constants, kobs, were determined using non-linear regression analysis, and second-order rate constants, k2, were calculated by dividing the kobs values by the concentration of 2PDS. Transients obtained on the apo K178A and apo-like T244S GAPNs were analysed according to eqn (2), reflecting the contribution of Cys302 and Cys382. Transients obtained on the apo-like T244S/C382S GAPN were analysed according to eqn (1). (A) Apo K178A GAPN: both data sets were fitted against a single pKa model, which provided pKapp values of 8.80 and 9.15, and k2 values of 3.3×105 M−1·s−1 (●) and 3.05×104 M−1·s−1 (▲) respectively. (B) Main panel: data sets obtained on the apo-like T244S/C382S GAPN were fitted against a single pKa model, which provided pKapp and k2 values of 9.0 and 2×105 M−1·s−1 respectively. Inset: data sets obtained on the apo-like T244S GAPN were fitted against a single pKa model, which provided pKapp values of 8.50 and 9.00 and k2 values of 3.3×103 M−1·s−1 (▲) and 9.5×104 M−1·s−1 (■) respectively. S.E.M. on each experimental point was less than 5%.
Table 2. Parameters for the pH-dependence of the second-order rate constants for the reaction of 2PDS with K178A, T244S and T244S/C382S GAPNs.
The experimental conditions are described in the Experimental section. Transients obtained on the apo and apo-like K178A and apo-like T244S GAPN were best described by a double-exponential expression reflecting the contribution of Cys302 and Cys382 and data were analysed according to eqn (2). For the apo-like T244S/C382S GAPN, experimental data were best described by a mono-exponential expression according to eqn (1). The second-order rate constants at each pH were fitted to a monosigmoidal equation to determine the pKapp values in Sigmaplot (SPSS). The error estimates are those provided by the program. ND, not determined.
| GAPN | k2 (M−1·s−1) | pKapp |
|---|---|---|
| Apo-GAPNs | ||
| Wild-type* | 1350±28 | 8.84±0.04 |
| C382A* | 225±9 | 8.49±0.07 |
| C302A* | 588±14 | 9.07±0.02 |
| K178A | (3.30±0.25)×105 | 8.80±0.07 |
| (3.05±0.25)×104 | 9.15±0.15 | |
| Holo-GAPN | ||
| Wild-type* | >2×105 | ND |
| Wild-type† | 182±2 | 6.1±0.2 |
| Apo-like-GAPNs‡ | ||
| K178A§ | (3.0±0.1)×105 | 8.85±0.05 |
| (2.9±0.2)×104 | 9.25±0.05 | |
| T244S§ | (3.0±0.3)×103 | 8.5±0.1 |
| (9.5±2)×104 | 9.0±0.2 | |
| T244S/C382S§ | (2.0±0.4)×105 | 9.0±0.1 |
*From [9].
†This pKapp value of 6.1 for Cys302 was determined using iodoacetamide as a kinetic probe (from [9]).
‡Apo-like forms refer to GAPN/NADP binary complexes after incubation at 4 °C of the apo forms (or partially apo forms in the case of T244S and T244S/C382S GAPNs) in the presence of saturating concentration of NADP for 5 h at pH 5.5 under conditions where the conformational rearrangement occurred in the wild-type enzyme (from [9]) but not in the mutated GAPNs.
§Although no additional experimental data can be obtained beyond the inflection point of the curves, non-linear regression analysis provides accurate pKa values.
Kinetic properties of T244S, T244V and K178A GAPNs
In a previous study, it was shown that acylation and deacylation steps of wild-type GAPN could be kinetically resolved and that deacylation was rate-limiting under steady-state conditions [2]. It was also demonstrated that the rate-determining step within deacylation was associated with hydrolysis, whereas hydride transfer was rate-determining in acylation. Therefore, before interpreting the steady-state kinetic data, we first determined whether the rate-limiting step was still associated with the deacylation step for mutated GAPNs. For that, experiments were carried out at pH 8.2 and 25 °C under the following conditions, i.e. T244S GAPN (4 and 8 μM), NADP (0.03 mM), D,L-G3P (1.5 mM); K178A GAPN (4 and 8 μM), NADP (1 mM), D,L-G3P (0.15 mM); T244V GAPN (4 and 8 μM), NADP (1 mM), D,L-G3P (0.2 mM). No burst of NADPH formation was observed irrespective of the mutated GAPN and the measured rate constants were similar to the rate values determined under steady-state conditions (see next paragraph). These results showed that the rate-limiting step is associated with the acylation step for T244S, T244V and K178A GAPNs in contrast with the wild-type (Table 3). More precisely, as shown below, the rate-limiting step in T244S and K178A GAPNs is associated with hydride transfer. Therefore, to analyse a possible effect of the mutations introduced at positions 178 and 244 in terms of change in the apparent affinity constant for the G3P substrate and in rate constants, only the kac (acetylation constant) value and the Km value for G3P of the wild-type in the acylation step have been considered.
Table 3. Kinetic parameters of the acylation step for the T244S, T244V and K178A GAPNs.
Initial velocities of the reaction catalysed by mutated GAPNs were measured in 50 mM Tes buffer, pH 8.2, and 5 mM 2-mercaptoethanol at 25 °C. Previous enzyme activity measurements with either D,L-G3P or D-G3P showed that both enantiomeric forms equally behaved as efficient substrates for the wild-type GAPN [2]. Therefore D,L-G3P was used as a substrate for mutated GAPN-catalysed reactions. Km values for NADP and D,L-G3P were determined by fitting the experimental data to the Michaelis–Menten equation.
| NADP | D,L-G3P | |||
|---|---|---|---|---|
| GAPN | Km (μM) | Km (μM) | kcat (s−1) | Rate-limiting step |
| Wild-type* | 25±2* | 490±50* | 790±20* | Deacylation |
| T244S | 10±3 | 120±14 | 0.27±0.03 | Acylation |
| T244V | − | − | ∼0.002† | Acylation |
| K178A | 70±6 | 12±5‡ | 0.020±0.002 | Acylation |
*Corresponding to rate constant and Km values for the cofactor and the substrate in the acylation step and determined with D-G3P [2]. Substrate inhibition was described to be observed for [D-G3P]>200 μM (see [2]). The acylation rate constant was determined at 10 °C. Therefore, to compare the kcat values of the mutated GAPNs and kac value of the wild-type GAPN, the reported kac value of the wild-type was extrapolated from the value measured at 10 °C, assuming a 2.5-fold increase of the rate constant kac per 10 °C (from [2]).
†The kcat value was too low to permit determination of Km values for NADP and D,L-G3P.
‡Substrate inhibition was observed for [D,L-G3P]>150 μM.
T244S GAPN
As shown in Table 3, substituting serine for Thr244 resulted in a 2-fold decrease in Km for NADP with a value of 10 μM. An inhibitory effect was observed for [NADP]>60 μM. The Km value for D,L-G3P decreased by a factor of 4 and the kcat value decreased drastically, by a factor of 2900, relative to the kac value of the wild-type (Tables 3 and 4). At optimum pH 8.2, the kcat value was decreased by a factor of 2.1 when D-[1-2H]G3P was used as a substrate (Table 4). This strongly suggested that hydride transfer is rate-limiting. Therefore the hydride rate constant is decreased 2900-fold compared with the wild-type. The pH–rate profile was then studied over a pH range 5–8.75 at a saturating concentration of 1.5 mM D,L-G3P and at a concentration of 30 μM NADP, where NADP does not behave as an inhibitor. As shown in Figure 3, the pH–kcat profile exhibited a sigmoidal curve that can be related to the contribution of one ionizable group of pKapp 6.8 that must be deprotonated for acylation, and characterized by a kcat of 0.27 s−1. Compared with wild-type, pKapp of 6.8 would correspond to that of catalytic Cys302. The fact that the pH–kcat profile depends on Cys302 pKapp and not on Glu268 pKapp, which was shown to be 7.6 in the wild-type [9], could be indicative of relative positioning of Cys302 and Glu268 in the T244S ternary Michaelis complex, slightly different from that found in the wild-type ternary complex.
Table 4. Parameters for the pH-dependence of the kcat values for the T244S, T244V and K178A GAPNs.
pKapp and kcat values were deduced from non-linear regression analysis of experimental data obtained at 25 °C against a single pKa model. The substrate isotope effects were determined as described in the Experimental section.
Figure 3. pH-dependence of the steady-state rate constant kcat for K178A and T244S GAPNs.

pH-dependence of kcat was studied over the pH range 5.25–8.75 for T244S GAPN or 5.25–9.00 for K178A GAPN with a mixed reaction buffer consisting of 120 mM Tricine, 30 mM imidazole and 30 mM ethanoic acid at a constant ionic strength of 0.15 M. The other experimental conditions were as follows: 0.15 mM D,L-G3P, 1 mM NADP and 1.65 μM K178A GAPN; and 1.5 mM D,L-G3P, 0.03 mM NADP and 0.5 μM T244S GAPN. Data for T244S (▲) and K178A (●) GAPNs were best fitted by non-linear regression analysis against a single pKapp model, identified by the best-fit theoretical curves (solid line and dotted line respectively). S.E.M. on each experimental point was less than 5%.
T244V and K178A GAPNs
The kinetic parameters are summarized in Table 3. The T244V substitution led to a drastic 250000-fold decrease of the kcat value. The kcat value of 2×10−3 s−1 was so low that determination of Km values for NADP and D,L-G3P was not possible. The kcat value for K178A GAPN was strongly reduced by a factor of 25000 and the Km values for NADP and D,L-G3P were 3-fold higher and 40-fold lower respectively, with respect to the wild-type. The large decrease in the D,L-G3P Km value remains to be explained. Under steady-state conditions at optimum pH 8.2, a substrate kinetic isotopic effect of 2.1 was observed with D-[1-2H]G3P (Table 4). Thus hydride transfer is also rate-limiting and is 25000-fold less efficient than with the wild-type. The pH–rate profile was also studied over a pH range 5–9. kcat values for K178A were determined at saturating concentrations of 0.15 mM D,L-G3P and 1 mM NADP respectively. As shown in Figure 3, the pH–kcat profile exhibited a sigmoidal curve that can also be related to the contribution of one ionizable group of pKapp 6.7 and characterized by a kcat value of 0.02 s−1. Compared with the wild-type, pKapp of 6.7 would correspond to that of catalytic Cys302, a value similar to that determined for T244S GAPN.
Structure of the T244S apo-like form
The (2Fo−Fc) electron-density map is well defined for all of the structure. Difference maps revealed the presence of NADP in the four monomers. Three sulfate ions coming from the crystallization medium have also been identified in monomer D, whereas only two were present in monomer B, and one in each of monomers A and C. The sulfate anion whose position is conserved in all four monomers occupies the same location as the sulfate anion SO4a found in the wild-type apo structure (Apo1 and Apo2 [15,12]), while the other is located on the protein surface.
The least-squares superimpositions involving the 474 Cα atoms of one subunit of the apo-like wild-type [15] with each subunit of the T244S GAPN structure result in r.m.s.d. values less than 0.29 Å, showing that the overall structures are very close. Local differences are limited to solvent-exposed regions, mobile loops and to the active-site region. In the wild-type structure, Thr244 is located at the end of a β-strand and its β-hydroxy group is hydrogen-bonded to the ϵ-amino group of Lys178, a residue located in an α-helix. In the T244S structure, the β-hydroxy group of Ser244 adopts the same conformation and its interaction with Lys178 is conserved. The quality of the (2Fo−Fc) electron-density map is good enough to model the NMN moiety of the NADP molecule (Figure 1B). In particular, the pyridinium ring adopts a conformation, clearly different from that depicted for wild-type GAPN–NADP and ALDH2–NAD binary complexes which was described to be suitable for an efficient hydride transfer [17]. In the T244S GAPN, the pyridinium ring is stabilized through van der Waals interactions with Cys302, Pro167 and Leu174. Cys302 is located after an α-helix, Pro167 lies at the end of a β-strand, and Leu174 is located on an α-helix. Moreover, the conformation of the side chain of catalytic Cys302 is closer to that found in the wild-type apo form than in the wild-type–NADP complex and the salt bridge between Arg459 and Glu268 remains present, albeit that NADP is bound (Figure 1A).
Finally, although the side chain of Arg301 was shown to bind the sulfate anion, called SO4a, in the wild-type apo structures (Apo1 and Apo2 [12,15]) and was shown to both contribute to G3P binding and be involved in the efficient positioning of G3P relative to Cys302 within the ternary complex GAPN–NADP–G3P [26], it is positioned opposite to the SO4a anion corresponding to the C-3 phosphate of G3P and interacts with the carbonyl groups of Gly294 and Ala293. It is probable that the conformation adopted by the side chain of Arg301 in the binary complex corresponds instead to an alternative conformation which is not representative of the positioning of its guanidinium group within the competent wild-type ternary complex. Indeed, no significant Km change for G3P accompanies the drastic decrease of the acylation rate observed for the T244S GAPN in contrast with what was observed for the R301L GAPN [26].
DISCUSSION
A key aspect of the chemical mechanism of the CoA-independent ALDH family is the requirement of a conformational isomerization of the NMN(H) portion of the cofactor after the oxidoreduction process to allow Glu268 to play its role in the deacylation step [18]. The fact that the acylation step in GAPN, and also many other CoA-independent ALDHs, is not rate-limiting and is efficient implies several prerequisites for this step. One of those relates to the optimal positioning of the nicotinamide ring towards the hemithioacetal within the ternary acylating complex. Several X-ray structures of binary ALDH–NAD(P) complexes reveal a great conformational flexibility of the NMN portion of the NAD(P) molecule, in particular of the nicotinamide ring (see, e.g., [11,14–15]). However, once the transient hemithioacetal intermediate is formed, the NMN moiety, in particular in GAPN, must be positioned such that an efficient and stereospecific transfer of hydride ion occurs towards C-4 of the nicotinamide ring. As indicated in the Introduction, invariant Thr244 is postulated to play a major role in the positioning of the nicotinamide ring within the ternary complex of CoA-independent ALDHs. The kinetic and structural data reported here on GAPN strongly favour such an assumption, in particular of the critical role of the β-methyl group of Thr244.
Role of the β-methyl group of Thr244
Substitution of serine for Thr244 led to a change in the nature of the rate-limiting step which becomes associated with the acylation step. Hydride transfer becomes rate-limiting and its efficiency is strongly decreased by a factor of 2900 compared with the wild-type. This drastic effect could be the consequence of changes occurring in the location and orientation of the nicotinamide ring relative to the hemithioacetal intermediate within the ternary complex, thereby preventing an efficient hydride transfer. The T244S GAPN structure in the presence of NADP clearly shows that the absence of the β-methyl group leads to a unique stabilized position of the nicotinamide ring clearly distinct from that hypothesized for the ‘hydride transfer conformation’ in CoA-independent ALDHs including GAPN [10,15–17]. The crystal structure presented herein is likely to be representative of the position occupied by the nicotinamide ring within the T244S GAPN–NADP complex in solution. This is in agreement with the fact that: (i) the formation of the binary complex T244S GAPN–NADP is not able to promote the chemical activation of Cys302 (its pKapp remains around 9) in contrast with what was observed for the wild-type [9], and (ii) the salt bridge between Arg459 and Glu268, which is considered typical of the apo form [12], is still present in the X-ray binary complex structure.
Determination of the structure of a hemithioacetal intermediate is not possible because of its transient nature. If we assume that the nicotinamide conformation in the T244S hemithioacetal intermediate–NADP complex is the one observed in the crystal structure of the binary complex T244S GAPN–NADP, the poor efficiency of the hydride transfer could be explained by non-optimal positioning of the nicotinamide ring relative to the hemithioacetal intermediate. The 0.6-unit increase in pKapp of Cys302 compared with the wild-type, which is similar to the contribution of the positively charged nicotinamide ring in lowering the pKapp of Cys302 in the wild-type [9], can be also interpreted as the consequence of positioning of the nicotinamide ring relative to Cys302 slightly different from that in the wild-type acylating ternary complex. Therefore the role of the β-methyl group of Thr244 in the wild-type would be to mainly allow the nicotinamide ring to adopt a conformation suitable for an efficient hydride transfer.
An alternative explanation would relate to the positioning of the C-1 hydroxylate of the hemithioacetal intermediate relative to the oxyanion hole rendering it unsuitable for hydride transfer. However, this latter assumption is rather unlikely. First, the apparent affinity constant of T244S GAPN for G3P is not significantly changed. Secondly, whatever the X-ray structures considered, including those of the binary complex T244S GAPN–NADP, the apo-like wild-type and the ternary thioacylenzyme intermediate, the side chain of Asn169 involved in the oxyanion hole formation is always similarly positioned in all the structures [15,18].
Role of the β-hydroxy group of Thr244
As mentioned in the Introduction, the Thr244 β-hydroxy group, which is hydrogen-bonded to the Lys178 ϵ-amino group, can help the β-methyl group of Thr244 to position the nicotinamide ring in a conformation suitable for hydride transfer. The T244V substitution was therefore intended to (i) determine the consequence of the loss of the hydrogen bond between the Thr244 β-hydroxy group and the Lys178 ϵ-amino group, and (ii) to introduce a valine residue invariant at this position in CoA-dependent ALDHs whose acylation step was shown to be as efficient as in GAPN [27]. Removing the β-hydroxy group in fact drastically alters the catalytic properties of the enzyme. The efficiency of the rate-limiting acylation step is decreased 250000-fold and 86-fold compared with the wild-type and T244S GAPNs respectively. Although it was not possible to determine whether hydride transfer remained rate-determining within the acylation step because of the very low kcat value, the larger decrease of kcat probably originates from the disruption of the hydrogen-bond interaction between the residues at positions 244 and 178, thus leading to formation of an inefficient T244V ternary complex. However, one cannot exclude the possibility that the very low kcat value of T244V GAPN is also due to additional structural changes induced by the presence of a valine residue in the hydrophilic environment of the GAPN active site.
The substitution of alanine for the counterpart residue Lys178 also appears to be informative and complementary to the studies carried out on T244S GAPN. In fact, the K178A and T244S GAPNs exhibited very similar catalytic properties. First, the rate-limiting step is associated with hydride transfer and a 25000-fold decrease of the kcat value was observed. Secondly, the pKapp values for Cys302 are equivalent in both ternary complexes, i.e. 6.7 compared with 6.8. Therefore it is tempting to interpret these results in a similar way to that developed in the preceding section for the T244S GAPN. So, the disruption of the Thr244–Lys178 hydrogen-bond interaction by substituting either valine for Thr244 or alanine for Lys178 would alter the positioning of Thr244, in particular of its β-methyl group and thus of the nicotinamide ring that could explain the very poor efficiency of the hydride transfer.
Relevance to the two-step catalytic mechanism
The major feature that has emerged from the present study lies in the critical contribution of the β-methyl group of invariant Thr244 in the efficiency of the GAPN acylation step. It probably contributes to formation of a transient hemithioacetal intermediate with an adequate positioning of the nicotinamide ring to allow an efficient hydride transfer. Such a role is expected to be general for all the members of the CoA-independent ALDH family. This is supported further by the presence of an invariant valine residue at position 244 in the CoA-dependent ALDH family. In these ALDHs, position 178 is mostly occupied by a methionine residue. Thus it is tempting to propose that one of the two methyl groups of the β-branched side chain of Val244 is positioned like that of β-methyl of Thr244 and therefore fulfils a similar function thanks to the other methyl group of Val244 which could interact via hydrophobic interactions with the side chain of residue 178. Such a hypothesis remains to be validated. In addition to Thr244, the side chain of invariant Glu399 was shown to play an essential role by anchoring the NMN ribose through hydrogen bonds with the hydroxy groups of the ribose [28]. Lastly, it was also hypothesized that the negative charge of the tetrahedral oxyanion intermediate participates in the efficient positioning of the positively charged nicotinamide ring within the ternary complex hemithioacetal intermediate–NAD(P) [18]. Taken together, this probably delineates a pattern of interactions that stabilizes an efficient ‘hydride transfer conformation’ of the NMN moiety of the cofactor, once the transient hemithioacetal intermediate is formed.
Finally, recent structural data on the thioacyl intermediate of GAPN suggested that a low energetic barrier exists between the conformations adopted by the NMN(H) moiety during the two-step catalytic cycle of the CoA-independent ALDHs [18]. Based on the structure of the crystalline binary complex T244S GAPN–NADP, we propose that the β-methyl group of Thr244 might also play a role in the efficiency of the conformational isomerization of the NMNH after hydride transfer.
Acknowledgments
We are grateful to S. Boutserin for her very efficient technical help. We also thank Dr Van Dorsselear and Dr G. Chevreux for mass determination and Dr S. Sonkaria for careful reading of the manuscript. A. P. was fellow of the French Ministère de la Recherche et des Nouvelles Technologies (MRNT). This research was supported by the Centre National de la Recherche Scientifique, the French Ministère de la Recherche et de l'Enseignement Supérieur, the University Henri Poincaré Nancy I, the IFR 111 Bioingénierie and local funds from the Région Lorraine.
References
- 1.Wang X., Weiner H. Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry. 1995;34:237–243. doi: 10.1021/bi00001a028. [DOI] [PubMed] [Google Scholar]
- 2.Marchal S., Rahuel-Clermont S., Branlant G. Role of glutamate-268 in the catalytic mechanism of nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 2000;39:3327–3335. doi: 10.1021/bi9914208. [DOI] [PubMed] [Google Scholar]
- 3.Vallari R. C., Pietruszko R. Kinetic mechanism of the human cytoplasmic aldehyde dehydrogenase E1. Arch. Biochem. Biophys. 1981;212:9–19. doi: 10.1016/0003-9861(81)90338-6. [DOI] [PubMed] [Google Scholar]
- 4.Shone C. C., Fromm H. J. Steady-state and pre-steady-state kinetics of coenzyme A linked aldehyde dehydrogenase from Escherichia coli. Biochemistry. 1981;20:7494–7501. doi: 10.1021/bi00529a026. [DOI] [PubMed] [Google Scholar]
- 5.Söhling B., Gottschalk G. Purification and characterization of a coenzyme-A-dependent succinate-semialdehyde dehydrogenase from Clostridium kluyveri. Eur. J. Biochem. 1993;212:121–127. doi: 10.1111/j.1432-1033.1993.tb17641.x. [DOI] [PubMed] [Google Scholar]
- 6.Farres J., Wang T. T., Cunningham S. J., Weiner H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry. 1995;34:2592–2598. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 7.Vedadi M., Szittner R., Smillie L., Meighen E. Involvement of cysteine 289 in the catalytic activity of an NADP+-specific fatty aldehyde dehydrogenase from Vibrio harveyi. Biochemistry. 1995;34:16725–16732. doi: 10.1021/bi00051a022. [DOI] [PubMed] [Google Scholar]
- 8.Vedadi M., Meighen E. Critical glutamic acid residues affecting the mechanism and nucleotide specificity of Vibrio harveyi aldehyde dehydrogenase. Eur. J. Biochem. 1997;246:698–704. doi: 10.1111/j.1432-1033.1997.t01-1-00698.x. [DOI] [PubMed] [Google Scholar]
- 9.Marchal S., Branlant G. Evidence for the chemical activation of essential Cys-302 upon cofactor binding to nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 1999;38:12950–12958. doi: 10.1021/bi990453k. [DOI] [PubMed] [Google Scholar]
- 10.Steinmetz C. G., Xie P., Weiner H., Hurley D. T. Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. Structure. 1997;5:701–711. doi: 10.1016/s0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 11.Johansson K., El-Ahmad M., Ramaswamy S., Hjelmqvist L., Jörnvall H., Eklund H. Structure of betaine aldehyde dehydrogenase at 2.1 Å resolution. Protein Sci. 1998;7:2106–2117. doi: 10.1002/pro.5560071007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobessi D., Tête Favier F., Marchal S., Branlant G., Aubry A. Structural and biochemical investigations of the catalytic mechanism of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol. 2000;300:141–152. doi: 10.1006/jmbi.2000.3824. [DOI] [PubMed] [Google Scholar]
- 13.Marchal S., Cobessi D., Rahuel-Clermont S., Tete-Favier F., Aubry A., Branlant G. Chemical mechanism and substrate binding sites of NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. Chem. Biol. Interact. 2001;130–132:15–28. doi: 10.1016/s0009-2797(00)00218-0. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z. J., Sun Y. J., Rose J., Chung Y. J., Hsiao C. D., Chang W. R., Kuo I., Perozich J., Lindahl R., Hempel J., Wang B. C. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 1997;4:317–326. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 15.Cobessi D., Tête Favier F., Marchal S., Azza S., Branlant G., Aubry A. Apo and holo crystal structures of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol. 1999;290:161–173. doi: 10.1006/jmbi.1999.2853. [DOI] [PubMed] [Google Scholar]
- 16.Moore S. A., Baker H. M., Blythe T. J., Kitson K. E., Kitson T. M., Baker E. N. Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure. 1998;6:1541–1551. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Miller S. J., Hurley T. D. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry. 2003;42:7100–7109. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 18.D'Ambrosio K., Pailot A., Talfournier F., Didierjean C., Benedetti E., Aubry A., Branlant G., Corbier C. The first crystal structure of a thioacylenzyme intermediate in the ALDH family: new coenzyme conformation and relevance to catalysis. Biochemistry. 2006;45:2978–2986. doi: 10.1021/bi0515117. [DOI] [PubMed] [Google Scholar]
- 19.Corbier C., Mougin A., Mely Y., Adolph H. W., Zeppezauer M., Gerard D., Wonacott A., Branlant G. The nicotinamide subsite of glyceraldehyde-3-phosphate dehydrogenase studied by site-directed mutagenesis. Biochimie. 1990;72:545–554. doi: 10.1016/0300-9084(90)90119-2. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunsteleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22. Reference deleted.
- 23.Laskowsky R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 24.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 25.Esnouf R. M. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 26.Marchal S., Branlant G. Characterization of the amino acids involved in substrate specificity of nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutans. J. Biol. Chem. 2002;277:39235–39242. doi: 10.1074/jbc.M205633200. [DOI] [PubMed] [Google Scholar]
- 27.Stines-Chaumeil C., Talfournier F., Branlant G. Mechanistic characterization of the MSDH (methylmalonate semialdehyde dehydrogenase) from Bacillus subtilis. Biochem. J. 2006;395:107–115. doi: 10.1042/BJ20051525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni L., Sheikh S., Weiner H. Involvement of glutamate 399 and lysine 192 in the mechanism of human liver mitochondrial aldehyde dehydrogenase. J. Biol. Chem. 1997;272:18823–18826. doi: 10.1074/jbc.272.30.18823. [DOI] [PubMed] [Google Scholar]