

Abstract
Cell cycle checkpoints contribute to survival after exposure to ionizing radiation (IR) by arresting the cell cycle and permitting repair. As such, yeast and mammalian cells lacking checkpoints are more sensitive to killing by IR. We reported previously that Drosophila larvae mutant for grp (encoding a homolog of Chk1) survive IR as well as wild type despite being deficient in cell cycle checkpoints. This discrepancy could be due to differences either among species or between unicellular and multicellular systems. Here, we provide evidence that Grapes is needed for survival of Drosophila S2 cells after exposure to similar doses of IR, suggesting that multicellular organisms may utilize checkpoint-independent mechanisms to survive irradiation. The dispensability of checkpoints in multicellular organisms could be due to replacement of damaged cells by regeneration through increased nutritional uptake and compensatory proliferation. In support of this idea, we find that inhibition of nutritional uptake (by starvation or onset of pupariation) or inhibition of growth factor signaling and downstream targets (by mutations in cdk4, chico, or dmyc) reduced the radiation survival of larvae. Further, some of these treatments are more detrimental for grp mutants, suggesting that the need for compensatory proliferation is greater for checkpoint mutants. The difference in survival of grp and wild-type larvae allowed us to screen for small molecules that act as genotype-specific radiation sensitizers in a multicellular context. A pilot screen of a small molecule library from the National Cancer Institute yielded known and approved radio-sensitizing anticancer drugs. Since radiation is a common treatment option for human cancers, we propose that Drosophila may be used as an in vivo screening tool for genotype-specific drugs that enhance the effect of radiation therapy.
IONIZING radiation (IR) is damaging to cells and this property underlies its use as a leading anticancer therapy. However, cells and tissues of organisms are exposed to radiation naturally as well and as such have evolved mechanisms to counter its effects. In particular, DNA damage is a key effect of IR and cells respond by (i) activating cell cycle checkpoints to pause cell division, presumably to allow time for DNA repair, (ii) inducing DNA repair pathways, and (iii) stimulating apoptosis that can cull damaged cells (Zhou and Elledge 2000). The ultimate purpose of these responses is the preservation of genetic integrity. Passage through subsequent generations of genetic abnormalities is associated with and can lead to disease in humans. For this to happen, however, a cell with damaged DNA has to survive and reproduce. Hence we have been interested in DNA damage responses that determine how well a cell survives and reproduces after suffering DNA damage.
Classical studies in budding yeast showed that cell cycle checkpoints are required for cells to survive exposure to DNA-damaging agents (Weinert and Hartwell 1988). This requirement can be circumvented by artificially inducing a reversible cell cycle arrest following DNA damage. Therefore, cell cycle regulation by checkpoints likely affords the damaged cell a necessary reprieve during which repair can occur. More recently, however, genes needed for checkpoints are also found to induce other responses such as transcriptional and post-transcriptional regulation of genes needed for DNA repair. Comparative analysis of checkpoint mutants showed that even mutants that show comparable misregulation of cell cycle have different sensitivity to the same genotoxin, indicating that responses other than cell cycle regulation also contribute to the requirement for checkpoint genes. For example, stabilization of replication forks is found to be crucial for surviving the alkylating agent MMS in budding yeast whereas the ability to inhibit mitosis appears less important (Tercero and Diffley 2001).
The DNA damage checkpoint in eukaryotes is mediated by a conserved set of four kinases encoded by ATM, ATR, Chk1, and Chk2 (Zhou and Elledge 2000). In fission yeast, Chk1 acts by phospho-inhibition of Cdc25, an activator of Cdk1 and mitosis (Furnari et al. 1997). In budding yeast, Chk1 acts by maintaining the abundance of Pds1, an anaphase inhibitor to block metaphase-to-anaphase transition (Sanchez et al. 1999). Yeast chk1 mutants are sensitive to DNA-damaging agents; fission yeast chk1/rad27 was first isolated as a radiation-sensitive (rad) mutant that showed increased killing by UV radiation and was later found to be hypersensitive to IR (Walworth et al. 1993; al-Khodairy et al. 1994), whereas budding yeast chk1 mutants are mildly sensitive to IR and UV radiation (Sanchez et al. 1999). Targeted elimination of Chk1 in avian DT40 cells increased the sensitivity of cells to IR (Zachos et al. 2003). UCN-01, a potent inhibitor of Chk1 kinase (IC50 11–25 nm) (Busby et al. 2000; Graves et al. 2000), increases the radiation sensitivity of human cells, suggesting that Chk1 is also required to ensure survival after irradiation in this system.
In contrast to the contribution of Chk1 homologs to survival after DNA damage in the above experimental systems, Drosophila grp1 homozygous mutant larvae (hereafter called grp larvae) survive ionizing radiation as well as wild type do (Jaklevic and Su 2004). This was a surprise to us because grp larvae are deficient in cell cycle checkpoints that delay progress through S phase and the entry into mitosis. Thus, we found that cell cycle regulation by checkpoints was dispensable for survival after irradiation in Drosophila larvae. This difference could be because there were inherent differences between Drosophila and other eukaryotes. Alternatively, what cells (used in the above-described studies) need to survive irradiation is different from what multicellular organisms (used in our previous studies) need to survive irradiation. To distinguish between these possibilities we compared the requirement for Grp in single and multicellular contexts in Drosophila.
We report here that unlike larvae, Drosophila S2 cells depleted for Grp, previously shown to have defective cell cycle checkpoints (de Vries et al. 2005), show decreased survival after exposure to comparable doses of X rays. Thus, cells and organisms appear to have different requirements for surviving irradiation. We hypothesize that this difference stems from the ability of organisms to undergo compensatory cell proliferation and hence overcome cell-killing effects of radiation. In support for this hypothesis, treatments that are expected to interfere with compensatory proliferation are found to reduce the radiation survival of Drosophila larvae, with the effect of some treatments being greater on grp mutants than on wild type. The greater sensitivity of grp mutants allowed us to design a screen for small molecules that act as genotype-specific radiation sensitizers. In other words, such molecules would specifically kill grp mutant larvae but not wild type when applied in conjunction with IR. A screen through a molecule library from the National Cancer Institute (NCI)–Developmental Therapeutic Program yielded molecules that reduced the survival of grp mutants when combined with radiation. We propose that such molecules could be powerful anticancer agents when used in combination with radiation to treat checkpoint-deficient tumors. Consistent with this proposal, two of the molecules isolated are known anticancer agents.
MATERIALS AND METHODS
Fly stocks:
All mutant alleles used in this work have been described before: grp1, cdk43, chico2, dmycp0, and dmycp1 (Fogarty et al. 1997; Bohni et al. 1999; Johnston et al. 1999; Meyer et al. 2000). grp1 is a genetic null; there is a small (<3% of wild-type level) amount of Grp protein in 6- to 7-hr-old homozygous embryos but no detectable protein in larval discs (Purdy et al. 2005 and data not shown).
RNAi:
S2 cells were cultured at 25° throughout the experiment in Schneider's insect medium supplemented with 10% fetal bovine serum. Cells in log phase growth (0.5–5 × 106 cells/ml) were diluted to 106 cells/ml with fresh media containing double-stranded RNA (10 μg/ml) or water (negative control). One and one-half milliliters of cells were immediately transferred to a T12.5 tissue culture flask (Becton-Dickinson, Franklin Lakes, NJ) and allowed to incubate for 1 hr. One and one-half milliliters of fresh media were then added and the cells were allowed to grow for 4 days. The cells were diluted to 7–8 × 105/ml with fresh media containing either double-stranded RNA (5 μg/ml) or water. Three milliliters of cells at 0.5 × 106 cells/ml were transferred to a new T12.5 tissue culture flask and irradiated. At various times after irradiation, cells were loosened by gentle pipetting and counted. All cell counts were done using a hemacytometer (Hausser Scientific, Horsham, PA). Western blotting was performed as previously described (de Vries et al. 2005).
Drosophila S2 cells expressing GFP–H2AB have been described before (de Vries et al. 2005). Five days prior to irradiation, the Grp RNAi treatment was started and depletion of Grp was verified by Western blotting (de Vries et al. 2005). Cells were irradiated at room temperature using a 137Cs source in an IBL 637 irradiator (CIS Biointernational, Bagnols-sur-Cèze, France) at a dose rate of 0.73 Gy/min. At various times after irradiation, cells were harvested, washed once with phosphate-buffered saline (PBS), and centrifuged and cell pellets were resuspended in 0.2–0.5 ml of PBS for FACS analysis. The percentage of H2B–GFP-expressing cells was determined using a FACSCalibur (Becton Dickinson, San Jose, CA) and 10,000 events were analyzed for each sample. Data analysis was done with WinList (version 5.0) from Verity Sofware House (Topsham, ME). In one typical experiment every condition was performed and measured in triplicate. Three independent experiments were performed.
To visualize nuclear fragmentation, cells were fixed by mounting in an equal volume of PBS that contains 48% glycerol, 10% formaldehyde, 0.2% Tween, and 1 μg/ml Hoechest33258. Images were obtained on a Leica DMR microscope using Slidebook software (Intelligent Imaging).
Irradiation:
Irradiations were performed at room temperature in a TORREX X-ray generator (Astrophysics Research) set at 5 mA and 115 kV. To assay for radiation sensitivity of growth mutants, embryos were collected for 4 hr and aged for 4 days. At the time of irradiation, larvae were 97–107 hr after egg deposition. Larvae were placed in plastic petri dishes and irradiated as above. Irradiated larvae were placed on fresh food and allowed to develop for 10 days before eclosion was determined.
Drosophila food:
Standard Drosophila food contains 1487 g cornmeal, 2344 g molasses, 164 g agar, 938 g yeast, 375 ml Tegosept mold inhibitor (10% methyl-4-hydroxy benzoate in 95% EtOH), 94 ml proprionic acid, and 21,100 ml water. Starvation diet is 20% sucrose (w/v) in PBS. Colchicine and small molecules were mixed into cornmeal–agar food (7.7% w/v yellow cornmeal and 1.2% w/v agar; autoclaved at 121° for 20 min). Detailed recipes are available upon request.
Screening for radiosensitizers of grp mutants:
Embryos were raised on standard food for 5 days, sized on stainless steel sieves, and irradiated with 4000 R of X rays. Approximately 100 larvae were transferred into a vial containing cornmeal–agar food supplemented with drug or DMSO. We screened 1990 molecules of the Diversity set (http://dtp.nci.nih.gov) supplied at 10 mm in DMSO. Larvae can tolerate up to 0.10% DMSO in cornmeal–agar food. Therefore, small molecules were diluted to a final concentration of 10 μm in food. Eclosion was determined 10 days after irradiation. Most if not all larvae formed pupae in our experiments. The primary screen was performed with irradiated grp larvae, in batches of ∼30–80 molecules. For each batch, average percentage of eclosion and standard deviation (SD) were calculated. Molecules that reduced percentage of eclosion by ≥2 standard deviations from the average were identified and designated as “potential positives.” Potential positives were retested, against a control population of 8–10 DMSO vials. Potential positives that produced a percentage of eclosion that was ≥2 SD lower than the average of the DMSO population were designated as “positives.” The positives were obtained from the NCI–Developmental Therapeutic Program (DTP) and tested on irradiated and unirradiated grp and wild-type larvae, at different concentrations. A detailed protocol is available upon request.
Statistical analysis:
Standard error of the means (SEM) was calculated using 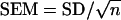 , where n is the number of measurements. Student's t-test (two-tailed, equal variance between samples) was used to determine statistical significance.
, where n is the number of measurements. Student's t-test (two-tailed, equal variance between samples) was used to determine statistical significance.
RESULTS
Most cells of the Drosophila larvae grow by endoreplication in which rounds of S phase alternate with gap phases. The exceptions are diploid cells of larval imaginal discs, sacs of epithelial cells that will differentiate into adult structures during metamorphosis. These cells proliferate by mitosis during larval growth and show canonical checkpoint responses that are dependent on Drosophila ATR (Mei-41) and Chk1 (Grp) (Jaklevic and Su 2004). While Chk1 is universally required for cells and unicellular organisms to survive radiation, grp mutant larvae survive as well as wild type upon irradiation. Interestingly, grp mutant larvae are deficient in checkpoints, suggesting that they use other mechanisms to counter the effects of radiation. We wanted to investigate whether this difference was attributable to disparate regulation in the Drosophila species or differing requirements of uni- and multicellular entities. To do so, we turned to a cell culture system to monitor cellular responses to radiation in a more quantitative manner.
Grp-depleted cells fail to increase in number after irradiation:
We used RNAi to deplete Grp (Chk1) in Drosophila S2 cells (Figure 1) prior to and following irradiation. Cell density was quantified for control (−) and RNAi-treated (+) samples at various times after exposure to 0, 4000, or 8000 R of X rays. Sample data from two experiments are shown in Figure 1, A and B. Grp RNAi alone made little difference to cell number (compare 0− samples to 0+ samples). In conjunction with radiation, however, Grp-depleted cells increased their number less efficiently than did controls (compare 4000− samples to 4000+ samples, for instance). The number of cells in RNAi-treated samples was divided by the number of cells in controls in these and one additional experiment (a total of three experiments), averaged, and shown along with standard deviations in Figure 1C. The success of Grp depletion was monitored by Western blotting in these experiments (Figure 1D). We conclude that irradiation reduced the number of Grp-depleted cells in a dose-dependent manner, with the difference becoming more apparent at longer times after irradiation. The X-ray doses used here (40 and 80 Gy or 4000 and 8000 R) are comparable to those in previous studies on larvae (4000 R) (Jaklevic and Su 2004).
Figure 1.—

Grp depletion reduces the number of irradiated S2 cells. (A and B) S2 cells were treated with grp double-stranded RNA (+) or left untreated (−) for 4 days prior to irradiation with 0, 4000, or 8000 R of X rays. Cell number (×104/ml) is plotted against time after irradiation (IR). Data from two independent experiments are shown. (C) Cell density in RNAi-treated samples was normalized to cell density in untreated samples for each day after irradiation and averaged. The data are from three different experiments. Error bars represent standard error of the mean (SEM). (D) The extent of Grp depletion was monitored for each experiment by Western blotting at the time of irradiation (not shown) and on day 4 after irradiation. Arrowhead indicates Grp (predicted at 55 kDa). A cross-reacting protein (arrow) serves as a loading control between − and + RNAi lanes. The specificity of the antibody has been demonstrated before (de Vries et al. 2005). (E and F) S2 cells carrying a GFP–H2AB transgene were treated for grp RNAi, treated for RNAi for an unrelated gene (Stwl, a putative transcription factor; Akiyama 2002), or left untreated (control). GFP-positive (“fluorescent”) cells were mixed with an equal number of untreated cells lacking the GFP transgene. Mixed cells were irradiated at 0 R (E) or 4000 R (F). The fraction of cells that are GFP positive is shown. Depletion of grp by RNAi lowered the number of irradiated cells, with the effect being most pronounced at 4–6 days after irradiation (F). The effect of grp depletion was less pronounced on unirradiated cells (E) while control depletion of Stwl or no treatment had little effect on irradiated cells (F). The success of Grp RNAi was confirmed by Western blotting as in Figure 1D. The averages of triplicate samples are shown for each data point; the SEMs are smaller than the symbols. The results are similar in three independent experiments, although the absolute numbers varied. Results of a typical experiment are shown here. (G–I) S2 cells were stained with a DNA dye Hoechest33258 (red) to visualize nuclear condensation and fragmentation in dead cells. Background fluorescence in the Rhodamine channel (blue) was used to visualize cell outlines. (G) A cell scored as “alive.” (H) A cell scored as “dead.” (I) Quantification of cell death in control and grp RNAi-treated cells 24 hr after exposure to 0, 4000, and 8000 R of X rays. Similar results were obtained at 6 hr after irradiation also (not shown). S2 cells cultured in 100 μm cisplatin for 2.5 days (cisplatin) show robust cell death and serve as a positive control for the assay. From 167 to 282 cells were counted per sample shown. Bar, 5.5 μm. Error bar, 1 SD.
To control for experimental variations in culture conditions, we used another protocol in which RNAi-treated and untreated cells were mixed prior to and following irradiation such that the two populations experience identical culture conditions. An S2 cell line that stably expresses a GFP-tagged Histone H2B was used to distinguish RNAi-treated and untreated cells. Figure 1, E and F, shows data from an experiment in which the GFP-expressing fluorescent cells were treated for RNAi (to deplete Grp, transcription factor Stwl as a control, or no dsRNA). GFP-positive RNAi-treated cells were mixed with GFP-negative untreated cells in approximately equal numbers and irradiated. For each day after irradiation, GFP-positive cells were counted and normalized to the total number of cells (y-axis). Depletion of Grp has little consequence in unirradiated cells; the fraction of GFP-positive cells is similar among the three samples. In contrast, when irradiated with 4000 R, depletion of Grp reduced the fraction of GFP-positive cells relative to Swl-RNAi or no RNAi controls, with the extent of reduction becoming greater with increasing time. This effect was more pronounced with increasing radiation doses (data not shown). Converse experiments, in which control cells and not Grp-depleted cells expressed GFP–H2B, produced similar results (not shown). We conclude that Grp is needed to increase cell number (to stay at levels comparable to controls) following irradiation.
The failure of irradiated Grp-depleted cells to increase their number could be due to increased cell death, decreased cell proliferation, or both. Staining with Trypan Blue, a vital dye, or with Hoechest33258 to examine nuclear fragmentation, revealed no significant differences in the number of dead cells between control and Grp-depleted populations (Figure 1, G–I, and data not shown). These data are more consistent with a need for Grp in maintaining cell proliferation after irradiation rather than in preventing cell death.
Radiation survival of larvae relies on nutritional uptake and growth factor pathways:
We reported previously that irradiation of third instar larvae (both wild type and grp) with 4000 R of X rays results in the death of 60–75% of imaginal disc cells, but that adult flies that result are indistinguishable in size from unirradiated controls, indicating that many of the dead cells have been replaced (Jaklevic and Su 2004). The grp1 allele, a genetic null, was used in the previous work, but at least one other allele, grpfs(A)4, also shows a similar resistance to killing by IR (data not shown). We also reported that mitotic activity in wild type drops immediately after irradiation due to checkpoint activation, but elevates to levels higher than those in unirradiated controls at longer times after irradiation. Irradiated larvae also fed longer and delayed the next developmental stage (pupariation) in a radiation-dose-dependent manner. These results led us to hypothesize that surviving cells of imaginal discs undergo increased growth and proliferation to compensate for dead cells and that the process of compensatory proliferation is critical for larvae to survive irradiation. If this is true, interference with growth and compensatory proliferation should reduce the survival of irradiated larvae. This appears to be the case on the basis of three lines of evidence.
First, we irradiated white pupae instead of larvae and monitored their survival. White pupae are immobilized larvae that have ended the larval stage and are no longer able to feed. We find that wild-type white pupae are more radiation sensitive than wild-type larvae (Figure 2A). We note, however, that white pupae, in addition to not feeding, are also at a different developmental stage. Therefore, the differences in radiation sensitivity of larvae and white pupae may be attributed to factors other than nutritional uptake. For this reason, we limited nutritional uptake directly with a sucrose diet, a standard starvation diet for Drosophila. We find that starvation reduced the survival of wild-type larvae (Figure 2B). Finally, we tested mutants in growth factor signaling pathways or their downstream targets for sensitivity to radiation (Gallant et al. 1996; Bohni et al. 1999; Johnston et al. 1999; Datar et al. 2000; Meyer et al. 2000; Stocker and Hafen 2000): chico [in the insulin-like growth factor (IGF) pathway], dmyc (in the receptor tyrosine kinase–Ras pathway), and cdk4. Hypomorphic mutants in chico and dmyc are used here because null mutants in these genes are lethal. We find that mutants in each gene are more radiation sensitive than wild-type controls or heterozygous siblings (Figure 2, C–E). Increased radiation sensitivity is unlikely to be due to differences in cell death; IR-induced apoptosis was similar in larval imaginal discs of wild type, starved wild type, and cdk4 mutants (supplemental Figure S1 at http://www.genetics.org/supplemental/).
Figure 2.—

The effect of developmental stage, starvation, and growth factor pathways on radiation survival. (A) Feeding third instar larvae (larvae) or white pupae within 5 hr of pupation (pupae) were irradiated with 2000 or 4000 R of X rays. Survival into adulthood was determined by quantifying the percentage of pupae that eclosed. Data are from four separate experiments for wild-type (WT) larvae, six experiments for WT white pupae at 2000 R, four experiments for WT white pupae at 4000 R, seven experiments for grp white pupae at 2000 R, and three experiments for grp white pupae at 4000 R. Error bars represent SEM for each sample. From 335 to 1645 animals were counted per sample. WT white pupae were more sensitive to killing than WT larvae (compare shaded bars to solid bars). grp pupae were more sensitive to radiation than WT pupae (compare open bars to shaded bars). The difference between WT larvae and WT pupae was significant (P < 0.05 for 2000 R). The difference between WT pupae and grp pupae was significant at 2000 R (P < 0.001 for 2000 R) but not at 4000 R. (B) WT larvae at ∼96 hr after egg deposition (AED) were either kept on standard Drosophila food (full food) or moved to sucrose starvation medium (sucrose) for another 24 hr. Larvae were then irradiated with 0 or 4000 R of X rays and maintained on their respective medium. Survival to adulthood was quantified by determining the percentage of pupae that eclosed at 10 days after irradiation. Data are from four separate measurements for full food and five for sucrose. From 466 to 1640 animals were counted per sample. Starvation reduced the survival of irradiated larvae in a statistically significant manner (P < 0.05 between sucrose and full-food samples at 4000 R). (C) chico2 mutation increases radiation sensitivity. Eclosion was determined 10 days after irradiation with the indicated doses. chico homozygous mutants (chico/chico) show lower survival than chico/CyO heterozygous siblings (chico/+). The data are from three separate experiments each at 0, 2000, and 4000 R and from four separate experiments at 5000 R. From 535 to 2817 pupae were counted for each genotype at each dose of radiation. The difference in eclosion was significant at 4000 R (P < 0.05 with respect to 0 R samples) but not at 2000 R. (D) cdk4 mutation increases radiation sensitivity. Eclosion was determined 10 days after irradiation with the indicated doses. cdk4 homozygous mutants (cdk4/cdk4) show lower survival than cdk4/CyO heterozygous siblings (cdk4/+). The data are from five separate experiments each. From 410 to 1949 pupae were counted per genotype at each dose of radiation. The difference in eclosion was significant both at 2000 R (P < 0.05 with respect to 0 R) and at 4000 R (P < 0.001). (E) myc mutations increases radiation sensitivity. Eclosion was determined 10 days after irradiation with the indicated doses. Totals of 1709 (WT), 1355 (mycp1), and 1205 (mycp0) pupae were counted in four to five separate experiments. The difference in eclosion was significant for both p0 and p1 alleles at both 2000 and 4000 R (P < 0.05 with respect to 0 R for all samples). Error bars, SEM.
Collectively these results indicate that experimental inhibition of nutritional uptake and growth factor signaling, which otherwise allow the survival of larvae, can interfere with the survival of irradiated larvae. We infer that irradiation increases the need for nutritional uptake, growth factor signaling, and cell proliferation in such a way that disruption of these processes has more profound consequences on irradiated larvae than on unirradiated larvae. We conclude that these processes contribute to the survival of Drosophila larvae after irradiation.
grp mutant larvae are more radiation sensitive than wild type under some conditions:
Grp-deficient S2 cells are unable to increase cell number after irradiation when compared to cells that have Grp (Figure 1). Such a deficiency at the cellular level might reduce the ability of grp larvae to compensate for dead cells after irradiation. In this case, further interference with compensatory proliferation may reduce the survival of irradiated grp larvae. We provide two lines of evidence to support this idea. First, grp white pupae, unable to feed, are more radiation sensitive than wild-type white pupae (Figure 2A). As in previous experiments, we used the grp1 allele, which is a genetic null (Fogarty et al. 1997; Bohni et al. 1999; Johnston et al. 1999; Meyer et al. 2000), throughout this work. Second, we used a microtubule depolymerization agent, colchicine, to interfere with mitotic proliferation after irradiation. In these experiments, larvae were first irradiated and then fed food containing colchicine. If compensatory proliferation was more important to grp mutants than to wild type, we expect to find a (small) concentration of colchicine that reduced the survival of irradiated grp mutants specifically. We find that 1.3 μm of colchicine reduced the survival of irradiated grp larvae but had little effect on wild type with or without irradiation and on grp without irradiation (Figure 3A). A reduction in survival of ∼30% may appear modest but differences of similar magnitude, for example, in tumor size after radiation treatment, correlate with significant differences in patient survival (Harima et al. 2001).
Figure 3.—

Colchicine radiosensitizes grp larvae. (A) WT or grp larvae were raised as described for the screen for radiation sensitizers (materials and methods), irradiated with 0 or 4000 R X rays, and placed in cornmeal–agar food without or with a supplement of 1.3 μm colchicine (+col). Eclosion was determined 10 days after irradiation. These treatments had no significant effect on wild-type larvae. The combination of IR and colchicine reduced the survival of grp homozygous mutant larvae in a statistically significant manner; the P-value is 0.026 between “4000 R +col” and “0 R” samples. The data are from three separate experiments for each genotype. From 135 to 314 pupae were counted for each genotype at each dose of radiation. Error bars = SEM. (B) A schematic of a screen for radiation sensitizers of grp mutants. For maximum efficiency, the screen was performed by feeding molecules to only the last sample (grp + X rays). Molecules that reduced the survival of this sample were designated as potential positives and tested on all four samples depicted.
We found that starvation on a sucrose diet reduced the survival of irradiated grp mutant larvae. On average, this effect was greater on grp larvae (1.8-fold) than on wild type (1.6-fold in Figure 2B), but the standard deviations on grp samples were too large, making this difference not statistically significant (data not shown).
Our hypothesis also predicts that mutations in growth factor signaling or downstream effectors that reduced the survival of irradiated wild-type larvae should affect the survival of irradiated grp larvae even more profoundly. Double mutants of grp1 with chico2 or cdk43, however, are homozygous lethal (our unpublished data) despite the fact that single mutants are homozygous viable. Such strong interaction could be explained if the need for growth factor signaling is higher in grp1 mutants possibly due to naturally occurring DNA damage.
Data described above suggest that grp mutant larvae are more affected than wild type by treatments that interfere with nutritional uptake or cell proliferation after irradiation. If so, we should be able to take advantage of this difference to identify molecules (like colchicine) that, at appropriate concentration, specifically killed irradiated grp larvae while having less effect on unirradiated grp larvae or irradiated wild-type larvae (schematic in Figure 3B). Such molecules have the potential to enhance the effect of radiation on checkpoint-deficient multicellular structures while sparing their wild-type counterparts.
A pilot screen for radiation sensitizers of grp mutants:
To isolate genotype-specific radiation sensitizers in Drosophila, we screened through a library of small molecules from the NCI for those that reduce the survival of irradiated grp larvae (http://dtp.nci.nih.gov/). We used the 1990-member Structural Diversity Set library because each member represents a family of structurally related molecules; we reasoned that this library allows us to rapidly and efficiently sample through a vast number of structural variations. Third instar grp larvae were irradiated in bulk and then aliquoted into vials, each of which contained 10 μm of a given small molecule mixed into the food (materials and methods). For most small molecules, all larvae formed pupae. Larval survival was determined by counting empty pupae cases (indicating that the imago within had eclosed) as a percentage of total pupae. Figure 4A shows sample data for 80 small molecules supplied on a single plate: NCI samples are supplied as 80 apparently randomly chosen molecules per 96-well plate. In this presentation, 10 molecules, for example, produced survival between 60 and 65% (fourth bar from the left) and 20 molecules produced survival of 75–80% (fourth bar from the right). Most data conform to a bell-shaped distribution as expected for a population. The mean eclosion for the population was 67.7%, with a standard deviation of 8.75%. Molecules at the extreme ends of the distribution represent those producing extremely low or high survival relative to the majority of the molecules. In this example, two molecules produced survival that was >2 SD lower than the average (<52.4% survival) and were therefore designated as potential positives (indicated by arrows in Figure 4A). Potential positives were retested against a control population made of 8–10 controls that contain only the solvent carrier, DMSO. Those that reproducibly lowered the eclosion by >2 SD from the average were designated as positives. One thousand nine hundred ninety molecules of the Structural Diversity Set yielded nine positives. These were then tested against a complete panel of four larval samples (wild type ± IR and grp ± IR). Three of the nine positives were found to reproducibly (twice) and statistically significantly (>2 SD from the mean) reduce the survival of irradiated grp larvae (Figure 4B and supplemental Figure S2 at http://www.genetics.org/supplemental/). Of these, two have been previously characterized because of their anticancer properties. These are topotecan (NSC609699) and its derivative camptothecin (NSC639174) that inhibit topoisomerase I. We are in the process of characterizing the remaining one.
Figure 4.—

A screen for radiation sensitizers of grp larvae. (A) The effect of a sample of 80 small molecules on survival of grp larvae irradiated with 4000 R of X rays. The number of molecules is plotted against the interval of percentage of eclosion as shown. (B) Camptothecin radiosensitizes grp larvae. WT or grp larvae were cultured as for the screen, irradiated with 0 or 4000 R of X rays, and raised on 0 or 10 μm camptothecin. Survival into adulthood was determined by quantifying the percentage of eclosion 10 days after irradiation. The averages are from nine samples each for WT for each treatment, five samples for grp 0 R minus drug and 4000 R minus and plus drug, and six samples for grp 0 R plus drug, in two separate experiments. A total of 4244 WT and 573 grp animals were counted. Error bars, SEM.
Topotecan is currently approved for treatment of ovarian and small cell lung cancer (SCLC) and is also in clinical trials for non-SCLC (www.hycamtin.com). In addition, its effectiveness in combination therapy with radiation for brain, lung, and cervical cancers is being evaluated in phase I and II clinical trials (www.clinicaltrials.gov). In cell culture and in xenografts of human cancer cells, topotecan and its derivatives can act as radiation sensitizers, although this effect appears highly dependent on the cell line and experimental protocol used (e.g., Chastagner et al. 2001). In particular, whether topotecan analogs can radiosensitize in a genotype-specific manner remains to be determined. In other words, the effectiveness of topotecan analogs may be dependent on particular mutation(s) present in the cell and understanding this relationship could help design better therapeutic regimes in treatment of cancers.
We find that the topotecan derivative identified in our screen, 9-glycineamido-20(S)-camptothecin-HCl, is a radiation sensitizer that acts in a genotype-specific manner in Drosophila (Figure 4B). Without radiation, topotecan reduced the survival of grp mutant larvae over wild type. With radiation, wild-type survival was reduced from 79.5% (drug alone) to 31.0% (radiation plus drug), reflecting a 2.7-fold reduction. For grp mutants, survival was reduced from 42.5% (drug alone) to 5.6% (drug plus radiation), reflecting a 7.6-fold reduction in survival. Thus, topotecan appears to act in a synergistic manner with radiation to kill grp mutants preferentially over wild-type larvae.
DISCUSSION
Checkpoints arrest the cell cycle in response to DNA damage to allow repair. As such, in single cells and unicellular organisms, checkpoint proteins including Chk1 are critical for survival after exposure to DNA-damaging agents. Our previous data with Drosophila larvae revealed that checkpoint regulation of the cell cycle is not necessary for surviving exposure to ionizing radiation. Here we offer an explanation for the apparent difference in the requirement for checkpoints of cells and multicellular organisms.
Grp is required by cells to survive irradiation:
Although grp is dispensable for Drosophila larvae to survive irradiation, this is not the case for Drosophila cells; cells that lack Grp fail to increase their number relative to those that contain Grp. This could be caused by increased cell death, decreased cell proliferation, or both. We have not found evidence for increased cell death in Grp-depleted samples after irradiation (e.g., Figure 1G). Therefore, Grp-depleted cells likely survived irradiation as well as controls, but later showed proliferation defects. This interpretation is in agreement with the behavior of cells in larval imaginal discs. Irradiation of Drosophila larvae with 4000 R of X rays results in the death of up to 75% of cells in imaginal discs (Haynie and Bryant 1977; Jaklevic and Su 2004). In wild-type larvae, checkpoint arrest of the cell cycle is followed by a recovery period in which there is increased mitotic activity compared to unirradiated controls. Imaginal discs from irradiated grp larvae show comparable levels of cell death as wild type, but failed to show increased mitotic activity during the recovery period (Jaklevic and Su 2004).
The phenomenon in which cells are alive, yet unable to reproduce has been termed “reproductive death,” as opposed to apoptotic or necrotic death. Reproductive death is well documented in radiation biology of mammalian cells and is also described as senescence-like (Hall 2000; Tsai et al. 2005). Reproductive death is of clinical interest because its induction may help control tumor growth even when apoptotic death is prevented due to mutations. The basis for reproductive death is poorly understood but our results suggest that Drosophila may be a good model to dissect the genetic basis for this phenomenon.
Growth as a determinant for radiation survival:
Most of what we know of radiation responses in eukaryotes is derived from unicellular models such as yeast and mammalian cells. These studies identified cell cycle checkpoints and DNA repair as being essential for survival to DNA damage caused by radiation. Here we add to that list by demonstrating additional requirements in a multicellular context, namely nutritional uptake, growth, and compensatory proliferation. Because dmyc, cdk4, and chico are part of or act downstream of distinct growth factor signaling pathways, it is unlikely that there is a single growth pathway responsible for radiation survival. Rather, multiple pathways likely contribute. It is, however, also possible that each cell type or tissue relies on a single pathway, but that all contribute at the level of a multicellular organism.
An in vivo screen for radiation sensitizers that act on checkpoint mutants:
We find that multicellular organisms deficient in cell cycle regulation are at a disadvantage as illustrated by increased sensitivity of grp mutant larvae to treatments expected to interfere with growth and proliferation. This difference can be exploited in a screen for radiation sensitizers that preferentially affect checkpoint mutants. We find that such a screen can yield molecules (topotecan and camptothecin) that are effective therapeutic agents against human cancers, which are often associated with defects in cell cycle checkpoints. Topotecan and campothecin inhibit topoisomerase I, an enzyme needed for many DNA-dependent processes such as DNA replication and transcription. We speculate that inhibition of topoisomerase I interferes with compensatory proliferation that is necessary to replace cells lost by IR-induced apoptosis, leading to increased sensitivity to IR. This would be similar to the case of colchicine, a microtubule poison that interferes with mitosis. Note that the Structural Diversity Set we screened through includes molecules based on their structural properties and not on their anticancer activity. Therefore, the fact that the screen yielded two anticancer agents is encouraging.
Topoisomerase I inhibitors can induce DSBs. It may be argued that topotecan and campothecin are radiation sensitizers because their presence simply mimics an increased IR dose. In this case, however, we would expect the effects of drug and IR to be additive. This is not the case. Instead, our data indicate a synergistic effect, which could be explained by the inhibition of two different pathways, one of which can compensate for the other. Specifically, compensatory proliferation, we propose, is compensating for and consequently masking the effects of IR. Only when the compensatory process is inhibited should one see the effect of IR, which is what we believe is happening.
The differential effect on checkpoint mutant larvae vs. wild type, if translatable between Drosophila larvae and human cancers, would mean that we can potentially find agents that preferentially eliminate mutant (cancerous) tissue while leaving the noncancerous tissue less affected or unaffected. Radiation responses between Drosophila and mammals show close parallels. Conserved kinases such as ATR and Chk1 mediate checkpoint responses in both systems (Chehab et al. 2000; Hirao et al. 2000), while conserved molecules like p53 and Chk2 mediate apoptosis (Brodsky et al. 2000, 2004; Sogame et al. 2003). DNA repair is essential for radiation resistance in Drosophila (Jaklevic and Su 2004) and contributes to radiation resistance of cancer cells (Schwartz et al. 1988). Radiation itself induces extra proliferation in Drosophila; we detect increased mitotic activity beyond normal levels at ∼24 hr after irradiation (Jaklevic and Su 2004). Radiation also induces in mammalian tumors “accelerated repopulation,” where surviving cells divide faster than before (Hall 2000, pp. 404–405). Repopulation by survivors can confer resistance to radiation treatment in both systems. Finally, growth factor signaling is important for radiation resistance in Drosophila and possibly also in humans. For example, the role of the IGF pathway in radiation survival in Drosophila (this report) mirrors the role of IGF in radiation resistance of human tumors; tumors deficient in the phosphatase and tensin homolog (PTEN) tumor suppressor are more radiation resistant (Wick et al. 1999; Harima et al. 2001). PTEN is an inhibitor of IGF signaling, which is overactivated in PTEN mutants. On the basis of these parallels and results described herein, we suggest that Drosophila may be an effective in vivo screening tool for finding small molecule radiation sensitizers that have the potential to eliminate mutant tumors while sparing healthy wild-type tissues. Furthermore, we predict that these molecules would include inhibitors of processes that contribute to survival after irradiation: DNA repair, growth (via IGF, MYC, and CDK4), and compensatory proliferation and any other radioprotective processes yet to be identified. For example, compensatory proliferation may rely on extracellular signals or cell–cell and cell–matrix attachment. Inhibitors of these processes could be potent radiation sensitizers. Such molecules would have been missed in cell-based screens that generated most anticancer therapeutics in use.
Acknowledgments
We thank Nancy Mendoza, Chandra Kilburn, Jannie Guzman, Joy Boyd, Matthew Schenk, and Vondina Brown for their assistance with the screen; the Hafen, Gallant, and Lehner labs and the Bloomington Stock Center for fly stocks; P. H. O'Farrell for the H2B–GFP S2 cell line; the Su lab members and Larry Gold for suggestions on the design of the screen; and the National Cancer Institute–Developmental Therapeutic Program and R. Schultz for small molecule libraries. This work was supported by grants from the National Institutes of Health to T.T.S. (RO1 GM66441 and R21 GM073973), a Dutch Cancer Society grant (KWF RuG99-1949), and a grant from The Netherlands Organization for Scientific Research (VIDI 917-36-400) to O.S.
References
- Akiyama, T., 2002. Mutations of stonewall disrupt the maintenance of female germline stem cells in Drosophila melanogaster. Dev. Growth Differ. 44: 97–102. [DOI] [PubMed] [Google Scholar]
- al-Khodairy, F., E. Fotou, K. S. Sheldrick, D. J. Griffiths, A. R. Lehmann et al., 1994. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol. Biol. Cell 5: 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohni, R., J. Riesgo-Escovar, S. Oldham, W. Brogiolo, H. Stocker et al., 1999. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell 97: 865–875. [DOI] [PubMed] [Google Scholar]
- Brodsky, M. H., W. Nordstrom, G. Tsang, E. Kwan, G. M. Rubin et al., 2000. Drosophila p53 binds a damage response element at the reaper locus. Cell 101: 103–113. [DOI] [PubMed] [Google Scholar]
- Brodsky, M. H., B. T. Weinert, G. Tsang, Y. S. Rong, N. M. McGinnis et al., 2004. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol. Cell. Biol. 24: 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby, E. C., D. F. Leistritz, R. T. Abraham, L. M. Karnitz and J. N. Sarkaria, 2000. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 60: 2108–2112. [PubMed] [Google Scholar]
- Chastagner, P., S. V. Kozin and A. Taghian, 2001. Topotecan selectively enhances the radioresponse of human small-cell lung carcinoma and glioblastoma multiforme xenografts in nude mice. Int. J. Radiat. Oncol. Biol. Phys. 50: 777–782. [DOI] [PubMed] [Google Scholar]
- Chehab, N. H., A. Malikzay, M. Appel and T. D. Halazonetis, 2000. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 14: 278–288. [PMC free article] [PubMed] [Google Scholar]
- Datar, S. A., H. W. Jacobs, A. F. de la Cruz, C. F. Lehner and B. A. Edgar, 2000. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 19: 4543–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries, H. I., L. Uyetake, W. Lemstra, J. F. Brunsting, T. T. Su et al., 2005. Grp/DChk1 is required for G2-M checkpoint activation in Drosophila S2 cells, whereas Dmnk/DChk2 is dispensable. J. Cell Sci. 118: 1833–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty, P., S. D. Campbell, R. Abu-Shumays, B. S. Phalle, K. R. Yu et al., 1997. The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr. Biol. 7: 418–426. [DOI] [PubMed] [Google Scholar]
- Furnari, B., N. Rhind and P. Russell, 1997. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 277: 1495–1497. [DOI] [PubMed] [Google Scholar]
- Gallant, P., Y. Shiio, P. F. Cheng, S. M. Parkhurst and R. N. Eisenman, 1996. Myc and Max homologs in Drosophila. Science 274: 1523–1527. [DOI] [PubMed] [Google Scholar]
- Graves, P. R., L. Yu, J. K. Schwarz, J. Gales, E. A. Sausville et al., 2000. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J. Biol. Chem. 275: 5600–5605. [DOI] [PubMed] [Google Scholar]
- Hall, E., 2000. Radiation Biology for Radiologists. Lippincott Williams & Wilkins, Philadelphia.
- Harima, Y., S. Sawada, K. Nagata, M. Sougawa, V. Ostapenko et al., 2001. Mutation of the PTEN gene in advanced cervical cancer correlated with tumor progression and poor outcome after radiotherapy. Int. J. Oncol. 18: 493–497. [DOI] [PubMed] [Google Scholar]
- Haynie, J. L., and P. J. Bryant, 1977. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Wilhelm Roux's Arch. 183: 85–100. [DOI] [PubMed] [Google Scholar]
- Hirao, A., Y. Y. Kong, S. Matsuoka, A. Wakeham, J. Ruland et al., 2000. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287: 1824–1827. [DOI] [PubMed] [Google Scholar]
- Jaklevic, B. R., and T. T. Su, 2004. Relative contribution of DNA repair, cell cycle checkpoints, and cell death to survival after DNA damage in Drosophila larvae. Curr. Biol. 14: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, L. A., D. A. Prober, B. A. Edgar, R. N. Eisenman and P. Gallant, 1999. Drosophila myc regulates cellular growth during development. Cell 98: 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C. A., H. W. Jacobs, S. A. Datar, W. Du, B. A. Edgar et al., 2000. Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 19: 4533–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy, A., L. Uyetake, M. G. Cordeiro and T. T. Su, 2005. Regulation of mitosis in response to damaged or incompletely replicated DNA requires different levels of Grapes (Drosophila Chk1). J. Cell Sci. 118: 3305–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, Y., J. Bachant, H. Wang, F. Hu, D. Liu et al., 1999. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286: 1166–1171. [DOI] [PubMed] [Google Scholar]
- Schwartz, J. L., J. Rotmensch, S. Giovanazzi, M. B. Cohen and R. R. Weichselbaum, 1988. Faster repair of DNA double-strand breaks in radioresistant human tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 15: 907–912. [DOI] [PubMed] [Google Scholar]
- Sogame, N., M. Kim and J. M. Abrams, 2003. Drosophila p53 preserves genomic stability by regulating cell death. Proc. Natl. Acad. Sci. USA 100: 4696–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker, H., and E. Hafen, 2000. Genetic control of cell size. Curr. Opin. Genet. Dev. 10: 529–535. [DOI] [PubMed] [Google Scholar]
- Tercero, J. A., and J. F. Diffley, 2001. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557. [DOI] [PubMed] [Google Scholar]
- Tsai, K. K., E. Y. Chuang, J. B. Little and Z. M. Yuan, 2005. Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue microenvironment. Cancer Res. 65: 6734–6744. [DOI] [PubMed] [Google Scholar]
- Walworth, N., S. Davey and D. Beach, 1993. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 363: 368–371. [DOI] [PubMed] [Google Scholar]
- Weinert, T. A., and L. H. Hartwell, 1988. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241: 317–322. [DOI] [PubMed] [Google Scholar]
- Wick, W., F. B. Furnari, U. Naumann, W. K. Cavenee and M. Weller, 1999. PTEN gene transfer in human malignant glioma: sensitization to irradiation and CD95L-induced apoptosis. Oncogene 18: 3936–3943. [DOI] [PubMed] [Google Scholar]
- Zachos, G., M. D. Rainey and D. A. Gillespie, 2003. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 22: 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, B. B., and S. J. Elledge, 2000. The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439. [DOI] [PubMed] [Google Scholar]