

Abstract
Statistical methods for mapping quantitative trait loci (QTL) have been extensively studied. While most existing methods assume normal distribution of the phenotype, the normality assumption could be easily violated when phenotypes are measured in counts. One natural choice to deal with count traits is to apply the classical Poisson regression model. However, conditional on covariates, the Poisson assumption of mean–variance equality may not be valid when data are potentially under- or overdispersed. In this article, we propose an interval-mapping approach for phenotypes measured in counts. We model the effects of QTL through a generalized Poisson regression model and develop efficient likelihood-based inference procedures. This approach, implemented with the EM algorithm, allows for a genomewide scan for the existence of QTL throughout the entire genome. The performance of the proposed method is evaluated through extensive simulation studies along with comparisons with existing approaches such as the Poisson regression and the generalized estimating equation approach. An application to a rice tiller number data set is given. Our approach provides a standard procedure for mapping QTL involved in the genetic control of complex traits measured in counts.
MODERN biological techniques make it possible to detect the abundant variation of molecular polymorphisms that segregate in most species in nature. Consequently, it is possible to detect quantitative trait loci (QTL) underlying quantitative variation of certain traits and to map their chromosomal locations throughout the entire genome using statistical methods (Mackay 2001). Given the recent intriguing result of gene cloning from rice on the basis of QTL mapping results (Li et al. 2006), QTL mapping is still proven to be an important tool for gene discovery in the postgenomic era. Therefore, there is a great demand to develop efficient statistical methods to improve the precision and power of QTL mapping, not only for continuous traits, but also for discrete traits such as those measured in counts.
Most current statistical methods for QTL mapping in experimental crosses date back to the seminal mapping article of Lander and Botstein (1989). Since then, this work has been extended and improved by a number of statistical methods, for example, composite interval mapping (Zeng 1994) and multiple-interval mapping (Kao et al. 1999). However, most of the existing methods developed so far assume that the phenotypic trait be normally distributed. This assumption is easily violated when the phenotype of interest shows nonnormal characteristics, for instance, pertaining to survival time (Diao et al. 2004) or displaying a binary characteristic (Xu and Atchley 1996).
Another type of data often observed in real experiments is count data where the phenotype of interest is measured in counts. For example, the number of roots generated in a plant (Lall et al. 2004), CD4 T cell counts in a human study (Hall et al. 2002), and the number of cholesterol gallstones formed in mice (Wittenburg et al. 2003) are all examples of phenotypes measured in counts. The distribution of these types of data is generally skewed, especially when the mean is comparatively small. Tilquin et al. (2001) proposed to perform a mathematical transformation of count data and then apply a standard QTL mapping approach such as least squares (Haley and Knott 1992), maximum likelihood (Lander and Botstein 1989), or a nonparametric approach (Kruglyak and Lander 1995). However, the nature of the data distribution is still not incorporated and consequently the mapping power might be affected. Due to the lack of an efficient statistical method, the standard QTL mapping approach assuming normally distributed phenotypes is still being applied (Lall et al. 2004).
When the sampling variance of a count variable Y is significantly greater or less than that predicted by an expected probability distribution, Y is said to be over- or underdispersed, respectively. A natural way to analyze regular count data is to use a Poisson regression model where the Poisson mean can be modeled as a function of linear predictors through the log link function in a generalized linear model (GLM) setting (McCullagh and Nelder 1989). Using parametric approaches by applying Poisson distribution in QTL mapping has been previously proposed (Rebaï 1997; Shepel et al. 1998; Sen and Churchill 2001). These approaches were built on the maximum-likelihood framework (Shepel et al. 1998), least-squares-based regression framework (Rebaï 1997), and Bayesian framework (Sen and Churchill 2001), and each one displays its own merits in handling count data in QTL mapping. However, if dispersion occurs, ignoring it will result in biased parameter estimates, which may lead to incorrect conclusions and inferences (Wang 1994). Therefore, these approaches are greatly limited when the underlying data are potentially dispersed.
When count data are dispersed, one can apply a nonparametric approach (Kruglyak and Lander 1995) using its nice distribution-free property. However, one of its major disadvantages is that it does not provide QTL-effect estimation and hence greatly restricts its utility for inference. Moreover, it is based on the Wilcoxon rank-sum test and chooses to rank tied individuals at random. This also greatly restricts its application when the number of ties is high, especially for count data (Rebaï 1997). McCullagh and Nelder (1989) suggest modeling mean and dispersion jointly as a way to take possible dispersion into account. The GLM was later applied to a QTL mapping study using a generalized estimating equation (GEE) approach (Lange and Whittaker 2001; Thomson 2003). The GEE approach shows its merits in handling dispersion. However, since the GEE approach does not assume a full probability model, a misspecified variance may have an influence on the efficiency of the parameter estimates and a likelihood-based inference procedure cannot be applied directly. Wang et al. (1996) suggest modeling data with a mixed Poisson regression model to take data dispersion into account. Famoye (1993) proposed a generalized Poisson regression model in which the dispersion parameter can be directly estimated and tested. Neither of these two approaches has been applied in QTL mapping studies.
In this article, we propose a rigorous extension of the interval-mapping approach to count traits. We model the QTL effects through a generalized Poisson regression model (Famoye 1993) and develop efficient likelihood-based inference procedures. Residual analysis and goodness-of-fit tests are proposed to check the model fitting. This approach, implemented with the EM algorithm, allows for a genomewide scan for the existence of QTL throughout the entire genome. Extensive simulation studies are performed to evaluate the statistical behavior of the approach. Comparisons with the GEE approach and the Poisson regression are also given on the basis of simulations. An application to a rice tiller number data set is provided in which several QTL are detected to affect tiller growth. Our approach provides a standard procedure for mapping QTL involved in the genetic control of complex traits measured in counts.
MODELS
Generalized Poisson regression model:
Suppose that Yi is a count response variable that follows a generalized Poisson distribution (Famoye 1993). The probability function of Yi is given by
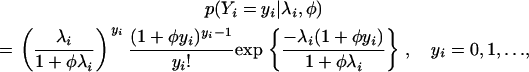 |
(1) |
where λi is the mean of the function and can be expressed as a function of genetic and nongenetic factors; i.e., 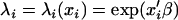 , where xi is a p-dimensional vector of covariates including genetic and nongenetic factors, β is a p-dimensional vector of regression parameters, and φ is a dispersion parameter.
, where xi is a p-dimensional vector of covariates including genetic and nongenetic factors, β is a p-dimensional vector of regression parameters, and φ is a dispersion parameter.
The generalized Poisson regression (GPR) model (1) is a generalization of the standard Poisson regression (PR) model. When the dispersion parameter φ = 0, the probability function in (1) reduces to the PR model. When φ > 0, the GPR model represents count data with overdispersion and when φ < 0, the GPR model represents count data with underdispersion. Therefore, the GPR model shows more flexibility in modeling count data when the underlying data show varying degrees of dispersion. Since the parameter φ is restricted to 1 + φλi > 0 and 1 + φyi > 0, the model is also called the restricted generalized Poisson regression model (Famoye 1993).
The mean of the response in the GPR model is given by E(Yi | λi, φ) = λi and the variance is given by V(Yi | λi, φ) = λi(1 + φλi)2. Clearly, when φ > 0, the variance is overdispersed and when −2/λi < φ < 0, the variance is underdispersed. The GPR model is very useful for modeling count data, especially when mean and variance differ.
Interval-mapping approach:
In this section, we develop an interval-mapping method for potentially dispersed count traits in a backcross population. Expanding the results to other crosses such as an F2 or a recombinant inbred line (RIL) is straightforward. Suppose that there is a putative QTL that is segregating with two alleles Q and q in a backcross population of size n, initiated with two contrasting inbred lines. The QTL is assumed to be responsible for the quantitative variation of the phenotype measured in counts. Data are randomly collected, which include a set of genetic markers with a known genetic linkage map and set of phenotype data.
For simplicity, we ignore nongenetic covariates and consider only the genetic covariates. Let xi = 1 or 0 according to whether the QTL genotype for the ith subject is QQ or Qq, respectively. We specify a GPR model for the effects of the QTL genotype on the count trait such that, conditional on the QTL genotype Gi, the mean of the GPR model can be expressed as
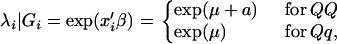 |
(2) |
where β = (μ, a) in which μ is the overall genetic effect, and a is the additive genetic effect.
Statistical methods for mapping QTL on the basis of a mixture model have been previously developed (Lander and Botstein 1989). In the mixture model, each observation y is assumed to have arisen from one of j components (QTL genotypes), with each component being modeled by a probability distribution function, for example, a generalized Poisson regression function in the current setting. At each locus, the conditional probability of QTL genotype j given on the flanking markers Mi for individual i can be calculated, which is expressed as πi|j = Pr(xi = j | Mi) (i = 1, . . . , n), where n is the total sample size and j takes value 1 or 0 depending on whether the QTL genotype is QQ or Qq (Lynch and Walsh 1998). The conditional probability is considered as the mixture proportion in the mixture model. For the backcross family, the mixture model has the form
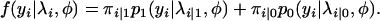 |
(3) |
From the mixture distribution, we can easily compute the unconditional mean and variance of Yi, which are expressed as
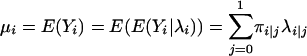 |
(4) |
and
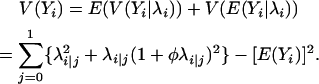 |
(5) |
Assuming independent observations, the log-likelihood function given the phenotype y and marker data M can be expressed as
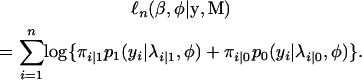 |
(6) |
The parameters specifying function p1 are (μ, a, φ) with λi|1 = exp(μ + a) and the parameters specifying function p0 are (μ, φ) with λi|0 = exp(μ).
Parameter estimation:
Define 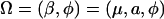 , which contains the genetic parameters and dispersion parameter. The maximum-likelihood estimate (MLE)
, which contains the genetic parameters and dispersion parameter. The maximum-likelihood estimate (MLE) 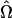 for Ω is such that it solves the partial-derivative equation with respect to the rth parameter contained in Ω: ∂ℓn(Ω)/∂Ωr = 0. In practice, we treat the positions of QTL, τ, as known parameters rather than unknown, although their MLEs can also be obtained through iterative steps. We can then use a grid search approach to estimate the QTL positions. By hypothesizing a QTL every 1 or 2 cM at marker intervals, the landscape of log-likelihood test statistics throughout the entire genome can be obtained. The positions corresponding to the peak of the landscape across a linkage group are the MLEs of the QTL positions. Therefore, a computational algorithm can be formulated as follows. For any fixed QTL position, τ, the EM algorithm (Dempster et al. 1977) is used to find the restricted MLE
for Ω is such that it solves the partial-derivative equation with respect to the rth parameter contained in Ω: ∂ℓn(Ω)/∂Ωr = 0. In practice, we treat the positions of QTL, τ, as known parameters rather than unknown, although their MLEs can also be obtained through iterative steps. We can then use a grid search approach to estimate the QTL positions. By hypothesizing a QTL every 1 or 2 cM at marker intervals, the landscape of log-likelihood test statistics throughout the entire genome can be obtained. The positions corresponding to the peak of the landscape across a linkage group are the MLEs of the QTL positions. Therefore, a computational algorithm can be formulated as follows. For any fixed QTL position, τ, the EM algorithm (Dempster et al. 1977) is used to find the restricted MLE 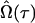 , with the Newton–Raphson algorithm employed in the M-step (detailed instructions are given in the appendix). Then obtain
, with the Newton–Raphson algorithm employed in the M-step (detailed instructions are given in the appendix). Then obtain 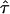 by varying τ over an interval with a small increment of 1 or 2 cM at a time.
by varying τ over an interval with a small increment of 1 or 2 cM at a time.
With a backcross design, we have two mixture components. For a fixed number of mixtures, asymptotic normality of 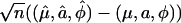 can be proved under standard regularity conditions (Lehmann 1983). However, as restricted by the condition 1 + φyi > 0, the parameter φ is bounded below by the observed data set. If φ reaches the lower bound, the asymptotic normality for φ may not be satisfied. The approximate standard errors of the estimates can be obtained from the by-product of the Newton–Raphson algorithm. Applying Wald's (1949) consistency argument and using the techniques developed in Chen and Chen (2005), we can prove the consistency of the MLEs of Ω under the GPR mixture model. If a QTL exists in an interval, i.e., a ≠ 0, the MLE of QTL position τ is also consistent.
can be proved under standard regularity conditions (Lehmann 1983). However, as restricted by the condition 1 + φyi > 0, the parameter φ is bounded below by the observed data set. If φ reaches the lower bound, the asymptotic normality for φ may not be satisfied. The approximate standard errors of the estimates can be obtained from the by-product of the Newton–Raphson algorithm. Applying Wald's (1949) consistency argument and using the techniques developed in Chen and Chen (2005), we can prove the consistency of the MLEs of Ω under the GPR mixture model. If a QTL exists in an interval, i.e., a ≠ 0, the MLE of QTL position τ is also consistent.
Hypothesis test:
The presence of QTL responsible for the variation of the count phenotype can be tested by using the following hypotheses:
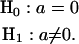 |
(7) |
The test statistic for testing the above hypotheses is calculated as the log-likelihood-ratio test statistic (LR) of the full model (H1) over the reduced model (H0),
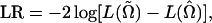 |
(8) |
where 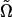 and
and 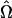 denote the MLEs of the unknown parameters under H0 and H1, respectively. Because of the mixture model, the regularity conditions for asymptotic χ2-distribution of the LR do not hold. To find the threshold value, we use the permutation test proposed by Churchill and Doerge (1994).
denote the MLEs of the unknown parameters under H0 and H1, respectively. Because of the mixture model, the regularity conditions for asymptotic χ2-distribution of the LR do not hold. To find the threshold value, we use the permutation test proposed by Churchill and Doerge (1994).
MODEL IMPLEMENTATION
Model comparison:
After specifying a regression function, different regression models such as the regular Poisson, the generalized Poisson, or the compound Poisson regression models may be applied to a given data set. A natural question arises: Which model should one adopt to fit the data for QTL analysis? This is essentially a model selection problem. Two widely used model selection criteria are Akaike's Information Criterion (AIC) (Akaike 1974) and the Bayesian Information Criterion (BIC) (Schwarz 1978). Quantitatively, the BIC puts more penalty on the log-likelihood function and the model selected by the BIC is more parsimonious. Here we define the AIC and the BIC criteria for the mixture model as
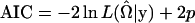 |
(9) |
and
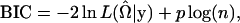 |
(10) |
where p is the number of free parameters in the defined model. The model with the smallest AIC or BIC value is selected as the best.
Dispersion test:
The GPR model reduces to the Poisson regression model when the dispersion parameter φ vanishes. To assess the adequacy of the GPR model over the Poisson regression model, and to determine whether the data are over- or underdispersed with respect to the generalized Poisson regression model, a test for the dispersion parameter can be formulated as follows:
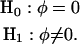 |
(11) |
When the lower bound for 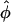 is not reached, a Wald-type test can be conducted in which
is not reached, a Wald-type test can be conducted in which 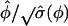 may asymptotically follow a standard normal distribution. Further theoretical investigation is needed to demonstrate the validity of the Wald test for φ under the mixture distribution framework. Alternatively we can apply the likelihood-ratio test in which the threshold is determined using permutation tests. The sign of significant test statistics suggests over- or underdispersion, where negative estimates indicate underdispersion and positive estimates indicate overdispersion. Meanwhile, significance of the test provides evidence of a better fit for the GPR model over the PR model.
may asymptotically follow a standard normal distribution. Further theoretical investigation is needed to demonstrate the validity of the Wald test for φ under the mixture distribution framework. Alternatively we can apply the likelihood-ratio test in which the threshold is determined using permutation tests. The sign of significant test statistics suggests over- or underdispersion, where negative estimates indicate underdispersion and positive estimates indicate overdispersion. Meanwhile, significance of the test provides evidence of a better fit for the GPR model over the PR model.
Residual analysis and goodness-of-fit:
After model selection and a GPR model is fitted, it is essential to check the quality of the fit. One way to check the quality of fits is to perform a residual analysis. For this purpose, we consider a Pearson or a deviance residual to check the model fit. The Pearson residual for the ith observation is defined as
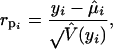 |
(12) |
where 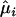 and
and 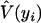 can be obtained from (4) and (5) by replacing the parameters by the MLEs. The sum of squared Pearson residuals, X2, gives the Pearson goodness-of-fit statistic for the GPR mixture model.
can be obtained from (4) and (5) by replacing the parameters by the MLEs. The sum of squared Pearson residuals, X2, gives the Pearson goodness-of-fit statistic for the GPR mixture model.
The deviance residual is defined as
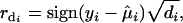 |
(13) |
where 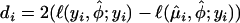 , and
, and 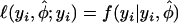 , and
, and 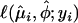 is the log likelihood of the generalized Poisson regression mixture model for yi. The goodness-of-fit of the generalized Poisson regression mixture model can be measured by the deviance
is the log likelihood of the generalized Poisson regression mixture model for yi. The goodness-of-fit of the generalized Poisson regression mixture model can be measured by the deviance 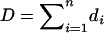 .
.
The above two residuals asymptotically follow the standard normal distribution as 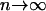 . Therefore, large residuals may indicate poorly fitting observations (Pierce and Schafer 1986). An index plot of these residuals may be used for detection of potential outliers.
. Therefore, large residuals may indicate poorly fitting observations (Pierce and Schafer 1986). An index plot of these residuals may be used for detection of potential outliers.
After calculating the residuals, we can also use these residuals to test the goodness-of-fit for the GPR mixture model. The Pearson's statistic X2 and the deviance statistic D are asymptotically distributed as 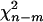 under the null hypothesis, where m is the number of free parameters under the alternative (Wang et al. 1996). A large value of X2 or D indicates poor fit.
under the null hypothesis, where m is the number of free parameters under the alternative (Wang et al. 1996). A large value of X2 or D indicates poor fit.
We can also apply the techniques developed by Wang et al. (1996) to evaluate how the ith observation affects a set of parameter estimates. We define the following quantity as the influential estimate (IE) for individual i, which has the form
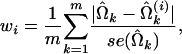 |
(14) |
where 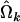 and
and 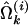 are the MLEs of the GPR mixture model based on the complete data set of n individuals and on the dataset of n − 1 individuals excluding the ith individual, respectively; and m is the total number of parameters in the model. The IE calculated for individual i can be interpreted as the average relative coefficient changes for a set of estimates and is useful for assessing the effect of parameter estimates by exclusion of the ith observation (Wang et al. 1996). Therefore, a relatively large value of wi indicates a potential influential observation that might cause instability in model fitting. An index plot of w may be used for detection of the influential point.
are the MLEs of the GPR mixture model based on the complete data set of n individuals and on the dataset of n − 1 individuals excluding the ith individual, respectively; and m is the total number of parameters in the model. The IE calculated for individual i can be interpreted as the average relative coefficient changes for a set of estimates and is useful for assessing the effect of parameter estimates by exclusion of the ith observation (Wang et al. 1996). Therefore, a relatively large value of wi indicates a potential influential observation that might cause instability in model fitting. An index plot of w may be used for detection of the influential point.
SIMULATION
To investigate the statistical behavior of the proposed methods in practical situations, we perform Monte Carlo simulations. The simulation is designed to evaluate the model performance considering the effects of sample sizes (n = 100, 200, and 400) and the pattern of dispersion (under-, non-, and overdispersion) on parameter estimation as well as the mapping power. The mapping power is defined as the proportion of simulations in which a significant QTL is identified. Consider a backcross population with which a 100-cM-long linkage group composed of six equidistant markers is constructed. A putative QTL that affects the phenotype of interest is located at 48 cM from the first marker on the linkage group. The Haldane map function is used to convert the map distance into the recombination fraction. To test the model performance, we simulate data with different specifications, namely different sample sizes (n = 100, 200, and 400), and different patterns of dispersion using the proposed GPR mixture model.
In each simulation scenario, 1000 Monte Carlo repetitions are performed. For each Monte Carlo sample, the EM algorithm is used to obtain the MLEs of parameters. Table 1 tabulates the MLEs of all parameter estimates. The square root of the mean square error (SMSE) is given in parentheses, which provides a measure of precision for each parameter estimate. The result listed in the first row for each given sample size is obtained using the GPR model, and the one in the second row is obtained using the regular PR model. In general, the GPR model can provide reasonable estimates of the QTL positions (τ) and effects of various kinds, with estimation precision depending on sample size and dispersion pattern. As expected, the precision of QTL parameters increases with increased sample size. For example, the SMSE of the mean parameter a decreases by more than twofold under different dispersion patterns when the sample size increases from 100 to 400. Meanwhile, as the sample size increases, the mapping power also increases (Table 1).
TABLE 1.
The mean MLEs with their square-root mean square errors (SMSEs) (in parentheses) of the parameters estimated from 1000 simulation replicates with different dispersion patterns
| n | τ = 48 cM | φ = −0.03 | μ = 2 | a = 0.2 | Power |
|---|---|---|---|---|---|
| Underdispersion | |||||
| 100 | 47.27 (14.19) | −0.0317 (0.0065) | 1.9966 (0.0425) | 0.2055 (0.0589) | 85 |
| 47.32 (15.16) | — | 2.0037 (0.0412) | 0.1918 (0.0517) | 81.3 | |
| 200 | 47.34 (8.29) | −0.0308 (0.0045) | 1.9978 (0.0289) | 0.2028 (0.0396) | 99 |
| 47.25 (8.58) | — | 2.0077 (0.0288) | 0.1851 (0.0399) | 94 | |
| 400 | 47.69 (5.145) | −0.0302 (0.003) | 1.9996 (0.0208) | 0.2004 (0.0279) | 100 |
| 46.59 (6.661) | — | 2.0089 (0.0218) | 0.1832 (0.0306) | 99.9 | |
| n | τ = 48 cM | φ = 0 | μ = 2 | a = 0.3 | Power |
| No dispersion | |||||
| 100 | 47.18 (13.323) | −0.0021 (0.0085) | 1.9963 (0.0545) | 0.3046 (0.0743) | 93.2 |
| 47.19 (13.389) | — | 1.9970 (0.0543) | 0.3033 (0.0739) | 93.5 | |
| 200 | 47.24 (6.893) | −0.0009 (0.0059) | 1.9972 (0.0377) | 0.3025 (0.0515) | 100 |
| 47.23 (6.885) | — | 1.9976 (0.0375) | 0.3018 (0.0510) | 99.9 | |
| 400 | 47.86 (4.116) | −0.0003 (0.0040) | 1.9995 (0.0272) | 0.3002 (0.0362) | 100 |
| 47.85 (4.117) | — | 1.9995 (0.0271) | 0.3001 (0.0360) | 100 | |
| n | τ = 48 cM | φ = 0.015 | μ = 1.6 | a = 0.3 | Power |
| Overdispersion | |||||
| 100 | 47.19 (15.754) | 0.0113 (0.0138) | 1.5912 (0.0740) | 0.3125 (0.1042) | 74.5 |
| 47.34 (17.327) | — | 1.5866 (0.0756) | 0.3207 (0.1078) | 71.2 | |
| 200 | 47.43 (10.596) | 0.0134 (0.0096) | 1.5943 (0.0496) | 0.3068 (0.0682) | 95.3 |
| 47.44 (10.874) | — | 1.5885 (0.0512) | 0.3171 (0.0714) | 93.5 | |
| 400 | 47.56 (6.405) | 0.0145 (0.0065) | 1.5988 (0.0358) | 0.3014 (0.0483) | 100 |
| 47.66 (6.518) | — | 1.5925 (0.0369) | 0.3125 (0.0507) | 99.9 | |
Data are simulated using the GPR model. Power is calculated as the percentage of all simulations in which the significant QTL is detected. The first and second rows for a given sample size correspond to the results analyzed using the GPR and the PR approaches, respectively.
Under different dispersion patterns, all the parameters can be reasonably estimated with high precision with the GPR model, which suggests the robustness of the model and good convergence rate of parameters. A minor difference is observed for the QTL location estimates in which slightly higher precision is observed when data show no dispersion. When the sample size is small (i.e., n = 100), high mapping power is observed when data show no dispersion compared to dispersed data (Table 1). For example, the power is 93.2% for no dispersion data compared to 74.5% for overdispersed data and 85% for underdispersed data with the sample size 100. The reduction of mapping power is possibly due to extra data variation caused by under- or overdispersion. The difference is not so notable when sample size increases to 200, which suggests that a reasonable sample size of 200 is needed in practice.
Comparisons between the GPR and PR model are summarized in Table 1, where the underlying data are simulated using the GPR model. When data are potentially dispersed, the GPR model outperforms the PR model with increased precision for QTL location and other genetic parameter estimation as well as increased testing power. The differences are more notable when sample size is small. For example, the power is 85% using the GPR model compared to 81.3% using the PR model with a sample size of 100 from underdispersed data. We also observe a larger bias for the additive effect a using the PR model compared to the GPR model, which could lead to biased inference. When data are not dispersed, both models perform similarly.
Comparisons of the current approach with the GEE-type approach (Lange and Whittaker 2001) are summarized in Figures 1 and 2. Figure 1 shows the comparisons based on the power and type I error rate. Data are simulated using the GPR model and are then analyzed using both approaches. The power and type I error are calculated on the basis of the 5% nominal level from the permutation test (Churchill and Doerge 1994). When data are dispersed and sample size is small (100 say), the GPR approach has higher power than the GEE approach. As sample size increases to ≥200, these two approaches are comparable. Both approaches underestimate the type I error rate when sample size is small and data are dispersed. When sample size increases to 400, the GEE approach overestimates the type I error and performs poorly compared to the GPR approach. When data are not dispersed, the difference in power is not significant, but it is not so for the type I error.
Figure 1.—

The comparison of power and type I error plots between the GEE and GPR approaches from 1000 simulation replicates. Data are simulated using the GPR model under different dispersion patterns of (A) underdispersion, (B) no dispersion, and (C) overdispersion with parameters listed in Table 1 and are analyzed using the GEE and GPR approaches separately.
Figure 2.—

The boxplot of the estimated QTL position from 1000 simulation replicates. Data are simulated using the GPR model under different dispersion patterns with parameters listed in Table 1 and are analyzed using the GEE and the GPR approaches separately. The numbers 1, 2, and 3 on the vertical axis indicate sample sizes 100, 200, and 400, respectively. The true QTL position is simulated at 48 cM away from the first marker indicated by the vertical dotted line.
A boxplot of the QTL position estimates is given in Figure 2, which displays the interquantile and the range of the estimated position. Outliers are indicated by asterisks. The notch indicates a robust estimate of the uncertainty about the median. The dotted vertical line represents the true QTL location. In all simulation studies, the GPR approach gives more efficient estimates of the QTL position than the GEE approach.
APPLICATION
The proposed model is employed to reanalyze a real data set of rice tiller number (Yan et al. 1998). Two inbred lines, semidwarf IR64 and tall Azucena, were crossed to generate an F1 progeny population. By doubling haploid chromosomes of the gametes derived from the heterozygous F1, a doubled-haploid (DH) population of 123 lines was founded (Huang et al. 1997), which is genetically equivalent to a backcross population. A genetic linkage map was constructed using 175 genetic markers, with a total length of 2005 cM, representing a good coverage of 12 rice chromosomes.
The 123 DH lines were planted in a completely randomized design with two replications. Each replicate was divided into different plots, each containing eight plants per line. Starting from 10 days after transplanting, tiller numbers were measured every 10 days for five central plants in each plot until all lines had headed. The tiller numbers were averaged from the two replicates. Given that tiller number can be only an integer, the averaged tiller number was rounded to the nearest integer for QTL analysis. Since the majority of individuals have only one tiller and the rest have two tillers at day 10 after rounding, the data do not provide enough variability to fit the GPR model. Only data beginning at day 20 were subject to QTL mapping study.
Three types of statistical model are applied, namely a model with regular Poisson regression, a model with the newly proposed generalized Poisson regression, and the GEE approach (Lange and Whittaker 2001). The PR and GPR approaches lead to a significantly different LR profile throughout the genome and consequently a larger number of QTL are identified by the GPR than by the PR model. There is only one genomewide significant QTL identified by the GEE approach, located on chromosome 5 before day 50. After day 50, no genomewide significant QTL are identified. Also, the location of the identified QTL by the GEE approach is completely different from the ones detected using the GPR model. Both the dispersion test and the goodness-of-fit test show that data are underdispersed. Therefore, we focus only on the results obtained by the GPR model in this section.
By genomewide scanning for QTL at every 2 cM within each marker interval across 12 rice chromosomes, our model has identified six major QTL that trigger effects on tiller growth. As shown by the genomewide LR profile plot in Figure 3, QTL located on chromosomes 2, 5, and 8 are significant only at the 5% chromosomewide level and QTL located on chromosome 4 (marker interval RZ565–RZ675) show genomewide significance on the basis of the critical thresholds determined from 1000 permutation tests. Both QTL located on chromosome 3 show genomewide significance at days 40 and 70 but show chromosomewide significance at the other periods. One of the possible reasons that these QTL do not show genomewide significance during the whole study period might be due to small sample size. As revealed by the simulation study, the mapping power is greatly affected by sample size when data are potentially dispersed.
Figure 3.—

The profiles of the log-likelihood ratios (LR) between the full and reduced (no QTL) models estimated from the GPR mixture model for tiller growth across the entire genome from chromosome 1 to 12 at different stages. The genomic positions corresponding to the peaks of the curves are the MLEs of the QTL positions. The genomewide threshold values for claiming the existence of QTL are given as the dotted horizonal lines, and the chromosomewide threshold values are marked as the dashed–dotted line.
It is noteworthy that different QTL are involved in the control of tiller growth during different stages of rice development (Figure 3). A QTL detected on chromosome 3 (marker interval CDO337–RZ337A) has triggered continuous effects on tiller growth since activation. A QTL detected on chromosome 8 is obviously an early locus that affects tiller growth only during the first 30 days. As this QTL is switched off, some other QTL are activated to regulate tiller development. For example, a QTL on chromosome 5 becomes operational at day 40 but only functions for a short period of time and is switched off after day 50. Another QTL on chromosome 2 is then switched on at day 50 and continuously functions. Following the turn-off of the QTL on chromosome 5, the QTL located on chromosome 4 starts to function from day 70.
To know more about the behavior of the detected QTL, we tabulate the MLEs of parameters, along with the approximate standard errors of the estimates (Table 2). All the parameters are estimated with reasonably high precision as shown by the small standard errors. QTL significant at the 5% chromosomewide level are marked by single asterisks and those significant at the 5% genomewide level are marked by double asterisks. Clearly shown in Table 2, for individuals carrying genotype QQ, QTL on chromosomes 2 and 5 trigger a positive effect on tiller growth while the rest of the QTL located on chromosomes 3, 4, and 8 exert a negative effect on tiller growth. Depending on the need in breeding, a scientist may pay particular attention to those QTL.
TABLE 2.
Estimated genetic effects and their asymptotic standard errors (in parentheses) of QTL detected for the tiller number of the DH population at different stages
| Chromosome | Marker interval | Day | μ | a | φ | LR |
|---|---|---|---|---|---|---|
| 2 | RG654–RG256 | 50 | 2.264 (0.038) | 0.144 (0.048) | −0.027 (0.005) | 8.90* |
| 60 | 2.194 (0.038) | 0.165 (0.048) | −0.031 (0.005) | 11.82* | ||
| 70 | 2.061 (0.044) | 0.168 (0.056) | −0.027 (0.006) | 8.94* | ||
| 80 | 1.871 (0.039) | 0.165 (0.048) | −0.056 (0.005) | 11.54* | ||
| 90 | 1.847 (0.039) | 0.169 (0.048) | −0.058 (0.005) | 12.06* | ||
| 3 | CDO337–RZ337A | 20 | 1.589 (0.032) | −0.168 (0.054) | −0.104 (0.006) | 9.49* |
| 30 | 2.258 (0.029) | −0.168 (0.048) | −0.037 (0.005) | 11.44* | ||
| 40 | 2.488 (0.028) | −0.193 (0.045) | −0.027 (0.004) | 17.13** | ||
| 50 | 2.419 (0.029) | −0.149 (0.046) | −0.028 (0.005) | 9.95* | ||
| 60 | 2.361 (0.029) | −0.161 (0.046) | −0.032 (0.005) | 11.73* | ||
| 70 | 2.258 (0.032) | −0.223 (0.052) | −0.033 (0.005) | 17.25** | ||
| 80 | 2.048 (0.029) | −0.163 (0.047) | −0.057 (0.005) | 11.9* | ||
| 90 | 2.018 (0.030) | −0.159 (0.049) | −0.057 (0.005) | 9.93* | ||
| 3 | RZ519–Pgi-1 | 30 | 2.299 (0.027) | −0.204 (0.048) | −0.039 (0.005) | 11.56* |
| 40 | 2.516 (0.026) | −0.226 (0.045) | −0.029 (0.004) | 17.41** | ||
| 50 | 2.447 (0.027) | −0.197 (0.047) | −0.029 (0.004) | 11.83* | ||
| 60 | 2.383 (0.028) | −0.178 (0.047) | −0.032 (0.005) | 10.37* | ||
| 70 | 2.274 (0.032) | −0.234 (0.055) | −0.029 (0.006) | 13.05** | ||
| 80 | 2.065 (0.027) | −0.184 (0.047) | −0.058 (0.005) | 10.86* | ||
| 90 | 2.029 (0.029) | −0.153 (0.048) | −0.058 (0.005) | 8.03* | ||
| 4 | RZ565–RZ675 | 70 | 2.276 (0.038) | −0.216 (0.056) | −0.035 (0.006) | 13.72** |
| 80 | 2.078 (0.033) | −0.195 (0.048) | −0.061 (0.005) | 14.25** | ||
| 90 | 2.056 (0.033) | −0.198 (0.050) | −0.059 (0.006) | 14.05** | ||
| 5 | RZ67–RZ70 | 40 | 2.332 (0.037) | 0.140 (0.046) | −0.025 (0.004) | 8.96* |
| 50 | 2.264 (0.037) | 0.166 (0.047) | −0.028 (0.004) | 12.27* | ||
| 8 | Amy3D/E–RZ66 | 20 | 1.576 (0.032) | −0.175 (0.060) | −0.099 (0.007) | 8.85* |
| 30 | 2.247 (0.026) | −0.166 (0.050) | −0.039 (0.005) | 10.75* |
The significance is at level 5% through 1000 permutation tests. *, chromosomewide significance; **, genomewide significance; LR, the log-likelihood ratio.
We pick one QTL located at chromosome 3 within marker interval RZ519–Pgi-1 at day 40 to demonstrate the model implementation. The sample mean is 11.07 and the sample variance is 6.33, which indicates potential underdispersion with respect to Poisson distribution. This conjecture is further confirmed by the dispersion test (11) as shown in Figure 4. As revealed by the real data analysis, the parameter φ never reaches the lower bound across all the linkage locations, hence we can apply Wald's test. The ratios of dispersion parameters with respect to their standard errors are all < −2 across all loci of the entire genome. The differences of the AIC and BIC values when fitting the data with the GPR mixture model and the PR mixture model are also calculated across the entire genome. As clearly shown in Figure 5, the differences are always negative for both criteria, which indicates that the GPR mixture model has better fit than PR mixture model at all loci.
Figure 4.—

The ratio of the dispersion parameter 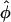 with its standard error across the entire 12 chromosomes for tillers measured at day 40.
with its standard error across the entire 12 chromosomes for tillers measured at day 40.
Figure 5.—

The differences of AIC (AICD) and BIC (BICD) information criteria between the generalized Poisson regression mixture model and the Poisson regression mixture model across the entire 12 chromosomes for tillers measured at day 40.
We also calculated the deviance and Pearson residuals as well as the influential estimates for the QTL detected on chromosome 3 at marker interval RZ519–Pgi-1 at day 40. The Pearson and the deviance goodness-of-fit statistic X2 and D are 3.45 and 86.48, respectively, with 88 d.f. These values are less than the upper 95% critical point of the 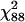 -distribution, suggesting that there is no evidence of lack of fit. The Pearson and the deviance residuals are displayed in Figure 6. The Pearson residuals appear to be normally distributed. However, the deviance residuals show that some of the data may be potential outliers such as the 47th and 53rd observations. On omitting these observations, the deviance is reduced by 0.2, while the X2 is reduced by 0.46. This implies that these observations are possible outliers, but they may not have a significant impact on the overall fit of the GPR model.
-distribution, suggesting that there is no evidence of lack of fit. The Pearson and the deviance residuals are displayed in Figure 6. The Pearson residuals appear to be normally distributed. However, the deviance residuals show that some of the data may be potential outliers such as the 47th and 53rd observations. On omitting these observations, the deviance is reduced by 0.2, while the X2 is reduced by 0.46. This implies that these observations are possible outliers, but they may not have a significant impact on the overall fit of the GPR model.
Figure 6.—

Real data, influential estimates, and residuals for tiller number observed at day 40. The influential estimates and the residuals are calculated only for QTL detected on chromosome 3 at marker interval RZ519–Pgi-1.
To check which observations are influential points on parameter estimates, we calculated the influential estimates w, which are displayed in Figure 6. As shown, observations 47 and 53 are potentially influential. If we omit these two observations, the overall genetic effect estimate μ does not change, but the additive and dispersion parameter estimates change by 11 and 17%, respectively. By further omitting three more observations (the 2nd, 71st, and 82nd), we observe a change of 33 and 16%, respectively, for additive and dispersion parameters. However, omitting these observations does not affect the likelihood-ratio test statistic as much.
EXTENSIONS
The interval-mapping approach considers only one QTL at a time (Lander and Botstein 1989). However, when the phenotypic variation is explained by more than one QTL, those QTL located elsewhere in the genome can have interfering effects. As a result, potential bias of QTL effects and location parameters may occur and the power of detecting QTL may be reduced. To overcome these problems, a number of approaches have been developed and here we consider only two popular approaches, namely composite-interval mapping (CIM) (Zeng 1994) and multiple-interval mapping (MIM) (Kao et al. 1999). We extend our single-QTL model to multiple-QTL analysis on the basis of these two approaches. An extension of the current approach to a random-mean model is also given.
Composite-interval mapping:
The basic idea of CIM is to incorporate multiple-regression analysis into interval mapping by conditioning on markers outside an interval of interest. By controlling the background markers effect, the precision and power of QTL mapping is improved. To extend the original CIM model to count traits, we consider the following mean function,
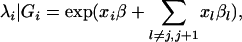 |
(15) |
where j and j + 1 represent two flanking markers bordering the putative QTL, and xl is the indicator variable for the selected background marker genotype, which takes values 1 and 0 corresponding to marker genotypes QQ and Qq, respectively. We can also model QTL interactions by considering interaction terms in model (15). Standard methods developed by CIM can be applied here to select background markers. The EM algorithm derived for interval mapping can be applied to estimate parameters.
Multiple-interval mapping:
When only one QTL is considered at a time, it could bias QTL identification and estimation if indeed multiple QTL are located in the same linkage group (Jansen 1993; Zeng 1994). To deal with these problems and to further improve QTL mapping precision, Kao et al. (1999) proposed using multiple marker intervals simultaneously to map multiple QTL of epistatic interactions throughout a linkage map. Consider s QTL, Q1,…,Qs, located on the genome. The mean function can be expressed as
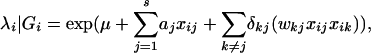 |
(16) |
where μ is the overall mean, xij is coded as 1 or 0 if the genotype of QTL Qj is QjQj or Qjqj, respectively, aj is the additive effect of Qj, wkj is the epistatic effect between Qj and Qk, and δkj is an indicator variable for epistasis between Qj and Qk. A stepwise or chunkwise selection procedure can be implemented to identify and separate linked QTL (Kao et al. 1999).
Random mean model:
The models we described so far are called fixed mean models in which the Poisson mean for each genotype is expressed as a linear function of covariates through log link function and hence is treated as fixed. A natural generalization of the model is to incorporate random effects in the linear predictor of each mixture component. When random effects are introduced, the relationship of Poisson means and the QTL genotypes can be described as
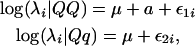 |
(17) |
where μ and a are defined the same as before, and ɛ1i and ɛ2i are two random terms that are assumed to be independent and distributed as 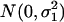 and
and 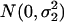 , respectively. We can also assume equal variance for the two random terms such that
, respectively. We can also assume equal variance for the two random terms such that 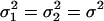 . Such a random mean model is also called the hierarchical Poisson mixture model (Wang et al. 2002). The incorporation of such random effects allows us to model the interindividual variation of Poisson means caused by the genetic effects of individuals carrying different QTL genotypes.
. Such a random mean model is also called the hierarchical Poisson mixture model (Wang et al. 2002). The incorporation of such random effects allows us to model the interindividual variation of Poisson means caused by the genetic effects of individuals carrying different QTL genotypes.
Following the GLMM formulation of McGilchrist (1994), the best linear unbiased prediction (BLUP)-type log-likelihood is given by ℓ = ℓ1 + ℓ2, where
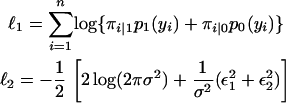 |
(18) |
The usual EM algorithm can be applied to estimate parameters. In the initial step of the M-step, dispersion parameters and coefficients in the linear predictors are estimated for fixed variance components, by maximizing the above BLUP log-likelihood (18). The variance components are then estimated using residual maximum-likelihood (REML) estimating equations. For a detailed estimation procedure, refer to Wang et al. (2002).
DISCUSSION
We have developed an efficient method in QTL mapping for count data. The generalized Poisson regression mixture model is derived on the basis of the generalized Poisson distribution proposed by Famoye (1993) and is implemented within the maximum-likelihood framework. With the incorporation of the dispersion parameter, the developed model has greater flexibility in modeling genetic count data showing different patterns of dispersion. Computer simulations demonstrate that the model has high power in mapping QTL for count data with reasonable sample size and is quite robust in various situations.
As shown by the simulation results (Table 1), the mapping power is affected by data dispersion. High power is observed when data show no dispersion. Also, the QTL location is more precisely estimated when data show no dispersion compared to over- or underdispersion. The information indicates that dispersion does affect QTL mapping precision and power.
The GPR approach outperforms the regular PR approach when the underlying data are potentially dispersed and performs similarly to the PR approach for count data with no dispersion (Table 1). As clearly demonstrated by the real data set, the theoretical information criteria such as AIC or BIC always favor the GPR model when data are potentially underdispersed. Correspondingly, more QTL are detected by the GPR model. Therefore, the GPR model should be always preferred over the PR model in real data analysis.
Given the fact that most current approaches do not account for data dispersion (Rebaï 1997; Shepel et al. 1998; Sen and Churchill 2001) and there are considerable drawbacks to implementing the nonparametric approach (Kruglyak and Lander 1995), a further comparison with the GEE approach is focused on in this article. Since the full probability model is specified, both simulation studies and real data analysis indicate that our approach shows a number of advantages over the GEE-type approaches for analyzing count data. For example, GPR is more efficient than GEE for estimating QTL location and other genetic parameters. Higher power is observed using the GPR than the GEE approach. Consequently, more QTL are detected using the GPR in real data analysis compared to the GEE. Moreover, the likelihood-based inference procedures can be easily applied under the current approach such as the goodness-of-fit test and residual analysis. These model diagnostic techniques are very useful for identifying which potential outliers and influential points affect QTL parameter estimation and inference. Also, our method is based on the maximum-likelihood estimator and is thus statistically efficient.
The same data were previously analyzed by Yan et al. (1998), assuming normality distribution for the tiller number. We obtained different results in which both methods do not agree on most QTL detected. However, their results were based on the composite-interval mapping approach, which could cause potential differences as our results are based on interval mapping. Another possible reason for the difference in the results might be due to the difference in the models fitted. A misfitting of nonnormal data with normal distribution could lead to spurious QTL.
We have described our methods in the context of a backcross population. Extensions to composite- and multiple-interval mapping are also proposed. The proposed methods can also be generalized to other populations such as F2, RIL, or combined crosses. A computer program written in MATLAB is available upon request.
Acknowledgments
We thank R. Doerge and the two anonymous referees who provided valuable suggestions that have improved every aspect of this article and who brought to our attention some important references. We also thank V. Melfi for his careful reading and comments on an earlier draft of this manuscript. The research of the first author was supported by a start-up fund from Michigan State University.
APPENDIX
The EM algorithm with the backcross population is derived as follows. Define ci = 1 or 0 if the QTL genotype is QQ or Qq, respectively, with its distribution function
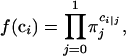 |
where πj = P(ci|j = 1). Thus,
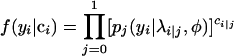 |
and
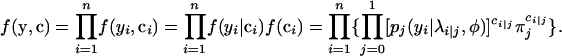 |
Then the complete log-likelihood function is given by
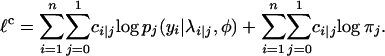 |
(A1) |
Since
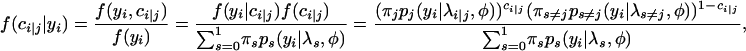 |
therefore, at the E-step of the (t)th iteration, we calculate
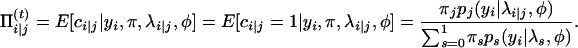 |
(A2) |
Replace the missing value ci|j by 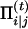 in the log-likelihood function with the complete data and then, in the M-step, we maximize
in the log-likelihood function with the complete data and then, in the M-step, we maximize
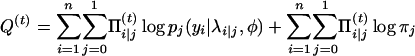 |
with respect to Ω = (μ, a, φ). To do so, we can use the Newton–Raphson iteration method, which needs the first and the second partial derivatives given below:
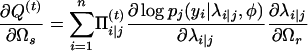 |
with
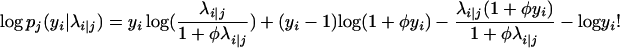 |
and
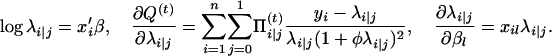 |
Thus,
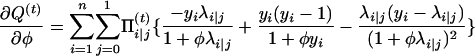 |
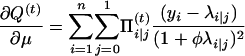 |
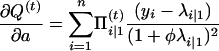 |
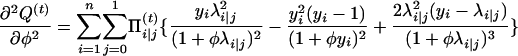 |
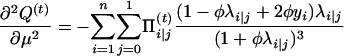 |
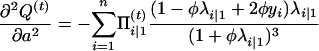 |
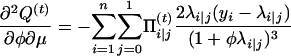 |
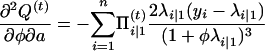 |
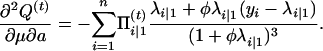 |
The Hessian matrix at the (t)th iteration is given by 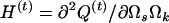 , which leads to the updated parameters Ω at the (t + 1)th iteration,
, which leads to the updated parameters Ω at the (t + 1)th iteration,
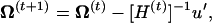 |
(A3) |
where u is a vector of the first derivative of Q(t) with respect to Ωr. The EM algorithm is repeated between Equations A2 and A3 until certain convergence criteria are satisfied.
References
- Akaike, H., 1974. A new look at the statistical model identification. IEEE Trans. Automat. Control 19: 716–723. [Google Scholar]
- Chen, Z., and H. Chen, 2005. On some statistical aspects of the interval mapping for QTL detection. Stat. Sin. 15: 909–925. [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via EM algorithm. J. R. Stat. Soc. Ser. B 39: 1–38. [Google Scholar]
- Diao, G. Q., D. Y. Lin and F. Zou, 2004. Mapping quantitative trait loci with censored observations. Genetics 168: 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famoye, F., 1993. Restricted generalized Poisson regression model. Commun. Stat. Theor. Methods 22: 1335–1354. [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Hall, M. A., P. J. Norman, B. Thiel, H. Tiwari, A. Peiffer et al., 2002. Quantitative-trait loci on chromosomes 1, 2, 3, 4, 8, 9, 11, 12, and 18 control variation in levels of T and B lymphocyte subpopulations. Am. J. Hum. Genet. 70: 1172–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, N., A. Parco, T. Mew, G. Magpantay, S. McCouch et al., 1997. RFLP mapping of isozymes, RAPD and QTL for grain shape, brown planthopper resistance in a doubled haploid rice population. Mol. Breed. 3: 105–113. [Google Scholar]
- Jansen, R. C., 1993. Interval mapping of multiple quantitative trait loci. Genetics 135: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C. H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak, L., and E. S. Lander, 1995. A nonparametric approach for mapping quantitative trait loci. Genetics 139: 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lall, S., D. Nettleton, R. DeCook, P. Che and S. H. Howell, 2004. Quantitative trait loci associated with adventitious shoot formation in tissue culture and the program of shoot development in Arabidopsis. Genetics 167: 1883–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, C., and J. C. Whittaker, 2001. Mapping quantitative trait loci using generalized estimating equations. Genetics 159: 1325–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, E. L., 1983. Theory of Point Estimation. Wiley, New York.
- Li, C. B., A. L. Zhou and T. Sang, 2006. Rice domestication by reducing shattering. Science 311: 1936–1939. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Mackay, T. F. C., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35: 303–339. [DOI] [PubMed] [Google Scholar]
- McCullagh, P., and J. A. Nelder, 1989. Generalized Linear Models. Chapman & Hall, London.
- McGilchrist, C. A., 1994. Estimation in generalized mixed models. J. R. Stat. Soc. Ser. B 56: 61–69. [Google Scholar]
- Pierce, D. A., and W. Schafer, 1986. Residuals in generalized linear models. J. Am. Stat. Assoc. 81: 977–986. [Google Scholar]
- Rebaï, A., 1997. Comparison of methods for regression interval mapping in QTL analysis with non-normal traits. Genetics 69: 69–74. [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6: 461–464. [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepel, L. A., H. Lan, J. D. Haag, G. M. Brasic, M. E. Gheen et al., 1998. Genetic identification of multiple loci that control breast cancer susceptibility in the rat. Genetics 149: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, P., 2003. A generalized estimating equations approach to quantitative trait locus detection of non-normal traits. Genet. Sel. Evol. 35: 257–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilquin, P., W. Coppieters, J. M. Elsen, F. Lantier, C. Moreno et al., 2001. Statistical power of QTL mapping methods applied to bacteria counts. Genet. Res. 78: 303–316. [DOI] [PubMed] [Google Scholar]
- Wald, A., 1949. Note on the consistency of the maximum likelihood estimate. Ann. Math. Stat. 20: 595–601. [Google Scholar]
- Wang, K., K. W. K. Yau and A. H. Lee, 2002. A hierarchical Poisson mixture regression model to analyze maternity length of hospital stay. Stat. Med. 21: 3639–3654. [DOI] [PubMed] [Google Scholar]
- Wang, P., 1994. Mixed regression models for discrete data. Ph.D. Dissertation, University of British Columbia, Vancouver, BC, Canada.
- Wang, P., M. L. Puterman, I. Cockburn and N. Le, 1996. Mixed Poisson regression models with covariate dependent rates. Biometrics 52: 381–400. [PubMed] [Google Scholar]
- Wittenburg, H., M. A. Lyons, R. Li, G. A. Churchill, M. C. Carey et al., 2003. FXR and ABCG5/ABCG8 as determinants of cholesterol gallstone formation from quantitative trait locus mapping in mice. Gastroenterology 125: 868–881. [DOI] [PubMed] [Google Scholar]
- Xu, S., and W. R. Atchley, 1996. Mapping quantitative trait loci for complex binary diseases using line crosses. Genetics 143: 1417–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, J. Q., J. Zhu, C. X. He, M. Benmoussa and P. Wu, 1998. Quantitative trait loci analysis for the developmental behavior of tiller number in rice. Theor. Appl. Genet. 97: 267–274. [Google Scholar]
- Zeng, Z-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]