

Abstract
We report here the resonance Raman spectra of the FeIII–NO and FeII–NO complexes of the bacterial NOSs (nitric oxide synthases) from Staphylococcus aureus and Bacillus subtilis. The haem–NO complexes of these bacterial NOSs displayed Fe–N–O frequencies similar to those of the mammalian NOSs, in presence and absence of L-arginine, indicating that haem-bound NO and L-arginine had similar haem environments in bacterial and mammalian NOSs. The only notable difference between the two types of NOS was the lack of change in Fe–N–O frequencies of the FeIII–NO complexes upon (6R) 5,6,7,8-tetrahydro-L-biopterin binding to bacterial NOSs. We report, for the first time, the characterization of NO complexes with NOHA (Nω-hydroxy-L-arginine), the substrate used in the second half of the catalytic cycle of NOSs. In the FeIII–NO complexes, both L-arginine and NOHA induced the Fe–N–O bending mode at nearly the same frequency as a result of a steric interaction between the substrates and the haem-bound NO. However, in the FeII–NO complexes, the Fe–N–O bending mode was not observed and the νFe−NO mode displayed a 5 cm−1 higher frequency in the complex with NOHA than in the complex with L-arginine as a result of direct interactions that probably involve hydrogen bonds. The different behaviour of the substrates in the FeII–NO complexes thus reveal that the interactions between haem-bound NO and the substrates are finely tuned by the geometry of the Fe-ligand structure and are relevant to the use of the FeII–NO complex as a model of the oxygenated complex of NOSs.
Keywords: L-arginine, haem, Nω-hydroxy-L-arginine (NOHA), nitric oxide synthase (NOS), resonance Raman spectroscopy
Abbreviations: 5C, 5-co-ordinated; 6C, 6-co-ordinated; bsNOS, Bacillus subtilis nitric oxide synthase; drNOS, Deinococcus radiodurans NOS; DTT, dithiothreitol; eNOS, endothelial NOS; FeIII, ferric form; FeII, ferrous form; gsNOS, Geobacillus stearothermophilus NOS; H4B, (6R) 5,6,7,8-tetrahydro-L-biopterin; iNOS, inducible NOS; nNOS, neuronal NOS; NOHA, Nω-hydroxy-L-arginine; NOSox, oxygenase domain of NOS; saNOS, Staphylococcus aureus NOS; THF, tetrahydrofolate
INTRODUCTION
NOSs (nitric oxide synthases) catalyse the oxidation of L-arginine to citrulline and NO. They are found in many organisms, including insects [1], invertebrates [2] and mammals [3]. The best-characterized enzymes are the three mammalian NOS isoforms, iNOS (inducible NOS), eNOS (endothelial NOS) and nNOS (neuronal NOS). Mammalian NOSs share a common arrangement, their N-termini contain the oxygenase domain and the C-termini contain the reductase domain, whose function is to supply electrons from NADPH to the haem in the oxygenase domain [4,5]. The oxygenase and reductase domains are separated by a central region that controls the flow of electrons in response to the binding of the Ca2+–calmodulin complex.
The active site of all NOSs contains a haem that is co-ordinated with the sulphur atom of a cysteine residue, a substrate-binding site on the opposite side of the Fe–thiolate bond and a cofactor-binding site on the proximal side of the haem. The substrate, L-arginine, forms several van der Waals and hydrogen bonds with surrounding amino acids and one of the haem propionates [4]. The cofactor H4B [(6R) 5,6,7,8-tetrahydro-L-biopterin] also forms van der Waals and hydrogen bonds with surrounding amino acids as well as a hydrogen bond with the same haem propionate as the substrate. Full-length mammalian NOSs, as well as the NOSox (NOS oxygenase domain), are found as dimers. Besides the contact area of the dimer, an N-terminal hook involving a helix from each monomer [6] and a Zn atom, which is co-ordinated with pairs of cysteine residues in each monomer, stabilize the dimer [7].
NO is synthesized by NOSs in two consecutive enzymatic reactions. First, the enzyme oxidizes L-arginine to the stable intermediate NOHA (Nω-hydroxy-L-arginine) with the concomitant consumption of two electrons from NADPH. This step is thought to occur via a P450-type reaction cycle, with the possible formation of a compound I species as the ultimate oxidizing species [5,8]. H4B supplies one electron, and possibly one proton, to activate the oxygen molecule bound to the haem during this first catalytic step [5,8,9]. Subsequently, the H4B is recycled on the protein with an electron provided by the reductase domain. The second reaction involves the oxidation of NOHA to L-citrulline and NO with the consumption of one electron from NADPH. A number of mechanisms have been proposed for this reaction [10,11] but the nature of the ultimate oxidizing species remains to be elucidated. It has, however, been shown that H4B may also provide an electron for this second reaction [12–14].
Genes encoding NOS-like proteins have been found in the genomes of several bacteria. The region similar to that of mammalian NOSs encompasses the catalytic oxygenase domain [15,16]. However, differences with respect to mammalian NOSs have been observed. The bacterial proteins do not have a reductase domain attached to the oxygenase domain and they lack the first ∼60 N-terminal amino acids of mammalian NOSs, including the cysteine residues that form the Zn-binding site at the dimer interface. Based on the crystal structures of the NOSs of Staphylococcus aureus (saNOS) [15], Bacillus subtilis (bsNOS) [16] and Geobacillus stearothermophilus (gsNOS) [17], it appears that many residues that form the substrate and cofactor binding sites of mammalian NOSs are present in these bacterial NOSs, suggesting a conserved function.
There is some evidence that the bacterial NOSs may have NO synthesis activity. saNOS [15] and gsNOS [17] synthesize nitrite from L-arginine in a peroxide assay. bsNOS and the NOS of Deinococcus radiodurans (drNOS) produce nitrite in a reconstituted system with the reductase domain of a mammalian NOS [18]. In addition, gsNOS, bsNOS and saNOS synthesize NO in single turnover experiments with NOHA in an H4B-dependent manner [17,19,20]. However, there is no conclusive evidence that these proteins can synthesize NO in equilibrium conditions, as measured using an NO electrode or a haemoglobin capture assay. One difficulty is that the native reductase that can provide electrons to bacterial NOSs has not been identified. In addition, reconstituted systems with the reductase domain of mammalian NOSs are inefficient. The second difficulty is that the cognate molecule that binds to the cofactor site is not known. Most bacteria may not synthesize H4B because they appear to lack the sepiapterin reductase gene [21]. However, bacteria synthesize THF (tetrahydrofolate), and this molecule has been shown to occupy the cofactor-binding site of bsNOS [16] and to stimulate nitrite production in a reconstituted system [18]. Besides the synthesis of free NO, another function has been proposed for bacterial NOSs based on the observed nitration of Trp at position 4 by drNOS. This activity is stimulated by a tryptophanyl-tRNA synthetase and is thought to occur at the cofactor site [22]. Similarly, the enzymatic activity of the NOS of Streptomyces turgidiscabies results in the nitration of a phytotoxin dipeptide required for pathogenicity [23]. Taken together, these results suggest that bacterial NOSs may synthesize NO-related compounds used for the nitration of specific molecules. However, more studies are required to decipher the role of bacterial NOSs.
Resonance Raman spectroscopy is a powerful tool for investigating the haem environment of haem proteins. It can provide information about haem co-ordination, spin and oxidation states. In addition, the haem environment can be probed with haem ligands to provide a detailed picture of steric and hydrophilic interactions between amino acids and haem-bound ligands, and between substrates, cofactors and haem-bound ligands [24,25]. In this study, we used NO, which is a physiological ligand of NOSs, to characterize the properties of the FeIII–NO and FeII–NO complexes of two bacterial NOSs (saNOS and bsNOS). Our goals were to determine the similarities and differences between the FeIII–NO and FeII–NO complexes of these bacterial NOSs and those of the mammalian NOSs, and to compare, for the first time, the NO-complexes obtained in the presence of NOHA with those with L-arginine, to determine whether these substrates interact in a similar manner with the haem-bound NO. As nitrosyl complexes are used as a model of the oxygenated complex of haem proteins [26], these studies are relevant with respect to the proposed stabilization of different oxygenated intermediates during the first and second parts of the catalytic cycle of NOSs, which involve L-arginine and NOHA as substrates respectively [11,26].
MATERIAL AND METHODS
Chemicals
L-arginine was from Alexis Biochemicals (QBiogene, Carlsbad, CA, U.S.A.), H4B, THF, sodium dithionite and NOHA were from Sigma–Aldrich (St Louis, MO, U.S.A.). 14N16O was from MEGS (Montreal, QC, Canada). 15N16O gas was from Icon Isotopes (Summit, NJ, U.S.A.).
Protein expression and purification
The recombinant saNOS protein was expressed and purified as described previously [27]. The bsNOS gene (yflM) was amplified by PCR from B. subtilis genomic DNA (strain 168). The translated bsNOS protein, starting at the initiating codon assigned for the yflM gene in the NCBI databank (NC_000964), has a shorter N-terminus compared with other bacterial NOSs. We considered the TTG codon, located 28 codons upstream of the assigned initiating codon, as the start codon, which generates a protein with a length similar to that used to obtain the crystal structure of bsNOS [16]. The PCR primer added an NdeI restriction site before the 5′ start codon and an XhoI restriction site after the 3′ stop codon. The PCR product was cloned into the pET30a (Novagen) expression vector in such a way that the His6-tag was not fused to the expressed protein. The bsNOS protein was overexpressed in Escherichia coli BL21(DE3) cells co-transformed with the POFX-tac-KJE2 plasmid for the expression of chaperones [28]. The recombinant bsNOS protein was purified by ammonium sulphate precipitation (40% and 60%) followed by anion exchange chromatography (DEAE Sepharose Fast-flow; GE Healthcare, Baie d'Urfé, QC, Canada). An NaCl gradient [0 to 1 M NaCl in 40 mM Hepes (pH 7.6) containing 1 mM DTT (dithiothreitol)] was used to purify the protein. Subsequently, the bsNOS was purified by size-exclusion chromatography on a Superdex 200PG column (GE Healthcare) equilibrated with 40 mM Hepes (pH 7.6) containing 1 mM DTT and 150 mM NaCl. The haem b content of the purified saNOS and bsNOS proteins was quantified by the pyridine haemochrome method [29]. The purified proteins were stored at −80 °C in 40 mM Hepes, pH 7.6, 150 mM NaCl, 10% (v/v) glycerol and 1 mM DTT until use. The purified saNOS and bsNOS preparations consisted mainly of dimeric protein with only a small amount of the monomer form as determined by gel filtration on a Superdex 200PG column with protein standards from Bio-Rad (Mississauga, ON, Canada).
Sample preparation for spectroscopy
Before use, the protein samples (150 μl) were desalted over a 2 ml column of P6DG (Bio-Rad) equilibrated in 40 mM Hepes pH 7.6 buffer containing 150 mM NaCl and 1 mM fresh DTT to remove the glycerol (the volume of the desalted sample was 200 μl). Where indicated, various concentrations of L-arginine (10 μM to 5 mM), NOHA (0.5–1 mM), THF (0.6–1 mM) and H4B (0.5–1.2 mM) were added to the protein samples.
Optical spectroscopy
Optical spectra were recorded with a Cary 3 spectrophotometer with samples placed in a 1 cm-path-length anaerobic cuvette (Hellma, Müllheim, Germany). The FeIII–NO complexes were obtained by equilibrating the protein solution with argon followed by the addition of gaseous NO that had been anaerobically bubbled through a solution of NaOH and then water. For the FeII–NO complexes, the proteins equilibrated anaerobically with NO were reduced using a small amount of freshly prepared dithionite solution. The protein concentration was 5 μM.
Affinity for L-arginine
The affinity of ferric saNOS for L-arginine was determined by the spectral changes caused by L-arginine binding to the ferric enzyme [30] using a non-linear regression equation (eqn 1) that takes the amount of ligand bound to the protein into account [31]. The protein concentration was 0.5 μM.
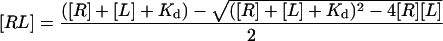 |
(1) |
where L is the ligand, R is the receptor, RL the receptor–ligand complex and Kd the dissociation constant for L-arginine. The affinity of the FeIII–NO complex for L-arginine was determined by laser flash photolysis (see below) using eqn 1.
Flash photolysis
Samples of 10 μM ferric saNOS containing known amounts of L-arginine (0 to 5 mM) were equilibrated first with argon and then with 25% NO gas to obtain the FeIII–NO complexes. A semi-micro fluorescence cuvette attached to a reservoir that allowed easy equilibration with gas was used. The experiments were carried out with a LKS-60 laser flash photolysis system (Applied-Photophysics, Leatherhead, U.K.) equipped with a frequency-doubled Brilliant laser from Quantel generating 5 ns pulses (Big Sky Lasers, Bozeman, MT, U.S.A.). The photolysis yield was calculated from the ratio of the measured variation of amplitude (ΔA) to the expected ΔA calculated from the difference spectra between the ferric sample and the FeIII–NO complex at equilibrium. The photodissociation yields of the FeIII–NO samples varied from 18% to 24%. The reactions were followed at 440 and 393 nm. A total of ten kinetic traces were acquired and averaged. The data were analysed using Olis GlobalWorks software (Olis, Bogart, GA, U.S.A).
The kinetic traces at 35–1000 μM L-arginine were best fitted with two exponential terms corresponding to fast and slow reactions. The kinetics followed single exponential behaviour at the extremes of the titration (0 and 5 mM L-arginine). The rate measured without L-arginine corresponded to the fast reaction observed at intermediate L-arginine concentrations. The rate at 5 mM L-arginine corresponded to the slow reaction observed at intermediate L-arginine concentrations. For each L-arginine concentration, the fit of the kinetic traces to two exponential functions allowed the slow and fast rates to be calculated. The amplitude of the slow reaction was used to calculate the concentration of L-arginine-bound saNOS. To calculate Kd, the concentration of L-arginine–saNOS was plotted as a function of the concentration of L-arginine, and the data were fitted to eqn 1.
Resonance Raman spectroscopy
The equipment used to acquire the resonance Raman spectra have been described previously [27]. The resonance Raman spectra of the FeIII–NO and FeII–NO (substrate-bound) complexes were acquired with the 441.6 nm line of a He/Cd laser (Liconix laser, Melles-Griot, Canada). The spectra of the FeII–NO complexes of the substrate-free saNOS and bsNOS, which were 5C (5-coordinated), were obtained with the 406 nm line of a krypton ion laser (Innova 302 laser, Coherent, Santa Clara, CA, U.S.A.). Typically, several 1 min spectra were recorded with a low excitation power (1.4–4.8 mW) at room temperature and averaged with Grams software (ThermoGalactic, Salem, NH, U.S.A.). The spectra were calibrated with the lines of indene in the 200–1900 cm−1 region. To verify the stability of the samples, optical spectra of the samples in the Raman cuvette were recorded prior to and after the resonance Raman spectra were obtained. Typically, samples containing 30–40 μM protein, based on the haem content, were used to acquire the resonance Raman spectra.
RESULTS
Optical absorption spectra
As shown in the inset of Figure 1 (dashed trace), ferric saNOS displays three absorption maxima at 377, 417 and 455 nm. The maxima at 377 and 455 nm indicated that DTT was co-ordinated with the haem [32]. The addition of gaseous NO caused the appearance of a new species with a Soret absorption band at 436 nm and absorbance maxima at 547 and 576 nm (Figure 1, inset), which are typical of 6C (6-co-ordinated) FeIII–NO complexes of haem proteins with a thiolate bond between the haem and the proximal ligand [24]. Similar FeIII–NO spectra were obtained with saNOS prepared in the presence of L-arginine (absorbance maxima at 438, 547 and 576 nm) and H4B (absorbance maxima at 439, 547 and 577 nm) (results not shown). For bsNOS, the FeIII–NO complexes of the substrate-free and pterin-free protein, the L-arginine-saturated protein and the H4B-saturated protein had the same optical spectra with a Soret absorption band at 439 nm and two maxima at 548 and 584 nm (results not shown) that are typical of 6C complexes and are in agreement with the findings of Santolini et al. [33].
Figure 1. High-frequency region of the resonance Raman spectra of the FeIII–NO complexes of saNOS.

The spectra were obtained in the absence of L-arginine and H4B (a), in the presence of 1 mM L-arginine (b) and in the presence of 1.2 mM H4B (c). The optical absorption spectra of the L-arginine-free and pterin-free ferric saNOS (dashed line) and the FeIII–NO complex (solid line) are shown in the inset panel.
The FeIII–NO complexes of saNOS were stable for several minutes but were converted slowly over time to FeII–NO complexes, unlike those of bsNOS, which remained stable over time. This property is reminiscent of the behaviour of many haem FeIII–NO complexes, including those of NOSs, that become reduced with time to FeII–NO complexes [34]. To obtain the resonance Raman spectra of the FeIII–NO complexes with saNOS, the acquisition time was kept as short as possible to avoid formation of the FeII–NO form.
High-frequency region of the FeIII–NO complexes of saNOS and bsNOS
The high-frequency region of the resonance Raman spectra of haem proteins is widely used to obtain information about the coordination, redox- and spin-states of the haem [35,36]. The FeIII–NO complexes of the substrate-free and pterin-free saNOS proteins (Figure 1, line a), of the sample with L-arginine (Figure 1, line b) and the sample with H4B (Figure 1, line c) were 6C and low-spin as shown by the ν3 line in the 1502–1503 cm−1 region and the ν2 line in the 1572–1575 cm−1 region. The frequency of the oxidation state marker line ν4 at 1375–1376 cm−1 indicated that the π electron density on the haem macrocycle was similar for all three NO complexes of saNOS [37], and was less than that of the ferric protein with no exogenous ligand, which displayed a ν4 line at 1369–1373 cm−1 [27]. The absence of a line near 1489 cm−1, which corresponds to the ν3 line of the high-spin form of the ferric enzyme [27], indicated that no significant photodissociation of the NO occurred. In accordance with the optical spectra, the resonance Raman spectra of the FeIII–NO complexes of bsNOS indicated that the haem was 6C and low spin as shown by the ν3 line at 1500–1502 cm−1, the ν2 line at 1580–1582 cm−1 and a ν4 line at 1372–1373 cm−1 [33]. From the analysis of the optical spectra and the resonance Raman spectra, we concluded that the FeIII–NO complexes of saNOS and bsNOS were 6C and low-spin with or without substrate and H4B.
The νN−O mode of the haem-bound NO should fall in the high-frequency region. However, as the νN-O mode is usually not resonance enhanced with Soret excitation [38], we could not localize this mode in the high-frequency region of the FeIII–NO spectra using isotope substitution (results not shown).
Low-frequency region of the resonance Raman spectra of the FeIII–NO complexes of saNOS and bsNOS
Figure 2 (left-hand panel) shows the resonance Raman spectra of saNOS (lines a, b and g), saNOS–L-arginine (lines c, d and h) and saNOS–L-arginine–H4B (lines e, f and i) obtained with 14N16O and 15N16O. The spectrum of the substrate-free and pterin-free saNOS with 14N16O showed a strong line at 539 cm−1 with a shoulder near 551 cm−1 that shifted to a symmetric line at 534 cm−1 with 15N16O (Figure 2, lines a and b). The difference spectrum shown in Figure 2 (line g) revealed two maxima at 541 cm−1 and 551 cm−1 and two minima at 526 cm−1 and 532 cm−1. We compared the spectra of the FeIII–NO complex of saNOS with those of P450cam for the assignment of the Fe–N–O modes [39]. For the latter, Hu and Kincaid assigned the line at 522 cm−1 (Δ2 cm−1 with 15N16O) to the Fe–NO stretching mode and the line at 546 cm−1 (Δ13 cm−1 with 15N16O) to the Fe– N–O bending mode of camphor-bound P450cam. While the νFe–NO and δFe−N−O modes may be of mixed character, we based the assignments of the modes of saNOS and bsNOS on those of the Fe–N–O modes for P450cam in order to compare the frequencies of the Fe–N–O modes of saNOS and bsNOS with those of nNOSox [24,40], iNOSox [24,41], P450nor [42] and P450BM3 [43]. The line at 539 cm−1 (Δ4 cm−1) of saNOS was thus assigned to the νFe−NO mode. The 4 cm−1 isotopic shift displayed by this line was similar in amplitude to that of the Fe–NO stretching modes of iNOSox and nNOSox (Table 1). The frequency of the νFe−NO mode remained at 539 cm−1 in the presence of H4B (Figure 3, line c) and THF (Figure 3, line b). The second isotope-sensitive line at 551 cm−1 (Δ∼16 cm−1) was assigned to the δFe−N−O mode. The isotope shift of this line was not easy to calculate precisely as the νFe−NO and δFe−N−O modes had a similar frequency in the 15N16O spectrum (534 cm−1, Figure 2, line b). However the amplitude of the shift of the δFe−N−O mode should be close to 16 cm−1 (Table 1).
Figure 2. Low-frequency region of the resonance Raman spectra of the FeIII–NO complexes of saNOS (left) and bsNOS (right).

The spectra were obtained with the L-arginine-free and pterin-free proteins with 14N16O (a and j) and 15N16O (b and k). The 14N16O minus 15N16O difference spectra (a minus b and j minus k) are shown in (g) and (p). The spectra of the L-arginine-bound proteins (1 mM) were obtained with 14N16O (c and l) and 15N16O (d and m). The 14N16O minus 15N16O difference spectra (c minus d and l minus m) are shown in (h) and (q). The spectra of the L-arginine- and H4B-bound proteins were obtained with 14N16O (e and n) and 15N16O (f and o). The 14N16O minus 15N16O difference spectra (e minus f and n minus o) are shown in (i) and (r).
Table 1. The frequencies of the resonance Raman modes of the 6C FeIII–NO complexes of NOSs.
The spectra of saNOS were acquired over a very short time (5 min) as the sample autoreduced over time to form a FeII–NO complex. −, absent; Arg, L-arginine; ND, not determined; sh, shoulder.
| Protein | νFe−NO (15N16O) cm−1 | δFe−N−O (15N16O) cm−1 | Reference |
|---|---|---|---|
| saNOS | 539 (Δ4) | 551 (Δ∼16), sh | This work |
| saNOS–H4B | 539 (Δ3) | 551 (Δ∼16), sh | This work |
| saNOS–THF | 539 (ND) | ND | This work |
| saNOS–Arg | – | 541 (Δ7) | This work |
| saNOS–Arg–H4B | – | 542 (Δ8) | This work |
| saNOS–NOHA–H4B | – | 543 (ND) | [20] |
| saNOS–citrulline–H4B | – | 549 (Δ12) | [20] |
| bsNOS | 540 (Δ3) | 551 (Δ∼16), sh | This work, [33] |
| bsNOS–H4B | 540 (ND) | ND | This work, [33] |
| bsNOS–THF | 540 (ND) | ND | This work |
| bsNOS–Arg | – | 541 (Δ7) | This work, [33] |
| bsNOS–Arg–H4B | – | 542 (Δ8) | This work, [33] |
| bsNOS–NOHA–H4B | – | 542 (Δ7) | This work |
| bsNOS–citrulline–H4B | – | 547 (Δ9) | This work |
| nNOSox | 535 (Δ3) | – | [40] |
| nNOSox–H4B | 542 (Δ5) | – | [40] |
| nNOSox–Arg–H4B | – | 546 (Δ10) | [40] |
| iNOSox | 537 (Δ4) | – | [41] |
| iNOSox–H4B | 541 (Δ4) | 550 (Δ13) | [41] |
| iNOSox–Arg | 537 (Δ4) | – | [41] |
| iNOSox–Arg–H4B | – | 545 (Δ8) | [41] |
Figure 3. Comparison of haem modes in the low-frequency region of the FeIII–NO complexes of saNOS and bsNOS.

The spectra were obtained in the presence of 1 mM L-arginine (a, which is the same spectrum as Figure 2, line c), 1 mM THF (b), H4B (c) and in the absence of both substrate and H4B for bsNOS (d, which is the same spectrum as Figure 2, line j) and saNOS (e, which is the same spectrum as Figure 2, line a). * represents a laser line.
In the presence of 1 mM L-arginine, a single isotope-sensitive mode was detected at 541 cm−1 (534 cm−1 with 15N16O) (Figure 2, lines c, d and h). Because the isotope shift was large (7 cm−1) and the frequency was similar to that of the δFe−N−O modes of iNOSox and nNOSox, the 541 cm−1 line of saNOS was assigned to the δFe−N−O mode (Table 1). The frequency of this line was 542 cm−1 in the presence of both H4B and L-arginine (Figure 2, lines e, f and i).
For bsNOS, the FeIII–NO complexes were studied by Santolini et al. [33], but without isotopic substitution. Here, we used isotopic substitution to identify the modes involving haem-bound NO (Figure 2 right-hand panel). In the absence of L-arginine and H4B, two isotope-sensitive lines were also observed for bsNOS. The isotope-sensitive line at 540 cm−1 (Δ4 cm−1 with 15N16O) was assigned to the νFe−NO mode and the isotope sensitive line at 551 cm−1 (Δ∼15 cm−1 with 15N16O) was assigned to the δFe−N−O mode (Figure 2, right-hand panel: lines j, k and p). With L-arginine present, alone (Figure 2, lines l, m and q) or in combination with H4B (Figure 2, lines n, o and r), a single isotope-sensitive line corresponding to the δFe−N−O mode was identified at 541–542 cm−1 (Δ7 cm−1 with 15N16O).
We also looked at the FeIII–NO complexes with NOHA and H4B and with citrulline and H4B. For saNOS, we have shown previously that the FeIII–NO complex prepared in the presence of NOHA and H4B displays a δFe−N−O mode at 543 cm−1 while the citrulline–H4B complex displays a δFe−N−O mode at 549 cm−1 (Table 1) [20]. With bsNOS, the resonance Raman spectrum of the FeIII–NO complex with NOHA and H4B displayed a single isotope-sensitive line at 542 cm−1 (Δ7 cm−1) that was assigned to a Fe–N–O bending mode (Table 1; supplementary Figure S1, which can be found at http://www.BiochemJ.org/bj/401/bj4010235add.htm). The citrulline/H4B sample displayed a single isotope-sensitive line at 547 cm−1 (Δ9 cm−1; Table 1; supplementary Figure S1), which was also assigned to a Fe–N–O bending mode. From all these results, we concluded that saNOS and bsNOS displayed nearly identical frequencies of the modes involving the Fe–N–O unit (Table 1). Additionally, in both proteins, the binding of L-arginine and NOHA generated a single line at 541–543 cm−1 that contained a significant contribution from a bending mode. The frequency of the bending mode induced by L-arginine and NOHA binding was different from that of the small intensity δFe−N−O mode of the substrate-free and pterin-free proteins at 550–551 cm−1 (Table 1) and of the δFe−N−O modes of the citrulline- and H4B-saturated proteins at 547–549 cm−1.
The intensities of the haem modes of saNOS, other than those of the νFe-NO and δFe-NO modes, were modified by the addition of L-arginine and pterin. The addition of H4B (Figure 3, line c), THF (Figure 3, line b) and L-arginine (Figure 2, line c and Figure 3, line a) caused the appearance of modes at 708 and 728 cm−1 and increased the intensities of the modes at 684, 745, 752 and 801 cm−1 compared with the spectrum of substrate- and pterin-free saNOS (Figure 2, line a and Figure 3, line e). For bsNOS, these modes were already present in the substrate- and pterin-free protein (Figure 2, line j and Figure 3, line d). Santolini et al. [33] observed these haem modes in the resonance Raman spectra of the FeIII–NO complexes of the substrate- and pterin-free bsNOS, a finding that is confirmed here. The intensity of these modes was only slightly modified by L-arginine and H4B binding.
Affinity of the FeIII–NO complex of saNOS for L-arginine
Because L-arginine binding caused a large increase of the intensity of the δFe−N−O modes of both saNOS and bsNOS and shifted their frequencies to lower values (541–542 cm−1) with respect to the frequency of the small intensity δFe−N−O modes (550–551 cm−1) of the substrate-free proteins, a direct interaction between NO and L-arginine may occur in both proteins. As this may reduce the affinity for L-arginine, we therefore sought to determine the affinity of the FeIII–NO complex of saNOS for L-arginine in order to compare it with the affinity of the ferric form of the protein for L-arginine, which is 4.2±0.2 μM (for details see supplementary Figure S2 at http://www.BiochemJ.org/bj/401/bj4010235add.htm). We reasoned that if L-arginine at the active site causes changes in the rate of NO binding to the haem, this property might be used to probe the presence of L-arginine at the active site of FeIII–NO complexes. A flash photolysis apparatus with a 5 ns laser pulse was used to determine the kinetics of NO rebinding to saNOS previously equilibrated with various concentrations of L-arginine. We used, as the starting material, the FeIII–NO complex prepared in the presence of various concentrations of L-arginine. Assuming that NO rebinding is fast compared with L-arginine binding, flashing off the NO molecule led to the measurement of NO recombination to L-arginine-containing saNOS and to L-arginine-free saNOS. The amplitudes of the two reaction rates were used to calculate the initial degree of saturation of the protein with L-arginine.
The rates of NO recombination measured for saNOS on the millisecond time scale corresponded to the bimolecular reaction of saNOS with NO, as the rates increased linearly with NO concentration (see supplementary Figure S3 at http://www.BiochemJ.org/bj/401/bj4010235add.htm). A single NO concentration was then used to evaluate the effect of L-arginine on the rate of NO recombination. As shown in Figure 4(A), the rate of NO recombination to ferric saNOS decreased in the presence of L-arginine. At L-arginine concentrations ranging from 35 μM to 1 mM, the results could be fitted to a two-exponential function with a fast rate (13250 s−1) and a slow rate (1200s−1) of NO recombination. The fast rate corresponded to the single rate measured in the absence of L-arginine. The slow rate corresponded to the single rate measured with a saturating concentration (5 mM) of L-arginine. For each L-arginine concentration, the variation of the absorbance (amplitude) of the slow reaction was obtained and used to calculate the concentration of L-arginine-bound saNOS. The plot of L-arginine-bound saNOS as a function of L-arginine concentration showed a hyperbolic function (Figure 4B). The best non-linear fit to these data indicated that the Kd was 102±8 μM. The Scatchard plot of these data (Figure 4B, inset panel) with n=0.92 indicated that there was one binding site. The Kd measured with NO bound to the haem was thus 24-fold higher than the Kd for the L-arginine of ferric saNOS determined directly from the change in the spin state of the haem upon L-arginine binding (4.2 μM±0.2 μM; supplementary Figure S2). These results clearly demonstrate that the presence of NO at the active site of the ferric saNOS considerably reduced the affinity of the protein for L-arginine.
Figure 4. Kinetics of NO rebinding following laser flash photolysis of FeIII–NO saNOS samples containing L-arginine.

(A) The kinetic traces were obtained with FeIII–NO complexes of saNOS equilibrated with 10 μM to 5 mM L-arginine.A total of ten kinetic traces were recorded at each L-arginine concentration and averaged. The traces shown are those recorded at 440 nm with 10 μM (no symbols), 100 μM (●), and 1 mM L-arginine (■). (B) Plot of the concentration of L-arginine-bound saNOS calculated from the amplitude of the slow kinetic phase as a function of L-arginine concentration. Kd (102±8 μM) was calculated using eqn 1. The inset shows the Scatchard plot of these results. (B is the fraction of saNOS bound to L-arginine). The linear fit to the data indicate that the number of binding sites was 0.92 (intercept on the y axis).
High-frequency region of the FeII–NO complexes of saNOS and bsNOS
Following the reaction of NO with reduced saNOS prepared in the absence of substrate and pterin, a complex with a Soret absorption band centred at 400 nm and a broad band near 560 nm was obtained (Figure 5, inset). This optical spectrum was typical of those displayed by 5C FeII–NO complexes of NOSs and other haem proteins [40]. The co-ordination state was confirmed by resonance Raman spectroscopy, which allowed the ν3 line at 1507 cm−1 for this complex to be identified (Figure 5, line b; Table 2). The addition of H4B or THF did not stabilize a 6C FeII–NO complex since a single ν3 line was observed at 1507 cm−1 (results not shown). In the presence of L-arginine, the FeII–NO complex of saNOS was 6C, as revealed by the absorption Soret band at 440 nm, the visible band near 565 nm (Figure 5, inset panel) and the resonance Raman spectrum with a single ν3 line at 1497 cm−1 (Figure 5, line a). The FeII–NO complex with NOHA was also 6C, given the similarity between its optical spectrum and that of the L-arginine sample (result not shown).
Figure 5. High-frequency region of the resonance Raman spectra of the FeII–NO complexes of saNOS.

The spectra of L-arginine-saturated saNOS (a) and substrate-free and pterin-free samples (b) are shown. The inset shows the optical absorption spectra of saNOS–L-arginine (dashed line) and substrate-free and pterin-free saNOS (solid line).
Table 2. The frequencies of the resonance Raman modes of the FeII–NO complexes of the NOSs.
Arg, L-arginine; ND, not determined.
| Protein | Co-ordination | ν4, ν3 cm−1 | νFe−NO (15N16O) cm−1 | Reference |
|---|---|---|---|---|
| saNOS | 5C | 1374, 1507 | 526 (Δ7) | This work |
| saNOS–Arg | 6C | 1374, 1497 | 550 (ND) | This work |
| saNOS–Arg–H4B | 6C | ND | 550 (Δ15) | This work |
| saNOS–NOHA–H4B | 6C | ND | 555 (Δ12) | This work |
| bsNOS | 5C/6C | 1374, 1508/1498* | ND | This work |
| bsNOS–Arg–H4B | 6C | 1374, 1498 | 550 (ND) | This work |
| bsNOS–NOHA–H4B | 6C | ND | 555 (Δ12) | This work |
| nNOSox | 5C | 1376, 1508 | ND | [40] |
| nNOSox–Arg–H4B | 6C | 1376, 1497 | 549 (Δ15) | [40] |
| iNOSox–Arg | 6C | ND | 540 (Δ17) | [41] |
| iNOSox–Arg–H4B | 6C | ND | 549 (Δ16) | [41] |
*The majority of the haem was 5C based on the relative intensities of the ν3 lines and the relative intensities of the Soret optical absorption bands.
With bsNOS, similar results were obtained. In the absence of L-arginine, the majority of the complex was 5C, as evidenced by the strong ν3 line at 1508 cm−1 and the weaker ν3 line of the 6C complex at 1498 cm−1 (see supplementary Figure S4 at http://www.BiochemJ.org/bj/401/bj4010235add.htm). With L-arginine present, the complex was 6C, as shown by the single ν3 line at 1498 cm−1 (supplementary Figure S4). The optical absorption spectra were consistent with these co-ordination state assignments as the complex in the absence of substrate had optical absorption bands at 403 and ∼562 nm, with a small intensity shoulder near 438 nm, and the complex in the presence of L-arginine had optical absorption bands at 438 and ∼567 nm (supplementary Figure S4, inset panel). The complex with NOHA had optical absorption bands at 435 and 569 nm, which was consistent with a 6C haem (result not shown).
Low-frequency region of the FeII–NO complexes of saNOS and bsNOS
Isotopic substitution was used to identify the modes involving the haem-bound NO in the low-frequency region of the resonance Raman spectra. Both the νFe−NO and δFe−N−O modes were observed in the resonance Raman spectra of P450cam [44]. However, the assignments of Fe–NO and N-O stretching modes were only approximations as these modes are of mixed character in the inherently bent FeII–NO complexes of haem proteins [44]. Nevertheless, the results of multiple isotope analyses, and normal mode calculations indicated that the line at higher frequency (547 cm−1, Δ16 cm−1 with 15N16O) of the substrate-free P450cam corresponded to a νFe−NO mode with some bending character and the line at lower frequency (444 cm−1, Δ3 cm−1 with 15N16O) corresponded to the δFe−N−O mode [44]. With isotope substitution, a single isotope-sensitive line was detected in the resonance Raman spectrum of the FeII–NO complex of L-arginine and H4B-bound saNOS at 550 cm−1 (Δ15 cm−1 with 15N16O; Figure 6, lines c, d and j). Through comparison with the frequencies and amplitudes of the isotope shifts displayed by the νFe−NO and δFe−N−O modes of the FeII–NO complexes of P450cam [44], this line was assigned to the νFe−NO mode of saNOS (Table 2). This frequency was nearly identical with that displayed by the FeII–NO complexes of nNOSox and iNOSox (Table 2). With L-arginine alone, the resonance Raman spectrum was identical to that of the sample containing L-arginine and H4B, and the νFe−NO mode was also at 550 cm−1 (Table 2). With NOHA and H4B, the νFe-NO stretching mode was at a higher frequency than with L-arginine at 555 cm−1 (Figure 6, lines e, f and k; Table 2). Like saNOS, the νFe−NO mode of the FeII–NO complex of bsNOS with NOHA and H4B was detected at 555 cm−1 (Figure 6, lines g, h and l), and the νFe−NO mode of the sample with L-arginine and H4B was at 550 cm−1 (Table 2). However, the δFe−N−O mode expected near 445 cm−1 [44] was not detected for either protein.
Figure 6. Low-frequency region of the resonance Raman spectra of the FeII–NO complexes of saNOS and bsNOS.

The spectra were obtained with substrate-free and pterin-free saNOS using 14N16O (a) and 15N16O (b). The 14N16O minus 15N16O difference spectrum (a minus b) is shown in (i). The spectra of L-arginine-bound and H4B-bound saNOS were obtained with 14N16O (c) and 15N16O (d). The 14N16O minus 15N16O difference spectra of L-arginine-bound and H4B-bound saNOS (c minus d) are shown in (j). The spectra of NOHA-bound and H4B-bound saNOS were obtained with 14N16O (e) and 15N16O (f). The 14N16O minus 15N16O difference spectra of NOHA-bound and H4B-bound saNOS (e minus f) are shown in (k). The spectra of NOHA-bound and H4B-bound bsNOS were obtained with 14N16O (g) and 15N16O (h). The 14N16O minus 15N16O difference spectra of NOHA-bound and H4B-bound bsNOS (g minus h) are shown in (l).
As mentioned above, in the absence of substrate and H4B, the FeII–NO complexes of saNOS and bsNOS were 5C. An isotope- sensitive line at 526 cm−1 (Δ7 cm−1 with 15N16O; Figure 6, lines a, b and i) was detected in the resonance Raman spectrum of substrate-free and pterin-free saNOS (Table 2). This frequency was similar to that of the νFe−NO modes of other haem proteins that form 5C FeII–NO complexes such as sGC [45]. This line probably corresponds to the νFe−NO mode of saNOS, although the isotope shift was smaller than the 13–16 cm−1 isotope shift displayed by other 5C complexes of haem proteins [46]. For bsNOS, good resonance Raman spectra of the 5C form could not be obtained.
DISCUSSION
A comparison of the environment at the active site of bacterial and mammalian NOSs
We used NO to probe the haem environment of the ferric saNOS and bsNOS proteins. The frequencies of the νFe−NO modes of saNOS (539 cm−1) and bsNOS (540 cm−1) in the absence of substrate and H4B were nearly identical, indicating that both proteins had very similar haem environments. These frequencies were compared with those of other haem proteins and were found to most resemble those of nNOSox (535 cm−1) and iNOSox (537 cm−1) rather than P450cam (528 cm−1; [39]) and P450BM3 (526 cm−1; [43]), indicating a similar environment at the active sites of bacterial and mammalian NOSs (Table 1). The frequencies of the νFe−NO modes of saNOS and bsNOS were insensitive to the addition of the pterins H4B and THF (Table 1). In contrast, the frequencies of the νFe−NO modes of mammalian iNOSox and nNOSox increased by 5–7 cm−1 upon H4B binding. H4B binding also induced the bending mode of iNOSox (Table 1), which is probably an electronic effect from the distortion of the haem caused by H4B binding [39]. The Fe–N–O bending modes of saNOS and bsNOS were observed as a shoulder (∼551 cm−1) of the main line at 539 cm−1 of the substrate-free and pterin-free enzymes and were clearly visible in the difference spectra (Figure 2). As already mentioned, the intensity of this bending mode did not increase with H4B or THF binding, and the frequency of the νFe-NO mode remained at 539–540 cm−1 for both proteins (Table 1). The absence of response to H4B binding may reflect the fact that H4B is probably not the native cofactor of these bacterial NOSs and that optimal interactions with the proteins may not be achieved. There is, however, no doubt that H4B bound to the FeIII–NO complex, at least for saNOS, as haem-deformation modes were enhanced in its presence (see below).
With either L-arginine or NOHA present, the isotope-sensitive line of FeIII–NO complexes had a higher contribution from the δFe−N−O bending mode for both saNOS and bsNOS (see below). The binding of L-arginine induces the Fe–N–O bending mode of H4B-containing nNOSox and iNOSox [24,41]. The similar response and frequencies of bacterial and mammalian NOSs following L-arginine binding suggest that the interaction between the haem-bound NO and L-arginine is comparable in both the mammalian and bacterial NOSs. This result was not surprising given that the crystal structures of several bacterial NOSs showed that the substrate-binding site is conserved with respect to those of the mammalian NOSs [15–17]. In addition, the spectroscopic characterization of bacterial NOSs using CO and oxygen as probes indicates that L-arginine interacts with haem-bound ligands in a similar manner to the mammalian NOSs, particularly nNOS [20,27,33].
The extent of the similarity of the haem environments of bacterial NOSs with those of iNOS and nNOS includes the conservation of haem-deformation modes observed in the FeIII–NO complexes of iNOSox and nNOSox [24,40,41]. With iNOSox, the intensity of the haem modes at 685 and 800 cm−1 is enhanced with H4B binding, and other haem modes at 352, 390, 710, 729 and 746 cm−1 appear [41]. With saNOS, a similar response to H4B binding was observed with the induction of lines at 708 and 728 cm−1 and the increase in the intensity of lines at 684, 745, 752 and 801 cm−1 (Figure 3). In addition to H4B, THF and L-arginine also induced these deformation modes for saNOS (Figures 2 and 3). The same modes were observed for bsNOS in the substrate-free and pterin-free protein as well as in the presence of substrate and H4B, as reported previously [33].
Several of these modes have been thoroughly analysed by Li et al. [41], who showed that they arise from distortion of the haem caused by H4B binding to iNOSox and that they are strongest for samples containing both L-arginine and H4B. The same modes appeared in the H4B-containing samples of the FeIII–NO complex of nNOSox [40]. Based on the assignment proposed for iNOSox, the modes of saNOS and bsNOS at 684 cm−1 may be assigned to γ15 with B2u symmetry, the mode at 708 cm−1 to γ11 with B1u symmetry, the mode at 728 cm−1 to γ5 with A2u symmetry, and the mode at 745 cm−1 to γ1 with A1u symmetry.
The fact that the association of H4B, THF and L-arginine with saNOS caused a distortion of the haem along many co-ordinates, as is the case for mammalian iNOSox and nNOSox, and that bsNOS already displayed these modes with or without substrate and pterin present, indicates that there is selective pressure to conserve these structural modifications of the haem. Haem distortion from planarity is generally believed to ease the oxidation of haem and make the reduction more difficult [47,48]. By analogy to nitrophorins, Li et al. [41] suggested that haem distortion of iNOSox may diminish the rate at which the FeIII–NO complex formed at the end of the catalytic cycle and may be reduced to the long-lived FeII–NO inhibitory complex. Haem distortion may thus help modulate the amount of enzyme inhibited by NO for a long period of time by interfering with the formation of the FeII–NO complex. In this regard, it has been shown that bsNOS is susceptible to inhibition as NO dissociates less rapidly from the ferric haem than in the mammalian NOSs [19].
Overall, there is little difference between the ferric haem environments of bacterial and mammalian NOSs when probed with NO given that the Fe–N–O frequencies and the response to L-arginine binding are similar (see below) and that most of the haem-deformation modes detected with the mammalian NOSs are observed in the spectra of the bacterial NOSs. However, the frequencies of the νFe−NO modes of the substrate-free bacterial NOSs were not modified in response to H4B binding.
Interactions between the substrates and the haem-bound NO of the ferric haem
The FeIII–NO complexes of both saNOS and bsNOS obtained with L-arginine displayed a single isotope-sensitive line that contained a significant contribution from the bending mode, as with the mammalian NOSs (Table 1). The frequency of this line (542 cm−1) was lower than the low-intensity bending mode detected in the absence of L-arginine at 551 cm−1 (Table 1). With P450cam, the δFe−N−O mode is enhanced in the presence of substrates, which is attributed to direct interactions between haem-bound NO and substrates that cause the Fe–N–O linkage to become slightly bent [39]. Similarly, the enhancement of the intensity of the bending mode and the shift of its frequency may indicate that with saNOS and bsNOS, L-arginine induces the bending and/or tilting of the Fe–N–O unit as with mammalian iNOSox and nNOSox containing H4B [24,41].
To determine whether the interaction between NO and L-arginine influences the affinity of the NO complex for L-arginine, we determined the Kd for L-arginine of saNOS using the kinetics of NO rebinding to L-arginine-repleted and L-arginine-depleted saNOS samples. The elevated Kd for L-arginine of the FeIII–NO complex (102±8 μM) with respect to the Kd for L-arginine of the ferric, resting enzyme (4.2±0.2 μM) indicated a less favourable environment for L-arginine when the haem was co-ordinated with NO. The elevated Kd for L-arginine when NO was bound to the haem may be interpreted as direct evidence for steric interactions between L-arginine and NO, which is consistent with the detection of the intense δFe−N−O mode. In addition, structural changes to the active site to allow L-arginine binding when haem is coordinated with NO may contribute to lower the affinity for L-arginine as well.
The δFe−N−O mode at 543 cm−1 was enhanced for both saNOS [20] and bsNOS with NOHA, as it was with L-arginine (Table 1). As the δFe−N−O frequencies of both proteins were nearly identical with NOHA and L-arginine, we concluded that these substrates had the same interactions with the haem-bound NO. Unlike the interactions with these substrates, the product of the reaction (L-citrulline) enhanced the intensity of the δFe−N−O mode for both saNOS and bsNOS, but the frequency shift with respect to the Fe–N–O bending mode without substrate was small (547–549 cm−1 compared with 551 cm−1) (Table 1). Given that the substrates differ substantially in the portion that binds near the haem, the similar δFe−N−O frequencies with L-arginine and NOHA indicate that the hydroxy group of NOHA was not involved in the interaction with the haem-bound NO. This interpretation is consistent with the NOHA-associated structures of bsNOS and other NOSs in which the hydroxy group of NOHA points away from the haem [26,49]. On the other hand, the frequency of the δFe−N−O mode with citrulline was different than that with L-arginine and NOHA (Table 1). This suggests that the Nω shared by L-arginine and NOHA may be involved in the interaction with the NO bound to the ferric haem. The crystal structures of the FeIII–NO complexes of bsNOS with L-arginine and NOHA at the active site are consistent with this proposal as the haem-bound NO and Nω are in close proximity, and partial nitrosation of Nω occurs in the NOHA structure [26].
Interactions between the substrates and the haem-bound NO of the ferrous haem
Unlike the FeIII–NO complexes, a dissimilar response to L-arginine and NOHA binding was observed with the FeII–NO complexes of saNOS and bsNOS. With L-arginine, the νFe−NO stretching mode was at 550 cm−1 for both saNOS and bsNOS, which is the same frequency as that of nNOSox and iNOSox (Table 2). With NOHA, the frequency of the νFe−NO mode was higher at 555 cm−1 in both bacterial NOSs (Table 2). One obvious difference between the FeIII–NO and FeII–NO complexes is their geometry. Unconstrained FeIII–NO complexes are nearly linear but become slightly bent if the NO interacts with nearby groups, while FeII–NO complexes are inherently bent [39,44,50,51]. The different geometries of FeIII–NO and FeII–NO complexes may explain the dissimilar effects of NOHA and L-arginine binding to the complexes. NOHA-saturated FeII–NO complexes of mammalian NOSs were not characterized by resonance Raman spectroscopy. However, it is noteworthy that the interactions of L-arginine and NOHA with the ferrous haem-bound NO of nNOS probed by EPR have been interpreted as changes in Fe–NO geometry imposed by the association of these substrates with the protein [52].
The higher frequency of the νFe−NO stretching mode with NOHA compared with that with L-arginine clearly shows that both substrates do not interact in the same way with the NO bound to the reduced haem. Direct interactions between the haem-bound NO and the substrates may be steric or polar or may involve hydrogen bonds. Steric interactions caused an increase in the νFe−NO frequency in P450cam in complex with substrates [44]. saNOS and bsNOS display higher frequencies in complex with NOHA than L-arginine, indicating that NOHA may exert higher steric constraint on the Fe–N–O unit. However, computational studies with Mb have revealed that a decreased Fe–N–O angle was associated with a lower frequency of the νFe−NO mode and that an increased Fe–N–O angle did not have much effect on the νFe−NO frequency [53]. In this regard, a decreased Fe–N–O angle of 126 ° versus 132 ° was reported for the recently characterized structures of the FeII–NO complexes of bsNOS with NOHA versus L-arginine [26]. Therefore, the increased νFe−NO frequency observed with NOHA as compared with that with L-arginine cannot be explained from increased steric interactions with the former if the Fe–N–O angle is smaller. Given that L-arginine and NOHA are both potential hydrogen bond donors, electrostatic interactions between the haem-bound NO of the reduced haem and these substrates may also contribute to the observed 5 cm−1 frequency difference. In this context, the structures of the FeII–NO complexes of bsNOS with L-arginine and NOHA recently reported by Crane et al. [26] offer an interesting insight. Overall, the structures show that both the O and N atoms of the haem-bound NO form hydrogen bonds with the Nω of L-arginine and NOHA. However, the hydrogen bond between Nω and the N atom of haem-bound NO appears to be stronger with NOHA than with L-arginine. Therefore, we suggest that the higher frequency of the νFe−NO mode with NOHA than with L-arginine may result from electrostatic interactions between NOHA and the NO of the reduced haem that are different to those with L-arginine. The different interactions of L-arginine and NOHA are interesting if one considers the use of the FeII–NO complexes as models of the FeIIO2 because different oxygenated species may participate in the catalysis of L-arginine and NOHA respectively. Recently, we characterized the oxygenated complex of saNOS in the presence of L-arginine and showed that the frequency of the O–O stretching mode was sensitive to L-arginine binding resulting in a decreased frequency from 1135 to 1123 cm−1, which was likely to be the result of hydrogen bonding interactions [20]. Hydrogen bonding interactions between L-arginine and the haem-bound dioxygen are also supported by the kinetics of oxygen dissociation that are 40 times faster without L-arginine present compared with L-arginine bound to saNOS [20]. We are currently investigating the oxygenated complex of saNOS with NOHA to determine whether the Fe–O and O–O stretching frequencies are the same as those for the L-arginine complex [20]. Preliminary results indicate that this is not the case reinforcing the idea that NO- and O2-bound to the reduced haem make substrate-specific interactions with L-arginine and NOHA.
While L-arginine and NOHA interacted differently with NO when the haem was reduced, they both stabilized the thiolate bond of the reduced saNOS and bsNOS complexes with NO (Table 2). In the absence of substrates, the FeII–NO complexes were 5C while L-arginine and NOHA binding stabilized the 6C structures of both proteins. These results indicate that for bacterial NOSs, as for the mammalian NOSs [54], binding of the substrate decreases the destabilizing effect on the proximal bond caused by NO binding in haem proteins [54].
Conclusions
In the present study, we have shown, with NO as a probe, that the haem environments of bacterial saNOS and bsNOS are similar to those of mammalian NOSs since the frequencies of the Fe–N–O modes were similar both for the FeII–NO and FeIII–NO complexes of the bacterial and mammalian NOSs. We noted that the response of the bacterial NOSs to H4B binding differed from that of the mammalian NOSs, while the response to substrate binding was similar. The interactions between the haem-bound NO and L-arginine and NOHA showed that these substrates interacted in a similar manner with the ferric haem, and that steric interactions between the haem-bound NO and substrates occurred as the affinity for L-arginine decreased very significantly as NO bound to the haem. When the haem was reduced, the substrates did not interact in the same way and, as such, electrostatic interactions between the haem-bound NO and the substrates are likely. The different behaviour of the substrates with the reduced and ferric NO complexes revealed that the fine-tuning of the interactions between the haem-bound ligand and the substrate depended on Fe-ligand geometry. The different interactions of NO with the L-arginine and NOHA of the FeII–NO complex may be relevant to the proposed formation of distinct oxidizing species during the first and second rounds of catalysis, which involve L-arginine and NOHA respectively [11,26].
Online data
Acknowledgments
This work was supported by National Sciences and Engineering Research Council of Canada (NSERC) grant 250073 and Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT) grant 78927 to M.C. We thank Gene Bourgeau for editorial assistance.
References
- 1.Sengupta R., Sahoo R., Mukherjee S., Regulski M., Tully T., Stuehr D. J., Ghosh S. Characterization of Drosophila nitric oxide synthase: a biochemical study. Biochem. Biophys. Res. Commun. 2003;306:590–597. doi: 10.1016/s0006-291x(03)01003-9. [DOI] [PubMed] [Google Scholar]
- 2.Kim H. W., Batista L. A., Hoppes J. L., Lee K. J., Mykles D. L. A crustacean nitric oxide synthase expressed in nerve ganglia, Y-organ, gill and gonad of the tropical land crab, Gecarcinus lateralis. J. Exp. Biol. 2004;207:2845–2857. doi: 10.1242/jeb.01117. [DOI] [PubMed] [Google Scholar]
- 3.Feldman P. L., Griffith O. W., Stuehr D. J. The surprising life of nitric oxide. Chem. Eng. News. 1993;71:26–38. [Google Scholar]
- 4.Raman C. S., Martasek P., Masters B. S. S. Structural haems determining function in nitric oxide synthases. New York: Academic Press; 2000. [Google Scholar]
- 5.Li H., Poulos T. L. Structure-function studies on nitric oxide synthases. J. Inorg. Biochem. 2005;99:293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh D. K., Crane B. R., Ghosh S., Wolan D., Gachhui R., Crooks C., Presta A., Tainer J. A., Getzoff E. D., Stuehr D. J. Inducible nitric oxide synthase: role of the N-terminal beta-hairpin hook and pterin-binding segment in dimerization and tetrahydrobiopterin interaction. EMBO J. 1999;18:6260–6270. doi: 10.1093/emboj/18.22.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raman C. S., Li H., Martasek P., Kral V., Masters B. S., Poulos T. L. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 8.Stuehr D. J., Santolini J., Wang Z. Q., Wei C. C., Adak S. Update on mechanism and catalytic regulation in the NO synthases. J. Biol. Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 9.Sorlie M., Gorren A. C., Marchal S., Shimizu T., Lange R., Andersson K. K., Mayer B. Single-turnover of nitric-oxide synthase in the presence of 4-amino- tetrahydrobiopterin: proposed role for tetrahydrobiopterin as a proton donor. J. Biol. Chem. 2003;278:48602–48610. doi: 10.1074/jbc.M305682200. [DOI] [PubMed] [Google Scholar]
- 10.Rosen G. M., Tsai P., Pou S. Mechanism of free-radical generation by nitric oxide synthase. Chem. Rev. 2002;102:1191–1200. doi: 10.1021/cr010187s. [DOI] [PubMed] [Google Scholar]
- 11.Cho K.-B., Gauld J. W. Second half-reaction of the nitric oxide synthase: computational insights into the initial step and key proposed intermediate. J. Phys. Chem. B. 2005;109:23706–23714. doi: 10.1021/jp054864o. [DOI] [PubMed] [Google Scholar]
- 12.Gorren A. C., Mayer B. Tetrahydrobiopterin in nitric oxide synthesis: a novel biological role for pteridines. Curr. Drug Metab. 2002;3:133–157. doi: 10.2174/1389200024605154. [DOI] [PubMed] [Google Scholar]
- 13.Stuehr D. J., Wei C. C., Wang Z., Hille R. Exploring the redox reactions between haem and tetrahydrobiopterin in the nitric oxide synthases. Dalton Trans. 2005;21:3427–3435. doi: 10.1039/b506355h. [DOI] [PubMed] [Google Scholar]
- 14.Wei C. C., Wang Z. Q., Hemann C., Hille R., Stuehr D. J. A tetrahydrobiopterin radical forms and then becomes reduced during Nω- hydroxyarginine oxidation by nitric-oxide synthase. J. Biol. Chem. 2003;278:46668–46673. doi: 10.1074/jbc.M307682200. [DOI] [PubMed] [Google Scholar]
- 15.Bird L. E., Ren J., Zhang J., Foxwell N., Hawkins A. R., Charles I. G., Stammers D. K. Crystal structure of SANOS, a bacterial nitric oxide synthase oxygenase protein from Staphylococcus aureus. Structure. 2002;10:1687–1696. doi: 10.1016/s0969-2126(02)00911-5. [DOI] [PubMed] [Google Scholar]
- 16.Pant K., Bilwes A. M., Adak S., Stuehr D. J., Crane B. R. Structure of a nitric oxide synthase haem protein from Bacillus subtilis. Biochemistry. 2002;41:11071–11079. doi: 10.1021/bi0263715. [DOI] [PubMed] [Google Scholar]
- 17.Sudhamsu J., Crane B. R. Structure and reactivity of a thermostable prokaryotic nitric-oxide synthase that forms a long-lived oxy-haem complex. J. Biol. Chem. 2006;281:9623–9632. doi: 10.1074/jbc.M510062200. [DOI] [PubMed] [Google Scholar]
- 18.Adak S., Bilwes A. M., Panda K., Hosfield D., Aulak K. S., McDonald J. F., Tainer J. A., Getzoff E. D., Crane B. R., Stuehr D. J. Cloning, expression, and characterization of a nitric oxide synthase protein from Deinococcus radiodurans. Proc. Natl. Acad. Sci. U.S.A. 2002;99:107–112. doi: 10.1073/pnas.012470099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adak S., Aulak K. S., Stuehr D. J. Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J. Biol. Chem. 2002;277:16167–16171. doi: 10.1074/jbc.M201136200. [DOI] [PubMed] [Google Scholar]
- 20.Chartier F. J., Blais S. P., Couture M. A Weak Fe–O bond in the oxygenated complex of the nitric-oxide synthase of Staphylococcus aureus. J. Biol. Chem. 2006;281:9953–9962. doi: 10.1074/jbc.M513893200. [DOI] [PubMed] [Google Scholar]
- 21.Werner-Felmayer G., Golderer G., Werner E. R. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr. Drug Metab. 2002;3:159–173. doi: 10.2174/1389200024605073. [DOI] [PubMed] [Google Scholar]
- 22.Buddha M. R., Tao T., Parry R. J., Crane B. R. Regioselective nitration of tryptophan by a complex between bacterial nitric-oxide synthase and tryptophanyl-tRNA synthetase. J. Biol. Chem. 2004;279:49567–49570. doi: 10.1074/jbc.C400418200. [DOI] [PubMed] [Google Scholar]
- 23.Kers J. A., Wach M. J., Krasnoff S. B., Widom J., Cameron K. D., Bukhalid R. A., Gibson D. M., Crane B. R., Loria R. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature. 2004;429:79–82. doi: 10.1038/nature02504. [DOI] [PubMed] [Google Scholar]
- 24.Rousseau D. L., Li D., Couture M., Yeh S. R. Ligand-protein interactions in nitric oxide synthase. J. Inorg. Biochem. 2005;99:306–323. doi: 10.1016/j.jinorgbio.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Spiro T. G., Wasbotten I. H. CO as a vibrational probe of haem protein active sites. J. Inorg. Biochem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 26.Pant K., Crane B. R. Nitrosyl–haem structures of Bacillus subtilis nitric oxide synthase have implications for understanding substrate oxidation. Biochemistry. 2006;45:2537–2544. doi: 10.1021/bi0518848. [DOI] [PubMed] [Google Scholar]
- 27.Chartier F. J., Couture M. Stability of the haem environment of the nitric oxide synthase from Staphylococcus aureus in the absence of pterin cofactor. Biophys. J. 2004;87:1939–1950. doi: 10.1529/biophysj.104.042119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castanie M. P., Berges H., Oreglia J., Prere M. F., Fayet O. A set of pBR322-compatible plasmids allowing the testing of chaperone-assisted folding of proteins overexpressed in Escherichia coli. Anal. Biochem. 1997;254:150–152. doi: 10.1006/abio.1997.2423. [DOI] [PubMed] [Google Scholar]
- 29.Appleby C. A. Purification of Rhizobium cytochromes P-450. Methods Enzymol. 1978;52:157–166. doi: 10.1016/s0076-6879(78)52018-1. [DOI] [PubMed] [Google Scholar]
- 30.McMillan K., Masters B. S. Optical difference spectrophotometry as a probe of rat brain nitric oxide synthase haem-substrate interaction. Biochemistry. 1993;32:9875–9880. doi: 10.1021/bi00089a001. [DOI] [PubMed] [Google Scholar]
- 31.Copeland R. A. Enzymes. New York: Wiley-VCH; 2000. A practical introduction to structure, mechanism, and data analysis. [Google Scholar]
- 32.Sono M., Ledbetter A. P., McMillan K., Roman L. J., Shea T. M., Masters B. S., Dawson J. H. Essential thiol requirement to restore pterin- or substrate-binding capability and to regenerate native enzyme-type high-spin haem spectra in the Escherichia coli-expressed tetrahydrobiopterin-free oxygenase domain of neuronal nitric oxide synthase. Biochemistry. 1999;38:15853–15862. doi: 10.1021/bi991580j. [DOI] [PubMed] [Google Scholar]
- 33.Santolini J., Roman M., Stuehr D. J., Mattioli T. A. Resonance Raman study of Bacillus subtilis NO synthase-like protein: similarities and differences with mammalian NO synthases. Biochemistry. 2006;45:1480–1489. doi: 10.1021/bi051710q. [DOI] [PubMed] [Google Scholar]
- 34.Li D., Hayden E. Y., Panda K., Stuehr D. J., Deng H., Rousseau D. L., Yeh S. R. Regulation of the monomer-dimer equilibrium in inducible nitric-oxide synthase by nitric oxide. J. Biol. Chem. 2006;281:8197–8204. doi: 10.1074/jbc.M507328200. [DOI] [PubMed] [Google Scholar]
- 35.Wang J., Caughey W. S., Rousseau D. L. Resonance Raman scattering: a probe of haem protein-bound nitric oxide. New York: John Wiley & Sons; 1996. [Google Scholar]
- 36.Hu S., Smith K. M., Spiro T. G. Assignment of protohaem resonance Raman spectrum by haem labeling in myoglobin. J. Am. Chem. Soc. 1996;118:12638–12646. [Google Scholar]
- 37.Spiro T. G., Li X. Y. Resonance Raman spectroscopy of metalloproteins. New York: Wiley; 1988. [DOI] [PubMed] [Google Scholar]
- 38.Tomita T., Haruta N., Aki M., Kitagawa T., Ikeda-Saito M. UV resonance Raman detection of a ligand vibration on ferric nitrosyl haem proteins. J. Am. Chem. Soc. 2001;123:2666–2667. doi: 10.1021/ja001431k. [DOI] [PubMed] [Google Scholar]
- 39.Hu S., Kincaid J. R. Resonance Raman characterization of nitric oxide adducts of cytochrome P450cam: The effect of substrate structure on the iron–ligand vibrations. J. Am. Chem. Soc. 1991;113:2843–2850. [Google Scholar]
- 40.Couture M., Adak S., Stuehr D. J., Rousseau D. L. Regulation of the properties of the haem–NO complexes in nitric-oxide synthase by hydrogen bonding to the proximal cysteine. J. Biol. Chem. 2001;276:38280–38288. doi: 10.1074/jbc.M105341200. [DOI] [PubMed] [Google Scholar]
- 41.Li D., Stuehr D. J., Yeh S. R., Rousseau D. L. Haem distortion modulated by ligand–protein interactions in inducible nitric oxide synthase. J. Biol. Chem. 2004;279:25489–25499. doi: 10.1074/jbc.M400968200. [DOI] [PubMed] [Google Scholar]
- 42.Obayashi E., Tsukamoto K., Adachi S., Takahashi S., Nomura M., Iizuka T., Shoun H., Shiro Y. Unique binding of nitric oxide to ferric nitric oxide reductase from Fusarium oxysporum elucidated with infrared, resonance Raman and X-ray absorption spectroscopies. J. Am. Chem. Soc. 1997;119:7807–7816. [Google Scholar]
- 43.Deng T. J., Proniewicz L. M., Kincaid J. R., Yeom H., Macdonald I. D., Sligar S. G. Resonance Raman studies of cytochrome P450BM3 and its complexes with exogenous ligands. Biochemistry. 1999;38:13699–13706. doi: 10.1021/bi991287j. [DOI] [PubMed] [Google Scholar]
- 44.Hu S., Kincaid J. R. Resonance Raman spectra of the nitric oxide adducts of ferrous cytochrome P450cam in the presence of various substrates. J. Am. Chem. Soc. 1991;113:9760–9766. [Google Scholar]
- 45.Deinum G., Stone J. R., Babcock G. T., Marletta M. A. Binding of nitric oxide and carbon monoxide to soluble guanylate cyclase as observed with Resonance raman spectroscopy. Biochemistry. 1996;35:1540–1547. doi: 10.1021/bi952440m. [DOI] [PubMed] [Google Scholar]
- 46.Lukat-Rodgers G. S., Rodgers K. R. Characterization of ferrous FixL-nitric oxide adducts by resonance Raman spectroscopy. Biochemistry. 1997;36:4178–4187. doi: 10.1021/bi9628230. [DOI] [PubMed] [Google Scholar]
- 47.Shelnutt J. A., Song S.-Z., Ma J.-G., Jia S.-L., Jentzen W., Medforth C. J. Nonplanar porphyrins and their significance in proteins. Chem. Soc. Reviews. 1998;27:31–41. [Google Scholar]
- 48.Walker F. A. Nitric oxide interactions with insect nitrophorins and thoughts on the electron configuration of the {FeNO}6 complex. J. Inorg. Biochem. 2005;99:216–236. doi: 10.1016/j.jinorgbio.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Crane B. R., Arvai A. S., Ghosh S., Getzoff E. D., Stuehr D. J., Tainer J. A. Structures of the Nω-hydroxy-L-arginine complex of inducible nitric oxide synthase oxygenase dimer with active and inactive pterins. Biochemistry. 2000;39:4608–4621. doi: 10.1021/bi992409a. [DOI] [PubMed] [Google Scholar]
- 50.Vogel K. M., Kozlowski P. M., Zgierski M. Z., Spiro T. G. Determinants of the FeXO (X=C, N, O) vibrational frequencies in haem adducts from experiment and density functional theory. J. Am. Chem. Soc. 1999;121:9915–9921. [Google Scholar]
- 51.Spiro T. G., Zgierski M. Z., Kozlowski P. M. Stereoelectronic factors in CO, NO and O2 binding to haem from vibrational spectroscopy and DFT analysis. Coordin. Chem. Rev. 2001;219–221:923–936. [Google Scholar]
- 52.Migita C. T., Salerno J. C., Masters B. S., Martasek P., McMillan K., Ikeda-Saito M. Substrate binding-induced changes in the EPR spectra of the ferrous nitric oxide complexes of neuronal nitric oxide synthase. Biochemistry. 1997;36:10987–10992. doi: 10.1021/bi970823+. [DOI] [PubMed] [Google Scholar]
- 53.Coyle C. M., Vogel K. M., Rush T. S., Kozlowski P. M., 3rd, Williams R., Spiro T. G., Dou Y., Ikeda-Saito M., Olson J. S., Zgierski M. Z. FeNO structure in distal pocket mutants of myoglobin based on resonance Raman spectroscopy. Biochemistry. 2003;42:4896–4903. doi: 10.1021/bi026395b. [DOI] [PubMed] [Google Scholar]
- 54.Huang L., Abu-Soud H. M., Hille R., Stuehr D. J. Nitric oxide-generated P420 nitric oxide synthase: characterization and roles for tetrahydrobiopterin and substrate in protecting against or reversing the P420 conversion. Biochemistry. 1999;38:1912–1920. doi: 10.1021/bi981954t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.