

Abstract
The X chromosome requires special treatment in the mapping of quantitative trait loci (QTL). However, most QTL mapping methods, and most computer programs for QTL mapping, have focused exclusively on autosomal loci. We describe a method for appropriate treatment of the X chromosome for QTL mapping in experimental crosses. We address the important issue of formulating the null hypothesis of no linkage appropriately. If the X chromosome is treated like an autosome, a sex difference in the phenotype can lead to spurious linkage on the X chromosome. Further, the number of degrees of freedom for the linkage test may be different for the X chromosome than for autosomes, and so an X chromosome-specific significance threshold is required. To address this issue, we propose a general procedure to obtain chromosome-specific significance thresholds that controls the genomewide false positive rate at the desired level. We apply our methods to data on gut length in a large intercross of mice carrying the Sox10Dom mutation, a model of Hirschsprung disease. We identified QTL contributing to variation in gut length on chromosomes 5 and 18. We found suggestive evidence of linkage to the X chromosome, which would be viewed as strong evidence of linkage if the X chromosome was treated as an autosome. Our methods have been implemented in the package R/qtl.
THERE is broad interest in genetic loci (called quantitative trait loci, QTL) that contribute to variation in quantitative traits, and so numerous statistical methods and computer programs have been developed to map QTL. Virtually all of this work has focused exclusively on autosomal loci. However, the X chromosome displays special behavior and must be treated differently in QTL mapping. Often crosses are set up to avoid recombination on the X chromosome. When the X chromosome is recombining in a cross, there are several possible patterns, and each requires a different analysis. For example, in a backcross in which the X chromosome is segregating, males are hemizygous A or B, while females have genotype AA or AB. Thus, rather than comparing, as for the autosomes, the phenotypic means between the AA and AB genotype groups, the X chromosome requires a comparison of the phenotypic means across four genotypic groups.
Both Rance et al. (1997) and Ahmadiyeh et al. (2003) described methods for the analysis of the X chromosome in the case of reciprocal F2 intercrosses. However, a number of important cases, and the issue of appropriate significance thresholds for the X chromosome, remain to be addressed.
Here, we describe methods for the treatment of the X chromosome in QTL mapping in experimental crosses that are suitable for routine use in QTL mapping. We develop our ideas in the context of rodent models, but our approach is general. We consider backcrosses and intercrosses derived from two inbred strains, A and B, and describe the necessary modifications to standard interval mapping. The most important modification concerns the formulation of the null hypothesis of no linkage to avoid spurious linkage to the X chromosome as a result of sex or cross-direction differences in the phenotype. Sex differences are observed in many phenotypes, and systematic phenotypic differences between reciprocal crosses may arise, for example, from parent-of-origin effects. If not taken into account, such systematic differences can lead to large LOD scores on the X chromosome even in the absence of X chromosome linkage. In addition, to account for the fact that the number of degrees of freedom for the linkage test on the X chromosome may be different from that on the autosomes, we propose a procedure to obtain an X chromosome-specific significance threshold while controlling the genomewide false positive rate at the desired level.
To illustrate our approach, we study data on gut length in a large mouse intercross. The phenotype exhibits a sex difference. Because of this, if the chromosome is treated as an autosome, there is a spurious inflation in the evidence for linkage to the X chromosome.
METHODS
X chromosome analysis:
Central to the appropriate treatment of the X chromosome is an examination of its segregation behavior, which is markedly different from that of the autosomes. In particular, the behavior of the X chromosome depends on the direction of the cross, as well as the sex of the progeny. We enumerate the possibilities in Figures 1 and 2 for backcross and intercross populations.
Figure 1.—

The behavior of the X chromosome in a backcross. Circles and squares correspond to females and males, respectively. Open and hatched bars correspond to DNA from strains A and B, respectively. The small bar is the Y chromosome.
Figure 2.—

The behavior of the X chromosome in an intercross. Circles and squares correspond to females and males, respectively. Open and hatched bars correspond to DNA from strains A and B, respectively. The small bar is the Y chromosome.
In Figure 1, the four possible backcrosses to a single strain are presented. For the backcrosses in Figure 1, c and d, in which the F1 parent is male, the X chromosome is not subject to recombination; thus, we omit these crosses from further consideration. In Figure 1, a and b, in which the F1 parent is female, the X chromosome does recombine and the order of the cross producing the F1 parent is seen to have no impact on the behavior of the X chromosome in the backcross progeny. Backcrosses to the other strain would be similar, and we do not consider the combined analysis of backcrosses to each strain, as we view it to be outside the scope of this article. The combined analysis of multiple types of crosses is feasible on the basis of the principles we outline below.
In Figure 2, the four possible intercrosses are presented. In all cases, F2 progeny have a single X chromosome subject to recombination, and male F2 progeny are, at any given locus, hemizygous A or B. In the cases that the F1 male parent was derived from a cross A × B (with the A parent being female; Figure 2, a and b), the female F2 progeny are either AA or AB. In the cases that the F1 male parent was derived from a cross B × A (with the B parent being female; Figure 2, c and d), the female F2 progeny are either BB or AB. Note that the direction of the cross giving the female F1 parent does not affect the behavior of the X chromosome in the F2 progeny. Thus, when we discuss the direction of the intercross, we consider only the direction of the cross that produced the male F1 parent. The crosses in Figure 2, a and b, are treated the same, and the crosses in Figure 2, c and d, are treated the same. We neglect the possibility of mitochondrial, Y chromosome, imprinting, or maternal effects; such parent-of-origin specific effects could be inspected through the inclusion of covariates that indicate the specific cross from which an animal was derived.
Our goal is to adapt standard interval mapping (Lander and Botstein 1989) for the X chromosome. As with standard interval mapping, we survey all single-QTL models contributing to the phenotype by considering a grid of genomic positions. At each putative locus we construct a likelihood-ratio test for the hypothesis of no linkage to the X chromosome. The likelihood is maximized under the null and alternative hypothesis using the EM algorithm (Dempster et al. 1977), and the ratio is presented in base 10 logarithms to give a LOD score. The two key ingredients are appropriate formulation of the null and alternative hypotheses and calculation of the genotype probabilities at the putative locus under the null hypothesis. We discuss these, in turn, below.
The choice of the null and alternative hypotheses requires careful thought. Since our goal is to develop a procedure for routine use in QTL mapping, our choice of genotype comparisons was based on the following basic principles. First, a sex or cross-direction difference in the phenotype should not lead to spurious linkage to the X chromosome. Second, the set of comparisons should be parsimonious but reasonable. Third, the null hypothesis must be nested within the alternative hypothesis. Our choices for all possible cases are presented in Table 1; we explain how we arrived at the choices through specific examples, below.
TABLE 1.
Proposed contrasts for analysis of the X chromosome in standard crosses
| Cross | Direction | Sexes | Contrasts | Null hypothesis | d.f. |
|---|---|---|---|---|---|
| BC | Both | AA:AB:AY:BY | Female:male | 2 | |
| BC | Females | AA:AB | Grand mean | 1 | |
| BC | Males | AY:BY | Grand mean | 1 | |
| F2 | Both | Both | AA:ABf:ABr:BB:AY:BY | Female forw:female rev:male | 3 |
| F2 | Both | Females | AA:ABf:ABr:BB | Female forw:female rev | 2 |
| F2 | Both | Males | AY:BY | Grand mean | 1 |
| F2 | One | Both | AA:AB:AY:BY | Female:male | 2 |
| F2 | One | Females | AA:AB | Grand mean | 1 |
| F2 | One | Males | AY:BY | Grand mean | 1 |
Forw, forward; rev, reverse.
Consider, for example, an intercross performed in one direction and including both sexes. Females then have X chromosome genotype AA or AB, while males are hemizygous A or B. Males that are hemizygous B should be treated separately from the AA or AB females, and so the null hypothesis must then allow for a sex difference in the phenotype, as otherwise the presence of such a sex difference would cause spurious linkage to the X chromosome. For the null hypothesis to be nested within the alternative, the alternative must allow separate phenotype averages for the genotype groups AA, AB, AY, and BY (see Table 1). We call this the contrast AA:AB:AY:BY. Note that there are 2 d.f. for this test of linkage, just as for the autosomes, as there are four mean parameters under the alternative and two under the null (the average phenotype for each of females and males).
A somewhat more complex example is for the case that both directions of the intercross were performed, but only females were phenotyped. As the AA individuals come from one direction of the cross (which we call the “forward” direction) and the BB individuals come from the other direction of the cross (the “reverse” direction), a cross direction effect on the phenotype would cause spurious linkage to the X chromosome if the null hypothesis did not allow for that effect. But then, for the null hypothesis to be nested within the alternative, the AB individuals from the two cross directions must be allowed to be different. Thus we arrive at the contrasts AA:ABf:ABr:BB for the alternative and forward:reverse for the null. Here, again, the linkage test has 2 d.f.
In the analogous case with males only, since both directions give rise to equal parts hemizygous A and hemizygous B individuals, we need not split the individuals according to the direction of the cross, as a cross-direction effect cannot cause spurious linkage to the X chromosome. In this case, the test for linkage has 1 d.f.
In the most complex case, of an intercross with both directions and both sexes, all four types of females must be allowed to be separate, whereas the males from the two directions may be pooled, and so the simplest comparison includes the contrasts AA:ABf:ABr:BB:AY:BY, with the null hypothesis using the contrasts female forward:female reverse:male. Thus, the linkage test has 3 d.f.
The actual statistical analysis is no different from what is done in standard interval mapping for autosomes. In the presence of a QTL, we first calculate, for individual i, the probability, piq, that the individual falls into QTL genotype group q, for each of the genotype groups under consideration, given the available marker data. Calculation of these QTL genotype probabilities at the putative QTL, given the available multipoint marker genotype data, is most efficiently done via a hidden Markov model (Lander and Green 1987). This can be constructed to allow for the presence of genotyping errors (Lincoln and Lander 1992). As each backcross or intercross individual has a single X chromosome that was subject to recombination (see Figures 1 and 2), the calculations here are identical to those for an autosome in a backcross, and so nothing new is needed.
We then assume that, given an individual's genotype, q, its phenotype follows a normal distribution with mean μq and common SD σ. Maximum-likelihood estimates (MLEs) of the parameters μq and σ are obtained via the EM algorithm (Dempster et al. 1977). The maximized likelihood is then 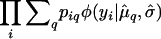 , where φ is the normal density. Under the null hypothesis, individuals are grouped as described in Table 1, with individual i assigned to, say, group ji, and the likelihood is
, where φ is the normal density. Under the null hypothesis, individuals are grouped as described in Table 1, with individual i assigned to, say, group ji, and the likelihood is 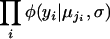 , for which MLEs may be obtained in closed form. A LOD score is calculated as the log (base 10) of the ratio of the maximized likelihoods under the alternative and the null hypotheses.
, for which MLEs may be obtained in closed form. A LOD score is calculated as the log (base 10) of the ratio of the maximized likelihoods under the alternative and the null hypotheses.
Chromosome-specific thresholds:
The usual approach for establishing statistical significance in a QTL genome scan is to calculate a single genomewide LOD threshold. At the 5% significance level, one calculates the 95th percentile of the distribution of the genomewide maximum LOD score, under the global null hypothesis that there are no QTL anywhere. This is best done via a permutation test (Churchill and Doerge 1994).
The number of degrees of freedom of the linkage test on the X chromosome can be different from that on the autosomes (see Table 1). In the case that the degrees of freedom for the X chromosome are larger than those for autosomes, the null distribution of the LOD score on the X chromosome will be stochastically larger than that for the LOD score on an autosome. Additionally, in intercrosses, only one X chromosome is potentially recombinant (compared to both, for autosomes), leading to an effectively smaller genetic length relative to autosomes. Thus, if we apply a constant threshold, our tests on the X chromosome may be too liberal.
One could assign chromosome-specific LOD thresholds, allowing for a chromosome-specific false positive rate of αi for chromosome i. We require, however, that the αi are chosen to maintain the desired genomewide significance level, α. Under the null hypothesis of no QTL and with the assumption of independent assortment of chromosomes, the LOD scores on separate chromosomes are independent, and so we must choose the αi so that
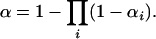 |
(1) |
Any choice of the αi satisfying Equation 1 will provide a genomewide false positive rate that is maintained at the desired level. For example, one could choose α1 = α and αi = 0 for i ≠ 1. A key issue, in choosing the αi, concerns the power to detect a QTL. In the preceding example, one would have high power to detect a QTL on chromosome 1, but no power to detect a QTL on any other chromosome. The usual approach, with a constant LOD threshold across the genome, provides high power to detect a QTL irrespective of its location: in the case of high and uniform marker density and the presence of a single autosomal QTL, the power to detect the QTL would be the same no matter where it resides.
In tackling the problem of how to assign a LOD threshold for the X chromosome, then, one might choose to ensure that the power to detect a QTL on the X chromosome is the same as that for the autosomes. This is tricky, however, in that the nature of the effect of an X chromosome QTL can be markedly different from one on the autosomes, as the individuals have a different set of possible genotypes. Thus, we have chosen to take a different approach.
It appears reasonable to choose the αi proportional to the genetic lengths of the chromosomes. Let Li denote the genetic length of chromosome i. We could choose αi = kLi for some k, subject to the constraint in Equation 1. (It is interesting to note that the analytical results of Lander and Botstein 1989, for the dense map case, give αi = a + bLi for a ≠ 0.) The solutions, unfortunately, cannot be written in closed form, but one may take 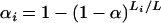 , where
, where 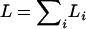 , and get results that are indistinguishable in practice. And so the latter is the approach that we recommend.
, and get results that are indistinguishable in practice. And so the latter is the approach that we recommend.
We recommend a constant LOD threshold for the autosomes and a separate threshold for the X chromosome. Taking LA to be the sum of the genetic lengths of the autosomes and LX to be the length of the X chromosome, we use 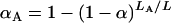 and
and 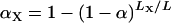 . In particular, in a permutation test to determine LOD thresholds, one would calculate, for permutation replicate j,
. In particular, in a permutation test to determine LOD thresholds, one would calculate, for permutation replicate j, 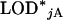 as the maximum LOD score across all autosomes and
as the maximum LOD score across all autosomes and 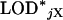 as the maximum LOD score across the X chromosome. The LOD threshold for the autosomes would be the 1 − αA quantile of the
as the maximum LOD score across the X chromosome. The LOD threshold for the autosomes would be the 1 − αA quantile of the 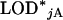 , and the LOD threshold for the X chromosome would be the 1 − αX quantile of the
, and the LOD threshold for the X chromosome would be the 1 − αX quantile of the 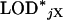 . Typical values for these thresholds are shown in the application section (see Table 2).
. Typical values for these thresholds are shown in the application section (see Table 2).
TABLE 2.
Genomewide 5% LOD thresholds
| Both sexes | Males | Females | |
|---|---|---|---|
| Commona | 3.45 | 3.44 | 3.45 |
| Autosomesb | 3.43 | 3.47 | 3.47 |
| X chromosomeb | 3.70 | 2.67 | 3.17 |
Thresholds derived in the standard way, constant for all chromosomes.
Thresholds derived allowing separate values for the autosomes and the X chromosome.
Genome-scan-adjusted P-values can be estimated from the permutation results as follows. For a putative QTL on an autosome, one would first calculate the proportion, call it p, of the 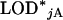 that were greater or equal to the observed LOD score. The adjusted P-value would then be
that were greater or equal to the observed LOD score. The adjusted P-value would then be 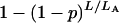 . Equations for a locus on the X chromosome are analogous, replacing the A's with X's.
. Equations for a locus on the X chromosome are analogous, replacing the A's with X's.
It is of some interest to compare, for autosomes, the chromosome-specific αi defined through 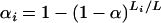 to those that are attained with an assumption of a constant genomewide LOD threshold. We would be concerned if they were not similar. To investigate this, we simulated data on a backcross of 200 individuals, with a genome modeled after the mouse, markers spaced at either 10 or 1 cM, and phenotypes following a normal distribution and independent of marker genotypes (thus under the global null hypothesis of no QTL). We used 5,000,000 simulation replicates. For each replicate, we calculated the maximum LOD score for each autosome, say LODij for chromosome i in simulation replicate j. The genomewide maximum LOD for each replicate was calculated as Mj = maxj LODij. The genomewide LOD threshold, call it T, was taken to be the 95th percentile of the Mj.
to those that are attained with an assumption of a constant genomewide LOD threshold. We would be concerned if they were not similar. To investigate this, we simulated data on a backcross of 200 individuals, with a genome modeled after the mouse, markers spaced at either 10 or 1 cM, and phenotypes following a normal distribution and independent of marker genotypes (thus under the global null hypothesis of no QTL). We used 5,000,000 simulation replicates. For each replicate, we calculated the maximum LOD score for each autosome, say LODij for chromosome i in simulation replicate j. The genomewide maximum LOD for each replicate was calculated as Mj = maxj LODij. The genomewide LOD threshold, call it T, was taken to be the 95th percentile of the Mj.
The chromosome-specific false positive rates were then taken as αi = Prop(j: LODij > T). These are shown in Figure 3, along with the curve following the chromosome-specific thresholds chosen according to our recommended procedure, 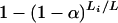 . The chromosome-specific false positive rates obtained with the assumption of a constant genomewide LOD threshold are somewhat higher than the procedure we have described for the smaller chromosomes and are somewhat lower for the larger chromosomes, but differ by no more than ∼0.0005. Thus, we conclude that the proposed procedure does not deviate too far from the use of a constant LOD threshold, but allows us to account for the difference in degrees of freedom between the autosomes and the X chromosome.
. The chromosome-specific false positive rates obtained with the assumption of a constant genomewide LOD threshold are somewhat higher than the procedure we have described for the smaller chromosomes and are somewhat lower for the larger chromosomes, but differ by no more than ∼0.0005. Thus, we conclude that the proposed procedure does not deviate too far from the use of a constant LOD threshold, but allows us to account for the difference in degrees of freedom between the autosomes and the X chromosome.
Figure 3.—

The attained chromosome-specific false positive rates for autosomes with the use of a constant 5% genomewide LOD threshold (based on computer simulations), for a genome modeled after the mouse and having markers spaced at 10 cM (circles) or 1 cM (x's). The curve corresponds to the chromosome-specific false positive rates from the formula 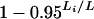 .
.
One final detail concerning the permutation procedure deserves emphasis. Sex and cross-direction differences in the phenotype must be preserved in the permutations. This could be accomplished by a stratified permutation test: permuting the phenotypes relative to the genotypes separately within the four strata (males and females in each cross direction). We use a slightly different strategy: sex and cross direction are viewed as additional phenotypes, and the rows in the phenotype matrix are shuffled relative to the genotype data. Thus the sex (and cross direction) attached to a particular phenotype is preserved. Autosomal genotypes are permuted across all individuals. For the X chromosome data, all individuals have one chromosome subject to recombination and one nonrecombinant chromosome, and we permute the data for the recombinant chromosome across all individuals. This approach will give results equivalent to the stratified permutation test, provided that the pattern of missing genotype data is similar in the different strata.
APPLICATION
As an illustration of our methods, we consider data on gut length from the cross of Owens et al. (2005). The aim of this study was to identify modifiers of Sox10Dom, a model of Hirschsprung disease. Lines of Sox10Dom mice congenic on the C57BL/6J background were crossed with C3HeB/FeJ mice to generate B6N15C3Fe.Sox10Dom (F1) progeny. Intercrosses were performed by crossing male F1 mice to wild-type B6C3Fe or C3FeB6 females as well as reciprocal intercrosses between female B6N15C3Fe.Sox10Dom (F1) mice and wild-type B6C3Fe or C3FeB6 males. From these crosses, 2210 F2 mice were collected, but only the 1068 mice carrying the Sox10Dom mutation were considered for the QTL analysis.
To avoid the technicalities associated with the selection of individuals carrying the mutation we omitted chromosome 15, which carries this mutation, from our analysis. The estimated genetic length of the X chromosome was 78 cM; the total length of the autosomes (excluding chromosome 15) was 1250 cM. Individuals were typed at a set of 117 markers on the remaining chromosomes, including 6 markers on the X chromosome. A selective genotyping strategy was used, with the most extreme 30% of individuals, phenotypically, genotyped at essentially all markers, and others genotyped at markers in regions showing an effect. Selective genotyping was based on the aganglionosis traits considered in Owens et al. (2005), but not considered here. Quantification of aganglionosis relies on measurement of total gut length from gastric sphincter to anus. The gut length phenotype was the focus of our analysis in this study.
The F2 mice comprised 563 males and 505 females. The gut length phenotype showed a clear sex difference, with average (SE) gut lengths of 16.6 (0.090) and 16.2 (0.095) cm in males and females, respectively.
For the QTL analysis, sex was included as an additive covariate, and the two sexes were also considered separately. Genomewide 5% LOD thresholds, estimated from a permutation test with 100,000 replicates, are displayed in Table 2. When a common LOD threshold is used for the entire genome, the threshold is 3.45 (approximately), whether analysis concerns both sexes, males only, or females only. If a separate threshold is allowed for the X chromosome, the threshold for the autosomes changed very little. For the analysis of both sexes combined, the threshold for the X chromosome increased to 3.70, due to the fact that the linkage test for the X chromosome has 3 d.f. while for the autosomes there are 2 d.f. For the analysis of males alone, the X chromosome-specific threshold decreased to 2.67; in this case, the linkage test on the X chromosome has 1 d.f., while that for the autosomes had 2 d.f. For the analysis of the females alone, the X chromosome-specific threshold decreased to 3.17, in spite of the fact that the tests for each of the X chromosomes and the autosomes has 2 d.f. The decrease in the LOD threshold in this case may be due to the fact that each individual has just one recombinant X chromosome, but two recombinant autosomes, and so the effective number of independent statistical tests is smaller for the X chromosome than for an autosome of equivalent length.
LOD curves for the gut length trait are displayed in Figure 4, with further details on the identified QTL shown in Table 3. In the analysis of both sexes, a strong QTL is seen on chromosome 5. A QTL near this position was previously observed to affect the extent of aganglionosis (Owens et al. 2005). The phenotype averages for each genotype group (Figure 5a), estimated by the multiple-imputation approach of Sen and Churchill (2001), indicate that the C3H allele is dominant and results in an increase in gut length.
Figure 4.—

LOD curves for analysis of gut length with both sexes (a), males only (b), and females only (c). Dashed horizontal lines are plotted at the estimated genomewide 95% LOD thresholds, allowed to be separate for the autosomes and the X chromosome.
TABLE 3.
Estimated QTL positions, LOD scores, and genome-scan-adjusted P-values
| Both sexes
|
Males
|
Females
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Pos (cM) | LOD | Pa | Pb | LOD | Pa | Pb | LOD | Pa | Pb |
| 5 | 22 | 6.31 | <0.001 | <0.001 | 2.61 | 0.259 | 0.270 | 4.34 | 0.008 | 0.008 |
| 18 | 48 | 1.25 | 0.995 | 0.993 | 0.46 | 1.000 | 1.000 | 3.54 | 0.043 | 0.044 |
| X | 57 | 3.33 | 0.065 | 0.110 | 1.47 | 0.954 | 0.552 | 2.32 | 0.423 | 0.332 |
Chr, chromosome; Pos, position.
Genome-scan-adjusted P-values calculated in the standard way, with a constant threshold for all chromosomes.
Genome-scan-adjusted P-values allowing separate thresholds for the autosomes and the X chromosome.
Figure 5.—

Plot of average gut length (±2 SE) as a function of sex and genotype at the inferred QTL on chromosomes 5, 18, and X. B and C denote the C57BL/6J and C3HeB/FeJ alleles, respectively. Estimates were derived by multiple imputation.
There is significant linkage to chromosome 18 in females but not in males. As shown in Figure 5b, the C3H allele again results in an increase in gut length and appears to be recessive. Little effect is seen in males, but a test for a sex difference in the effect of this locus was not significant: the lack of evidence for linkage in males is not sufficient to conclude the lack of an effect.
Potential linkage is seen on the X chromosome, and the support for an X chromosome QTL depends critically on whether an X chromosome-specific LOD threshold is used. When a constant threshold is used across the genome, the genome-scan-adjusted P-value for the putative X chromosome QTL is 6.5%, while if a separate threshold is used for the X chromosome, the P-value increases to 11%. [The latter P-value was obtained as follows: 0.68% of the permutations gave a maximum LOD score on the X chromosome equal to or greater than the observed LOD score of 2.32, and L/LX ≈ 17, and so the adjusted P-value was 1 − (1 − 0.0068)17 ≈ 0.11.]
Note that if the sex effect were not taken into account in the null hypothesis here, the LOD scores on the X chromosome would increase by 5.67, uniformly, resulting in greatly inflated evidence for X linkage. In the separate analyses of the two sexes, little evidence is seen for linkage to the X chromosome, but this may be due to the loss of power due to the halving of sample size.
DISCUSSION
We have described an approach for the treatment of the X chromosome in QTL mapping, focusing on the development of a procedure for routine use that will avoid spurious linkage in the presence of sex or cross-direction differences in the phenotype. We consider all possible cases for a backcross or intercross, identifying appropriate contrasts (sets of genotype groups that are to be compared) and the necessary covariates that must be included in the null hypothesis to avoid spurious linkage. While we have focused on the use of standard interval mapping (Lander and Botstein 1989), our approach may be applied with other methods for dealing with missing genotype information, such as Haley–Knott regression (Haley and Knott 1992) and multiple imputation (Sen and Churchill 2001). The extension to nonnormal models for the phenotype distribution given the QTL genotype is also straightforward.
As the number of degrees of freedom for the linkage test on the X chromosome differs from that for the autosomes, it is important to apply separate LOD thresholds for evaluating significance of the X chromosome vs. the autosomes. We have described an approach for deriving such chromosome-specific thresholds in the context of a permutation test.
We applied our methods to data on gut length in a large mouse intercross and identified QTL on chromosomes 5 and 18, plus a suggestion of a QTL on the X chromosome. Overly strong evidence for the X chromosome QTL would have been obtained had the sex difference in the phenotype not been taken into account. These loci may be relevant to gastroenterologists interested in short gut syndrome (Seashore et al. 1987; Kern et al. 1990), although it must be acknowledged that the QTL may affect gut length through body size; we made no attempt to account for body size in our analysis.
It is important to point out that the precise estimation of the X chromosome-specific LOD threshold will require considerably more permutation replicates. A first-order Taylor expansion of the adjusted P-value, 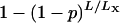 , indicates that its SE is, roughly, a factor L/LX larger than the SE of the unadjusted P-value. (Recall that LX is the genetic length of the X chromosome and L is the total genome length.) Further, we find that one must use roughly L/LX times more permutation replicates to get the same precision for an adjusted P-value for the X chromosome as one would typically need if a constant LOD threshold were used across the genome. (This result was confirmed via computer simulations, not shown.)
, indicates that its SE is, roughly, a factor L/LX larger than the SE of the unadjusted P-value. (Recall that LX is the genetic length of the X chromosome and L is the total genome length.) Further, we find that one must use roughly L/LX times more permutation replicates to get the same precision for an adjusted P-value for the X chromosome as one would typically need if a constant LOD threshold were used across the genome. (This result was confirmed via computer simulations, not shown.)
For the application to the data of Owens et al. (2005), if one were to use 1000 permutation replicates for the autosomes, ∼17,000 replicates would be required for the X chromosome, since L/LX ≈ 17. As each replicate of the X chromosome requires a scan over LX/L of the genome, the total computational effort is about double the usual amount. If one sought a separate threshold for each chromosome, one would want, for chromosome i, L/Li times the usual number of permutation replicates, and, in the case of 20 chromosomes, the overall effort would be 20 times the usual amount.
In our development we have neglected imprinting, maternal effects, the Y chromosome, and mitochondrial or cytoplasmic effects. Should these be deemed important for the phenotype of interest, modifications to our methods are necessary. These can be arrived at by careful consideration of Figures 1 and 2 and judicious use of the different cross options.
We have focused on single-dimensional, single-QTL genome scans. Analysis of two-dimensional, two-QTL genome scans (Haley and Knott 1992; Sen and Churchill 2001) must also be modified for the case of the X chromosome. There are two key issues. First, in cases where the two QTL are located on the X chromosome, it must be acknowledged that individuals can have only two of the possible genotypes at each locus. For example, in an intercross in one direction but with both males and females, one considers the 4 genotype groups AA, AB, AY, and BY, at each locus, but of the 16 two-locus genotypes, only 8 are observable. Care must be taken to avoid overparameterization. Second, in the case that one QTL is on the X chromosome and one is on an autosome, as the analysis of the X chromosome may require sex as a covariate in the null hypothesis, it must also be used for this portion of the two-dimensional, two-QTL scan.
The X chromosome, while comprising only ∼5% of the genome, requires a doubling of effort for QTL mapping. The appropriate treatment of the X chromosome is not conceptually difficult, but it does require some rather tedious bookkeeping. We are not aware of QTL mapping software that treats the X chromosome appropriately, except for our own software, R/qtl (Broman et al. 2003), an add-on package to the R statistical software (Ihaka and Gentleman 1996). The methods we have described are implemented in R/qtl.
Acknowledgments
The authors thank an anonymous reviewer for comments to improve the manuscript. This work was supported in part by National Institutes of Health grants GM074244 (to K.W.B.), GM070683 (to G.A.C.), NS43556 (to MS2), and DK60047 (to MS2) and by a National Science Foundation Graduate Research Fellowship (to A.M.).
References
- Ahmadiyeh, N., G. A. Churchill, K. Shimomura, L. C. Solberg, J. S. Takahashi et al., 2003. X-linked and lineage-dependent inheritance of coping responses to stress. Mamm. Genome 14: 748–757. [DOI] [PubMed] [Google Scholar]
- Broman, K. W., H. Wu, Ś. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890. [DOI] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B 39: 1–38. [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Ihaka, R., and R. Gentleman, 1996. R: a language for data analysis and graphics. J. Comp. Graph. Stat. 5: 299–314. [Google Scholar]
- Kern, I. B., A. Leece and T. Bohane, 1990. Congenital short gut, malrotation, and dysmotility of the small bowel. J. Pediatr. Gastroenterol. Nutr. 11: 411–415. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and P. Green, 1987. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 84: 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln, S. E., and E. S. Lander, 1992. Systematic detection of errors in genetic linkage data. Genomics 14: 604–610. [DOI] [PubMed] [Google Scholar]
- Owens, S. E., K. W. Broman, T. Wiltshire, J. B. Elmore, K. M. Bradley et al., 2005. Genome-wide linkage identifies novel modifier loci of aganglionosis in the Sox10Dom model of Hirschsprung disease. Hum. Mol. Genet. 14: 1549–1558. [DOI] [PubMed] [Google Scholar]
- Rance, K. A., W. G. Hill and P. D. Keightley, 1997. Mapping quantitative trait loci for body weight on the X chromosome in mice. I. Analysis of a reciprocal F2 population. Genet. Res. 70: 117–124. [DOI] [PubMed] [Google Scholar]
- Seashore, J. H., F. S. Collins, R. I. Markowitz and M. R. Seashore, 1987. Familial apple peel jejunal atresia: surgical, genetic, and radiographic aspects. Pediatrics 80: 540–544. [PubMed] [Google Scholar]
- Sen, Ś., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]