

Abstract
BHMT (betaine–homocysteine methyltransferase) remethylates homocysteine to form methionine. SAM (S-adenosylmethionine) inhibits BHMT activity, but whether SAM modulates BHMT gene expression is unknown. Transcriptional regulation of the human BHMT is also unknown. The present study examined regulation of the human BHMT gene by SAM and its metabolite, MTA (5′-methylthioadenosine). To facilitate these studies, we cloned the 2.7 kb 5′-flanking region of the human BHMT gene (GenBank® accession number AY325901). Both SAM and MTA treatment of HepG2 cells resulted in a dose- and time-dependent decrease in BHMT mRNA levels, which paralleled their effects on the BHMT promoter activity. Maximal suppression was observed with the BHMT promoter construct −347/+33, which contains a number of NF-κB (nuclear factor κB) binding sites. SAM and MTA treatment increased NF-κB nuclear binding and NF-κB-driven luciferase activities, and increased nuclear binding activity of multiple histone deacetylase co-repressors to the NF-κB sites. Overexpression of p50 and p65 decreased BHMT promoter activity, while blocking NF-κB activation increased BHMT expression and promoter activity, and prevented SAM but not MTA's ability to inhibit BHMT expression. The NF-κB binding site at −301 is responsible, at least in part, for this effect. Lower BHMT expression can impair homocysteine metabolism, which can induce ER (endoplasmic reticulum) stress. Indeed, MTA treatment resulted in increased expression ER stress markers. In conclusion, SAM and MTA down-regulate BHMT expression in HepG2 cells in part by inducing NF-κB, which acts as a repressor for the human BHMT gene. While SAM's mechanism is NF-κB-dependent, MTA has both NF-κB-dependent and -independent mechanisms.
Keywords: betaine–homocysteine methyltransferase (BHMT), endoplasmic reticulum stress, HepG2 cell, 5′-methylthioadenosine, nuclear factor κB (NF-κB), S-adenosylmethionine
Abbreviations: BHMT, betaine–homocysteine methyltransferase; ChIP, chromatin immunoprecipitation; Ct, threshold cycle value; EMSA, electrophoretic mobility-shift assay; ER, endoplasmic reticulum; FAM, 6-carboxy fluorescein; GADD153, growth-arrest and DNA-damage-inducible protein 153; GRP78, glucose-regulated protein 78; HDAC, histone deacetylase; HNF, hepatocyte nuclear factor; HPRT1, hypoxanthine–guanine phosphoribosyltransferase 1; MTA, 5′-methylthioadenosine; IκB, inhibitory κB; IκBSR, IκB super-repressor; NF-κB, nuclear factor κ B; RACE, rapid amplification of cDNA ends; RT, reverse transcriptase; SAM, S-adenosylmethionine; TAMRA, 6-carboxytetramethylrhodamine; UBC, ubiquitin C
INTRODUCTION
BHMT (betaine–homocysteine methyltransferase) is a zinc metalloenzyme that catalyses the transfer of a methyl group from betaine to homocysteine, forming dimethylglycine and methionine respectively [1]. BHMT is primarily expressed in the liver and kidney and is responsible, along with methionine synthase, for the remethylation of homocysteine to regenerate methionine [1]. In the rat, hepatic BHMT gene expression is induced by a methionine-deficient diet. This is further induced if the methionine-deficient diet contains dietary methyl groups such as betaine or choline [1]. In humans, betaine supplementation also increases homocysteine remethylation [2]. Despite these well-known effects of diet on BHMT gene expression, the molecular mechanism remains unknown.
Previously, we reported that hepatic BHMT mRNA levels are significantly increased in the MAT1A knockout mouse where hepatic SAM (S-adenosylmethionine; also known as AdoMet and SAMe) levels are reduced by 75% [3]. Another well-known condition where hepatic BHMT activity is induced is ethanol feeding in rodents [4]. We reported that ethanol feeding depleted both hepatic methionine and SAM levels [5]. In liver the bulk of methionine is metabolized to SAM so that methionine deficiency results in SAM deficiency [6]. This raises the possibility that the effects observed with a methionine-deficient diet may be exerted at least in part via SAM. Indeed, Finkelstein and Martin [7] showed that SAM inactivates BHMT. However, whether SAM can also influence BHMT at the mRNA level is unknown. Previously, Bose et al. [8] showed that the activity of recombinant human BHMT is not regulated by SAM, which challenges previous observations [7].
BHMT mRNA levels and activity are decreased in rat liver cirrhosis [9], which may contribute to the well-known hyperhomocysteinaemia that occurs in cirrhosis [10]. We have also shown that most of the patients with liver cirrhosis have either undetectable or reduced BHMT mRNA levels [11]. Given the importance of homocysteine in atherosclerosis and as an inducer of ER (endoplasmic reticulum) stress [12,13], it is important to better understand the regulation of the BHMT gene. The purpose of the current study is to examine whether SAM and its metabolite, MTA (5′-methylthioadenosine), can influence the expression of BHMT and, if so, define the molecular mechanisms.
MATERIALS AND METHODS
Materials
Cell culture medium and FBS (fetal bovine serum) were obtained from Gibco BRL Life Technologies (Grand Island, NY, U.S.A.). The Luciferase Assay System was obtained from Promega (Madison, WI, U.S.A.). All restriction endonucleases were obtained from either Promega or Gibco. [32P]dCTP (3000 Ci/mmol) was purchased from New England Nuclear (DuPont, Boston, MA, U.S.A.). SAM, in the stable form of sulfate-p-toluenesulfonate salt was from Knoll Farmaceutici (Milan, Italy). MTA and cycloleucine were from Sigma (St. Louis, MO, U.S.A.). All other reagents were of analytical grade and were obtained from commercial sources.
Cell culture
HepG2 cells were obtained from the Cell Culture Core of the USC Liver Disease Research Center and grown according to instructions provided by the American Type Culture Collection (Rockville, MD, U.S.A.).
Recombinant plasmid and expression vectors
NF-κB (nuclear factor κB)-luciferase construct and empty vector pluc-MCS were obtained from Stratagene (La Jolla, CA, U.S.A.). pCMV-p50 and pCMV-p65 expression plasmids were kindly provided by Dr Richard Rippe (University of North Carolina at Chapel Hill) as described in [14]. Recombinant, replication-defective adenoviruses expressing IκBSR [IκB (inhibitory κB) super-repressor], which expresses a mutant IκB that cannot be phosphorylated and therefore irreversibly binds NF-κB and prevents its activation, have been previously described [14].
Cloning of the 5′-flanking region of the human BHMT gene and construction of 5′-deletion constructs
Overlapping clones containing the BHMT gene were isolated from a human genomic library (Genome Walker, Clontech). The library was screened using a random 32P-labelled probe of the human BHMT cDNA [15]. Five positive plaques were selected and DNA was isolated and digested with EcoRI. The insert fragment was subcloned into a TA vector (Invitrogen) and sequenced in both directions using the automated ABI Prism dRhodamine Terminator Cycle Sequencer performed by the Sequencing and Genetic Analysis Core Facility, Norris Cancer Institute, Keck School of Medicine USC. The initial primers were universal primers for the TA vector, all subsequent primers were nested primers designed using the available sequence information and the MacVector software program. The nucleotide sequence was verified by multiple bi-directional sequencing reactions. Sequences were aligned and a consensus sequence was generated using the ASSEMBLIGN software program. A 2.7 kb 5′-flanking region of the human BHMT gene was cloned between MluI and SmaI sites of a promoterless pGL-3-basic vector (Promega) creating the recombinant plasmid −2698/+33 BHMT-LUC. The desired 5′-deletion constructs (−2386/+33, −1920/+33, −1471/+33, −1176/+33, −839/+33 and −347/+33) were generated using the upstream primers: GTGGATATTATCATCTATTATG, GTTTTGCCACATTTTGAGG, GTATTTCAAACACATGTCACTTTAG, GTTCAAGCGATTCTCCT, GTAACTACTGTTAACATCTC and GTGCAAGAGGCAAAAAGGGAC respectively. The universal downstream PCR primer is GGTGGCATCTTTGTGGTGTCCAG.
Transcription start sites of the human BHMT
Transcription start sites were determined using the GeneRacer™ kit (Invitrogen). The 5′ ends of cDNA using the reverse primer 5′-TGTCGGTGCCCATGGCAGCTGCC-3′, which is complementary to −10 to +13 of the human BHMT [15], were obtained. RACE (rapid amplification of cDNA ends) PCR products were cloned using TOPO TA cloning for sequencing and sequencing analysis was performed by the Sequencing Facility, Norris Cancer Centre, Keck School of Medicine USC.
Analysis of promoter constructs in cell culture
To study the relative transcriptional activities of the BHMT promoter fragments, HepG2 cells (0.8×106 cells in 2 ml of medium) were transiently transfected with 2.5 μg of BHMT promoter luciferase gene construct or the promoterless pGL3-basic vector (as negative control) using the Superfect transfection reagent (Qiagen, Valencia, CA, U.S.A.) as we described [14]. To control for transfection efficiency, cells were co-transfected with the Renilla pRL-SV40 vector (Promega, Madison, WI, U.S.A.), which is a plasmid containing the Renilla luciferase gene driven by a SV40 (simian virus 40) promoter. After 24 h, cells were harvested and lysed in 200 μl of reporter lysis buffer (Luciferase Assay System, Promega). Aliquots of the cell lysates were sequentially assessed for firefly and Renilla luciferase activities using a TD-20/20 luminometer (Promega). The luciferase activity driven by the BHMT promoter constructs was normalized to the Renilla luciferase activity. Each experiment was done with triplicate samples.
Effect of SAM and MTA on BHMT expression assessed by Northern-blot analysis and real-time RT (reverse transcriptase)–PCR
HepG2 cells were treated with SAM (0.25–5 mM) or MTA (0.25–1 mM) for up to 18 h. We had previously shown that these agents could induce apoptosis in HepG2 cells after 18 h of treatment [16]. Indeed, both 5 mM SAM and MTA did not cause apoptosis after 12 h of treatment but apoptosis was induced by 18 h (results not shown). Following treatment, total RNA was extracted and Northern hybridization analysis was performed using specific BHMT cDNA probes that correspond to 879–1090 of the human BHMT sequence [15] as described in [17]. To ensure equal loading of RNA samples and transfer in each of the lanes, prior to hybridization, membranes were rinsed with ethidium bromide and photographed and the same membranes were also rehybridized with a 32P-labelled β-actin cDNA probe as described in [17]. Auto-radiography and densitometry (Gel Documentation System, Scientific Technologies, Carlsbad, CA, U.S.A. and NIH Image 1.60 software program) were used to quantify relative RNA. Results of Northern-blot analysis were normalized to β-actin.
Real-time quantitative PCR was also carried out on the above RNA samples from HepG2 cells treated with SAM (0.25–5 mM) or MTA (0.25–1 mM) for 12 h. Total RNA (1 μg) was used in a 20 μl reverse transcription reaction volume. Following reverse transcription (M-MLV RT; Life Technologies, Gaithersburg, MD, U.S.A.), the real-time PCR was run in the Mx3005PTM thermo-cycler (Stratagene, La Jolla, CA, U.S.A.) in triplicates with the thermo-cycle profile of stage 1: 95 °C for 10 min, stage 2: 95 °C for 15 s, 60 °C for 1 min, 40 cycles. The primer and TaqMan probe [BHMT 00156084, HPRT1 (hypoxanthine–guanine phosphoribosyltransferase 1) Hs99999909, UBC (ubiquitin C) Hs00824723, ready-in-use mixture] and Universal PCR Master mix were purchased from ABI (Foster City, CA, U.S.A.). HPRT1 and UBC were used as housekeeping genes as described in [18].
The expression of BHMT RNA was checked by normalizing the Ct (threshold cycle value) of BHMT to that of the control housekeeping gene (HPRT1 or UBC) [19]. The ΔCt obtained was used to find the relative expression of BHMT in treated cells compared with untreated or empty vector-treated cells according to the formula:
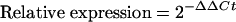 |
where ΔΔCt=(ΔCt of BHMT in treated cells)−(ΔCt of BHMT in control cells).
Effect of SAM and MTA on recombinant BHMT promoter and NF-κB-driven reporter activity
To assess the effect of SAM or MTA on BHMT promoter activity, HepG2 cells were transfected with recombinant human BHMT promoter constructs and treated with SAM (5 mM) or MTA (1 mM) during the last 12 h of the transfection. In some experiments, cells were pretreated with cycloleucine (20 mM) for 2 h in order to block the conversion of MTA back to SAM [16]. This was followed by MTA (1 mM) or SAM (5 mM) treatment for another 12 h. Luciferase activity driven by these promoter luciferase gene constructs was measured as described above.
To assess the effect of SAM or MTA on NF-κB-driven luciferase activity, HepG2 cells were transfected with recombinant NF-κB-LUC (contains five κB sequences linked to the reporter) and treated with SAM (5 mM) or MTA (1 mM) for 12 h. Luciferase activity driven by this NF-κB promoter was measured as described above.
Effect of p50 and p65 expression vectors on BHMT promoter activity
To see if overexpression of p50 or p65 can influence the BHMT promoter activity, HepG2 cells were first transfected with either a p50 or p65 expression vector (1.5 μg per well for 12 h) and then transfected with the recombinant BHMT promoter luciferase construct −347/+33-LUC or the pGL-3-basic vector for 24 h. Luciferase activity was measured as described above.
Effect of blocking NF-κB on BHMT expression, promoter activity and effects of SAM and MTA
To study the effect of blocking NF-κB activation, HepG2 cells were infected with recombinant adenoviruses carrying IκBSR or empty vector for 12 h as described previously [14]. After 12 h of infection, the viruses were removed and replaced with fresh medium for SAM (5 mM) or MTA treatment (1 mM) for 12 h and RNA was isolated for real-time PCR. In other experiments, following infection with adenoviruses carrying IκBSR or empty vector for 12 h, HepG2 cells were transfected with the BHMT promoter construct −347/+33-LUC, the pGL-3-basic vector, or the NF-κB-LUC for 12 h. Luciferase activity was measured as described above.
Effect of an NF-κB site mutation on BHMT promoter activity
Site-directed mutagenesis was performed to examine the functional roles of two putative NF-κB binding sites located at −301 and −135 of the BHMT gene (relative to the translation start site, see Figure 1). Each site was mutated (−301 site, from 5′-GTGGCAACCT-3′ to 5′-CTCGCAACCT; −135 site, from 5′-GGGAGGCCCT-3′ to 5′-AGTAGGCCCT-3′), where the mutated sequence is underlined. The mutation was performed by PCR and confirmed by restriction enzyme digestion and sequencing. HepG2 cells were transfected with −347/+33-LUC that contained the wild-type, a mutation at the −301 site (Mut−301), a mutation at the −135 site (Mut –135), or a double mutant (both NF-κB sites are mutated) and the effect of the mutation was assessed by measuring luciferase activity. HepG2 cells transfected with these constructs were also treated with SAM (5 mM) or MTA (1 mM) for 12 h and luciferase activity was measured as described above.
Figure 1. Nucleotide sequence of the 5′-flanking region of the human BHMT gene.

Sequence is numbered relative to the translational start site. The putative regulatory elements are indicated in bold letters above the underlined sequences. The transcriptional start site was determined using 5′-RACE as described in the Materials and methods section and indicated by the forward arrow.
EMSA (electrophoretic mobility-shift assay) and supershift assay
EMSAs for consensus (obtained from Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and putative NF-κB binding sites were performed as described in [14]. The probes are 32P-end labelled double-stranded DNA fragments (−142 to −114, −271 to −246 and −310 to −285 of BHMT) or containing a consensus NF-κB binding site (5′-AGTTGAGGGGACTTTCCCAGGC-3′). The effect of SAM (0–5 mM) and MTA (0–2 mM) on nuclear binding activity to the consensus NF-κB probe was first determined following treatment of HepG2 cells with these agents for 12 h. The effect of SAM (5 mM for 12 h), MTA (1 mM for 12 h), or p50 overexpression for 24 h on binding to the putative NF-κB binding sites of the BHMT promoter was next determined. Supershift assays were performed with anti-p50 and anti-p65 antibodies (Santa Cruz Biotechnology) as described in [14].
Western-blot analysis
A time course of NF-κB activation was determined using Western-blot analysis to assess the level of nuclear p65 and p50 as described in [20].
ChIP (chromatin immunoprecipitation) assay
To see if SAM or MTA treatment of HepG2 cells induces NF-κB and HDAC (histone deacetylase) binding to the BHMT promoter in an endogenous chromatin configuration, a ChIP assay was carried out following the ChIP assay kit protocol provided by Upstate (Waltham, MA, U.S.A.). HepG2 cells were treated with SAM (5 mM), MTA (1 mM) or vehicle control and processed for the ChIP assay as per the protocol except for minor modifications. Briefly, proteins were cross-linked to DNA by treating cells with 1% formaldehyde at 37 °C for 10 min. After fixation, cells were washed with ice-cold PBS, resuspended and lysed in SDS lysis buffer (Upstate) and centrifuged for 5 min at 110 g. Cell lysates were sonicated at 25–30% power, 5×10 s using a Sonic Dimembrator Model F60 (Fisher Scientific, Pittsburgh, PA, U.S.A.) to fragment the chromatin to approx. 1 kb or less. The sonicated cell lysates were spun in a microcentrifuge at 13000 g for 10 min at 4 °C. The supernatant containing the soluble chromatin sample was treated with 1 μl of 5 M NaCl and heated at 65 °C for 4 h to reverse the protein–DNA cross-links. The reversed soluble chromatin sample (20 μl) was removed and used as the input control (total chromatin fraction) for the final PCR reaction. The remaining chromatin solution was split into equal fractions and subjected to immunoprecipitation in the presence or absence of anti-p50, anti-p65, anti-HDAC1, anti-HDAC2 and anti-HDAC3 antibodies (Santa Cruz Biotechnology). PCR of the BHMT promoter region across the three NF-κB sites within −301 to −135 of the BHMT promoter was performed using the forward primer 5′-AAATCTGAGCGAGTGGCAACCTCGC-3′ (bp −313 to −289 relative to the ATG start codon) and the reverse primer 5′-ATCTTCGTGGTGTCCAGACAGGTGG-3′ (bp −23 to +2 relative to the ATG start codon). All PCR products were run on 8% (w/v) acrylamide gels and stained with ethidium bromide for 15–30 min.
SAM levels
SAM levels were measured in HepG2 cells treated with cycloleucine (20 mM, 2 h pretreatment) or vehicle followed by SAM (5 mM) or MTA (1 mM) with or without cycloleucine (20 mM) for another 12 h as we described previously [16].
Markers of ER stress
To study the effect of SAM and MTA on the ER stress response, the expression of the ER stress markers, GRP78 (glucose-regulated protein 78) and GADD153 (growth-arrest and DNA-damage-inducible protein 153) [13], was analysed in HepG2 cells treated with these agents for 12 h. Real-time PCR quantification of GRP78 and GADD153 RNA from SAM- or MTA-treated HepG2 cells was performed using the following custom made primers and probes (GenScript Corporation): GRP78 forward primer: TCCTGCGTCGGCGTGT; GRP78 reverse primer: GTTGCCCTGATCGTTGGC; and GRP78 probe labelled with FAM (6-carboxy fluorescein) and TAMRA (6-carboxytetramethylrhodamine): AAGAACGGCCGCGTGGAGATCAT. GADD153 forward primer: GTGAATCTGCACCAAGCATGA; GADD153 reverse primer: AAGGTGGGTAGTGTGGCCC; and GADD153 probe labelled with FAM and TAMRA: CAATTGGGAGCATCAGT-CCCCCACT. HPRT1 was used as a control housekeeping gene for these experiments. The real-time PCR was run in triplicate with the thermo-cycle profile of stage 1: 95 °C for 10 min; stage 2: 95 °C for 15 s, 60 °C for 1 min for 40 cycles.
Statistical analysis
Data are given as means±S.E.M. Statistical analysis was performed using ANOVA followed by Fisher's test for multiple comparisons. For changes in mRNA levels, ratios of BHMT to β-actin densitometric values were compared with ANOVA. Significance was defined by P<0.05.
RESULTS
Cloning and sequencing of the 5′-flanking region of the human BHMT
The sequence of the 2.7 kb product is shown in Figure 1. Analysis of the transcription factor binding site was performed using Transcription Factor Search (http://www.cbrc.jp/research/db/TFSEARCH.html) and MatInspector (http://www.genomatix.de/cgi-bin/eldorado/main.pl). The 5′-flanking region of the human BHMT contains multiple consensus binding sites for NF-κB, Sp1 (stimulating protein-1), SREBP (sterol-regulatory-element-binding protein), C/EBP (CCAAT/enhancer-binding protein) and HNF (hepatocyte nuclear factor). In addition, consensus binding sites for GRE (glucocorticoid receptor), CREB (cAMP-response-element-binding protein), STAT (signal transducer and activator of transcription), ISRE (interferon-stimulated response element), c-Myc, c-Myb and transcription factor E2F are also present.
Transcriptional start site
The transcriptional start site was determined by cloning products of 5′-RACE using cDNA from HepG2 cells, followed by sequencing. The primer corresponding to −10 to +13 of the human BHMT cDNA was used and yielded a product beginning 80 nt upstream of the translational start site (Figure 1, indicated by forward arrow).
Effect of SAM and MTA on BHMT expression
We next examined the effect of SAM and MTA on BHMT expression in HepG2 cells using Northern-blot analysis (Figure 2A) and real-time RT–PCR (Figures 2B and 2C). Both agents exerted a dose-dependent down-regulation of BHMT mRNA levels, with the maximum inhibitory dose of SAM at 5 mM (50% fall) and MTA at 1 mM (84% fall) at the 12 h time point (Figure 2B). At these doses, no apoptosis was evident (results not shown). However, treatment with 5 mM MTA for 12 h induced apoptosis (results not shown). Up to 12 h, the effect of MTA and SAM (results not shown) treatment on BHMT mRNA levels was time-dependent (Figure 2C). All subsequent studies were done using non-toxic regimens, namely SAM (5 mM) or MTA (1 mM) for 12 h.
Figure 2. Effect of SAM and MTA treatment on BHMT expression in HepG2 cells.

(A) RNA (20 μg/lane) samples from HepG2 cells treated with SAM (0–5 mM) or MTA (0–5 mM) for 12 h were analysed by Northern-blot analysis with a 32P-labelled BHMT cDNA probe as described in the Materials and methods section. The same membranes were then rehybridized with a 32P-labelled β-actin cDNA probe. Representative Northern blots are shown. (B) Effect of SAM (5 mM) and MTA (1 mM) treatment for 12 h on BHMT expression in HepG2 cells was examined by real-time RT–PCR as described in the Materials and methods section. Results represent means±S.E.M. from four experiments. *P<0.05, †P<0.001 versus control. (C) Time course of MTA's inhibitory effect on BHMT expression in HepG2 cells was examined using real-time RT–PCR as described in the Materials and methods section. HepG2 cells were treated with MTA (1 mM) for 0–12 h. *P<0.05 versus zero time.
Effect of SAM and MTA on BHMT promoter activity
To delineate sequences that drive the expression of the human BHMT gene, deletion mutants ranging from −2698 to +33 were cloned into the promoterless luciferase reporter gene vector pGL3-basic. The promoterless construct pGL3-basic served as the background control. Luciferase activity was measured after transient transfection of HepG2 cells with these constructs. Figure 3 shows that the human BHMT promoter was able to efficiently drive luciferase expression in HepG2 cells. Construct −347/+33 has maximal promoter activity. Treatment of transfected HepG2 cells with SAM (5 mM) or MTA (1 mM) during the last 12 h of the transfection led to a significant inhibition in promoter activity, which was most pronounced with the construct −347/+33 and closely paralleled the effect of these agents on endogenous BHMT gene expression (Figure 2B).
Figure 3. Effect of SAM or MTA treatment on luciferase activity driven by the human BHMT promoter.

Progressive 5′ deletions of the BHMT promoter extending from −2698 to +33 bp were generated and fused to the promoterless luciferase pGL-3-basic vector as described in the Materials and methods section. HepG2 cells were transfected transiently with these promoter constructs or pGL-3-basic vector alone and treated with SAM (5 mM for 12 h) or MTA (1 mM for 12 h) or vehicle control. Cells were co-transfected with Renilla luciferase for control of transfection efficiency. Results represent means±S.E.M. from four independent experiments performed in triplicate. Data are expressed as relative luciferase activity to that of pGL-3-basic vector control, which is assigned a value of 1.0. *P<0.05, †P<0.005 versus respective control.
To see if the effect of MTA may be mediated by its conversion into SAM, cells were pretreated with cycloleucine (20 mM for 2 h) prior to treatment with MTA or SAM. Indeed, cycloleucine treatment increased the reporter activity driven by the BHMT promoter construct −347/+33 but did not block the ability of MTA to inhibit the BHMT promoter activity (see supplementary data at http://www.BiochemJ.org/bj/401/bj4010087add.htm.). The effectiveness of cycloleucine treatment is demonstrated by measuring cellular SAM levels. Cycloleucine lowered SAM levels since it is an inhibitor of methionine adenosyltransferase; while MTA treatment raised SAM levels since it can be converted back to SAM [16]. Cycloleucine treatment blocked the MTA-mediated increase in cell SAM levels, which is consistent with its ability to block the conversion of MTA into SAM (see supplementary data).
SAM and MTA treatment induces NF-κB nuclear binding in HepG2 cells
The −347/+33 region of the human BHMT gene contains three NF-κB binding sites (Figure 1). To see if SAM and MTA treatment affects NF-κB nuclear binding, we first examined this using a consensus NF-κB probe. Indeed, treatment with these agents led to a significant increase in NF-κB nuclear binding (Figure 4A). SAM and MTA activated NF-κB in a time-dependent fashion, with appearance of p65 and p50 in the nuclear compartment by 2 h after treatment (Figure 4B). We next examined whether binding to each of the three NF-κB sites (at −301, −271 and −135) of the BHMT gene is affected by SAM or MTA treatment. Figure 4(C) shows that SAM and MTA treatment led to an increase in NF-κB binding to −135 and −301 sites, while binding to −271 site is minimally affected (results not shown). Overexpression with p50 led to increased p65/p50 and p50/p50 nuclear binding (Figure 4C), and overexpression with p65 also led to increased p65/p50 nuclear binding (results not shown). Increased NF-κB nuclear binding occurred following MTA or SAM treatment not only with recombinant probes for NF-κB (EMSA), but also in the endogenous chromatin configuration as demonstrated using the ChIP assay (Figure 5). Furthermore, both SAM and MTA also induced increased nuclear binding of HDAC1, HDAC2 and HDAC3 to the region spanning the NF-κB sites (Figures 5A and 5B).
Figure 4. Effect of SAM and MTA treatment on NF-κB nuclear binding activity in HepG2 cells.

HepG2 cells were treated with varying doses of SAM or MTA for 12 h and EMSA was performed using a consensus NF-κB probe as described in the Materials and methods section (A). In (B), time course of NF-κB activation by SAM (5 mM) was assessed by performing Western-blot analysis measuring nuclear p65 and p50 content and cytoplasmic actin for loading control. Numbers below refer to densitometric changes as % of baseline. In (C), EMSA was performed using the NF-κB probes that correspond to the two putative NF-κB binding sites following treatment with SAM (5 mM for 12 h), MTA (1 mM for 12 h), expression vector for p50 (24 h), or empty vector for 24 h. Note that SAM and MTA increased NF-κB nuclear binding to a concensus NF-κB probe (A) and they also increased NF-κB nuclear binding (both p65/p50 and p50/p50) to NF-κB sites at −135 and −301 (probes spanning −142 to −114 and −310 to −285 respectively). Overexpression with p50 also increased both p65/p50 and p50/p50 nuclear binding. Arrows point to supershifted bands.
Figure 5. Effect of SAM or MTA treatment on protein binding to the human BHMT NF-κB sites in vivo.

HepG2 cells were treated with SAM (5 mM) or MTA (1 mM) for 12 h and the ChIP assay was used to assess nuclear binding of p50, p65 and HDAC3 (A), or HDAC1 and HDAC2 (B) to the NF-κB sites within the −313 to +2 region of the human BHMT in an endogenous chromatin configuration as described in the Materials and methods section. PCR products from amplification of the NF-κB sites following immunoprecipitation with antisera against p50 or p65 demonstrate that SAM and MTA treatment increased p50 and p65 binding to the human BHMT NF-κB sites (A). In addition, SAM and MTA also increased HDAC1, HDAC2 and HDAC3 binding to the SAM region (A, B). Input genomic DNA (gDNA input) was used as a positive control and a no antibody immunoprecipitation (no Ab) was used as a negative control.
Role of NF-κB in SAM- and MTA-mediated inhibition of BHMT expression in HepG2 cells
To see if increased NF-κB nuclear binding translated to a functional change, we determined the effect of SAM and MTA on NF-κB-driven reporter activity. Figure 6(A) shows that treatment of HepG2 cells with either SAM or MTA nearly doubled the NF-κB-driven reporter activity. To demonstrate that increased NF-κB nuclear binding can impact on BHMT promoter activity, we determined the effect of overexpression of p50 or p65 on BHMT promoter activity. Figure 6(B) shows that overexpression of either p50 or p65 lowered the BHMT promoter activity by nearly 50%.
Figure 6. Effect of SAM and MTA on NF-κB-driven reporter activity (A) and the effect of NF-κB overexpression on BHMT promoter activity (B).

In (A), HepG2 cells were transfected with NF-κB-driven reporter construct and treated with SAM (5 mM) or MTA (1 mM) for 12 h. Results represent means±S.E.M. from four to six experiments done in triplicates. *P<0.005 versus control NF-κB-LUC. In (B), HepG2 cells were co-transfected with BHMT promoter construct −347/+33 and either p50, p65 expression vectors or empty vector control. Results represent means±S.E.M. from three independent experiments done in triplicates. *P<0.05 versus −347/+33-LUC+empty vector control.
To further examine the role of NF-κB in regulating human BHMT gene expression, HepG2 cells were infected with adenoviruses carrying IκBSR to block NF-κB activity. Infection with IκBSR reduced the NF-κB-driven luciferase activity to 4±0.4% (mean±S.E.M. from two experiments performed in duplicate) and led to an increase in BHMT promoter activity by 215±51% (mean±S.E.M. from four experiments, P<0.05 versus control BHMT promoter activity). Blocking NF-κB activation led to a slight increase in endogenous BHMT mRNA levels, prevented the ability of SAM to lower BHMT expression and blunted the inhibitory effect of MTA (Table 1).
Table 1. Effect of blocking NF-κB activation on baseline and SAM-/MTA-mediated inhibition of BHMT expression.
BHMT mRNA levels were determined using real-time PCR after HepG2 cells were infected with adenoviruses carrying empty vector or IκBSR for 12 h and treated with SAM (5 mM) or MTA (1 mM) for another 12 h. Results represent means±S.E.M. from four to eight experiments. *P<0.005 versus control, **P<0.05 versus adenoviral vector control, ***P<0.05 versus SAM or MTA.
| Treatment | BHMT mRNA level (% of control) |
|---|---|
| Control | 100±0 |
| Adenoviral vector control | 98±6 |
| SAM | 61±6* |
| MTA | 30±2* |
| IκBSR | 120±7** |
| +SAM | 134±16*** |
| +MTA | 70±6*** |
To conclusively demonstrate the importance of the two NF-κB sites present in the −347/+33 region of the human BHMT (at −301 and −135), we performed site-directed mutagenesis of these sites and determined the effect of these mutations and the effect of SAM and MTA treatment on BHMT promoter activity. Table 2 shows that, when the NF-κB site at −301 was mutated, the BHMT promoter activity increased, SAM no longer inhibited but MTA was still able to inhibit promoter activity. However, the effect of MTA is slightly blunted, from 63% inhibition with wild-type or the Mut–135 construct to 50% inhibition with the Mut–301 construct.
Table 2. Effect of mutating NF-κB binding sites at −301 and −135 on basal BHMT promoter activity and SAM- and MTA-mediated inhibition.
NF-κB binding sites were mutated at −135 site alone, −301 site alone, or both (double mutant) as described in the Materials and methods section. HepG2 cells were transfected with wild-type or mutant BHMT promoter constructs −347/+33 and treated with SAM (5 mM), MTA (1 mM) or vehicle control for 12 h. Results represent means±S.E.M. from four to eight experiments performed in triplicate. *P<0.05 versus respective controls, **P<0.05 versus −347/+33-BHMT-LUC.
| Sample | Luciferase activity (fold that of pGL-3 control) |
|---|---|
| −347/+33-BHMT-LUC | 33.2±1.9 |
| +SAM | 22.2±2.4* |
| +MTA | 12.9±2.7* |
| Mutation at −135 | 33.2±2.3 |
| +SAM | 23.9±4.1* |
| +MTA | 12.3±2.9* |
| Mutation at −301 | 40.0±1.4** |
| +SAM | 39.2±4.9 |
| +MTA | 20.2±3.9* |
| Double mutant | 38.4±1.0** |
| +SAM | 37.0±3.5 |
| +MTA | 19.2±3.3* |
Effect of SAM and MTA on ER stress markers
Homocysteine has been linked to increased ER stress [21]. To see if inhibition of BHMT expression by SAM and MTA can lead to increased ER stress, the expression of two ER stress markers GRP78 and GADD153 was examined. MTA treatment increased GRP78 and GADD153 mRNA levels to more than 350 and 500% of control levels respectively (GRP78 was increased to 362±63% and GADD153 was increased to 524±30% of control values; results represent means±S.E.M. from three to four experiments, P<0.005). However, SAM treatment did not increase these ER stress markers (results not shown).
DISCUSSION
Homocysteine is a risk factor for atherosclerosis [12] and an inducer of ER stress that may participate in the pathogenesis of alcoholic liver disease [13]. In the liver, homocysteine lies at the junction of two intersecting pathways, the transsulfuration pathway (the pathway that converts the sulfur atom of methionine into cysteine and glutathione), and the remethylation pathway (the pathway that conserves homocysteine as methionine), which is coupled with cobalamin, folate and betaine metabolism [22]. In the liver and kidney, BHMT is responsible, along with methionine synthase, for remethylation of homocysteine [1]. Hyperhomocysteinaemia commonly occurs in liver cirrhosis due to impaired homocysteine metabolism [10]. Betaine, a cofactor for BHMT, has been shown to prevent the hyperhomocysteinaemia and liver injury in rodent models of alcoholic liver disease [13,23].
Previous studies in the rat have shown that hepatic BHMT expression is induced by a methionine-deficient diet [1]. However, the molecular mechanism(s) are unknown. SAM has been shown to inactivate BHMT [7]; however, whether it influences BHMT at the mRNA level has not been studied. In our current study, we examined the influence of SAM and its metabolite, MTA, on BHMT gene expression. The rationale for studying MTA is that MTA is a product of SAM metabolism in the polyamine pathway [7] as well as non-enzymatic hydrolysis [24]. We have shown that SAM's effect on cell growth, apoptosis and TNFα (tumour necrosis factor α) expression can be mimicked by MTA [16,25,26]. In contrast with SAM, MTA does not contribute to GSH synthesis, is not a methyl donor and inhibits methyltransferases [27]. Thus additional insights can be gained by comparing the effect of these two agents.
We began these studies by cloning the 2.7 kb 5′-flanking region of the human BHMT. One transcriptional start site was identified 26 nt downstream from a putative TATA box and 80 nt upstream of the translational start site. These results are in agreement with the results published by Park and Garrow [1]. In the work of Park and Garrow [1], the human BHMT promoter was not functionally characterized. The sequence of the BHMT 5′-flanking region contains numerous NF-κB binding sites as well as HNF binding sites, which may play a role in conferring high expression in the liver. Transfection studies showed that maximal promoter activity was obtained with the BHMT promoter construct −347/+33-LUC, denoting the presence of enhancer element(s) in this region. Additional enhancer element(s) are likely present between −1920 and −1471. In contrast, the presence of repressor element(s) is suggested between −1471 and −347, as well as −2698 and −2386 as the promoter activity fell significantly.
Treatment of HepG2 cells with SAM and MTA led to a dose- and time-dependent decrease in BHMT mRNA levels. MTA is a more potent inhibitor than SAM, which is reflected by both dose–response curves as well as the magnitude of inhibition. We had also observed this in our other studies [16,25], and it raises the possibility that the effect of SAM may be mediated at least in part by MTA. The effect of these agents on the endogenous BHMT gene expression is comparable with their effects on the recombinant BHMT promoter activity. The maximum inhibitory effect occurred with the shortest promoter construct −347/+33.
Since MTA can be converted back to SAM via the methionine salvage pathway [16], we examined whether blocking this conversion by using cycloleucine (an inhibitor of methionine adenosyltransferase) would have any effect. Cycloleucine treatment lowered cellular SAM levels and increased BHMT promoter activity. SAM and MTA treatment raised cellular SAM levels by 268 and 160% respectively. Cycloleucine pretreatment completely blocked the effect of MTA on cellular SAM levels but had no influence on MTA's ability to inhibit BHMT promoter activity. These results are consistent with the following: (i) MTA's effect is not mediated via SAM, and (ii) there is no direct correlation between cellular SAM levels and BHMT expression. While we cannot exclude that cellular SAM levels may influence BHMT expression, our results suggest that the inhibitory effects of SAM and MTA occurred by some other mechanism(s). Furthermore, BHMT expression falls in human and rat liver cirrhosis [9,11], where SAM biosynthesis is inhibited [6]. Thus it is unlikely that the intracellular SAM level is the main regulatory control for BHMT expression.
Struck by the presence of multiple NF-κB binding sites in the −347/+33 region, we examined whether SAM and MTA treatment may influence NF-κB nuclear binding activity. Indeed, treatment with either of these agents led to an increase in NF-κB nuclear binding to two of the three NF-κB sites present within this region of the BHMT gene. Increased NF-κB nuclear binding to this region of the BHMT gene in its endogenous chromatin configuration was also demonstrated using the ChIP assay. Increased nuclear binding translated to a functional change as SAM and MTA treatment increased the NF-κB-driven reporter assay in HepG2 cells.
Overexpression of NF-κB inhibited BHMT promoter activity, whereas blocking NF-κB activation increased BHMT expression and promoter activity, supporting the notion that NF-κB acts as a repressor for this gene. Blocking NF-κB activation completely prevented the ability of SAM to inhibit BHMT expression, but it only blunted MTA's inhibitory effect. These results suggest that SAM exerts its effect on BHMT expression via NF-κB but MTA exerts its effect on BHMT expression by both NF-κB-dependent and -independent mechanisms. Finally, mutagenesis experiments suggest that SAM exerts its inhibitory effect via transactivation of the NF-κB site at −301, which is functioning as a repressor. However, we cannot rule out participation of other NF-κB sites in the regulation of BHMT. Much more work will be required to elucidate the NF-κB-independent mechanisms of BHMT transcriptional regulation.
While NF-κB typically acts as an enhancer to transactivate promoters, it has also been shown to act as a repressor [28,29]. In this regard, several co-repressor complexes for NF-κB have been described, including SMRT (silencing mediator for retinoic acid receptor and thyroid hormone receptor), NcoR (nuclear receptor co-repressor), HDAC1, HDAC2, HDAC3 and HDAC6 [28,30]. Interestingly, both SAM and MTA also induced recruitment of HDAC1, HDAC2 and HDAC3 to the BHMT promoter region containing the NF-κB binding sites. HDAC1 has been shown to physically interact with NF-κB and HDAC2 is known to associate with HDAC1 [29]. Presumably these HDAC proteins regulate the activity of NF-κB through their deacetylase activity since their effect can be blocked by HDAC inhibitors [28]. The mechanisms regulating the interaction between NF-κB and HDAC proteins are largely unknown and results from the present study suggest that SAM and MTA may enhance this interaction. This possibility will require further examination. Even though SAM and MTA may inhibit BHMT expression by recruiting HDACs to the BHMT promoter, the fact that overexpression of p50 and p65 can inhibit BHMT promoter activity would suggest that participation of these co-repressors may not be necessary to inhibit BHMT expression. The ability of NF-κB to repress BHMT gene expression may help to explain the marked down-regulation of BHMT expression in human hepatocellular carcinoma [11], where we have described increased NF-κB nuclear binding activity [31]. It may also help to explain decreased BHMT expression in liver cirrhosis [11], where inducers of NF-κB are often up-regulated (i.e. oxidative stress).
Since impaired homocysteine metabolism can lead to ER stress and apoptosis [13], we also examined whether treatment with SAM or MTA can induce ER stress in HepG2 cells. Consistent with this notion, MTA treatment more than tripled two ER stress markers. However, SAM treatment did not induce the SAM markers. This may be due to the fact that SAM's inhibitory effect on BHMT gene expression is only 40–50% at best, while MTA is much more potent in its inhibitory effect. This also suggests that blocking homocysteine metabolism via BHMT and inducing ER stress may be another mechanism for these agents (at least in the case of MTA) to exert a pro-apoptotic effect on liver cancer cells [16].
Castro et al. [32] have published in a meeting proceeding characterization of the human BHMT promoter and demonstration of SAM's ability to inhibit BHMT promoter activity. Our results of the basal human BHMT promoter activity and SAM's ability to down-regulate BHMT promoter are in full agreement with this work. These investigators found that methionine and ethionine did not affect BHMT promoter activity and had no influence on SAM's inhibitory effect, suggesting a direct effect of SAM on BHMT promoter. However, the molecular mechanism of SAM's inhibitory effect was not pursued in their work. Our current study extends these observations, shows an even more pronounced inhibition of BHMT expression by a SAM metabolite, MTA, and elucidates the molecular mechanism of SAM's inhibitory effect.
It should be stressed that our work used pharmacological concentrations of SAM. The plasma concentration of SAM is extremely low (∼0.05 μM) [33] and can increase up to 1 mg/l or 2.5 μM following oral administration of 1000 mg of SAM [34]. However, liver SAM levels can reach 0.5 mM following a methionine load [35]. Thus our data are likely relevant for the liver, particularly after a methionine load or for patients taking large doses of SAM. Nevertheless, the key finding of the present paper is that using SAM and MTA at pharmacological doses can inhibit BHMT expression by activating NF-κB, which acts as a repressor for this gene. While SAM's inhibitory effect is NF-κB-dependent, MTA exerts its inhibitory effect by both NF-κB-dependent and -independent mechanisms. Inhibiting BHMT expression can lead to increased ER stress and contribute to the pro-apoptotic effects of these agents on liver cancer cells.
Online data
Acknowledgments
This work was supported by NIH (National Institute of Health) grants DK51719 (S.C.L.), AA12677, AA13847 and AT1576 (S.C.L. and J.M.M.) and Plan Nacional of I+D SAF2002-00168 of the Ministerio de Educación y Ciencia (to J.M.M.). HepG2 cells were provided by the Cell Culture Core of the USC Research Center for Liver Diseases (DK48522). K.R. is a recipient of the Postdoctoral Fellowship of the Training Program in Alcoholic Liver and Pancreatic Diseases (T32 AA07578), and A.I.A. is a recipient of the Postdoctoral Fellowship supported by the CIC bioGUNE, Centro de Investigación Cooperativa en Biosciencías.
References
- 1.Park E. I., Garrow T. A. Interaction between dietary methionine and methyl donor intake on rat liver betaine–homocysteine methyltransferase gene expression and organization of the human gene. J. Biol. Chem. 1999;274:7816–7824. doi: 10.1074/jbc.274.12.7816. [DOI] [PubMed] [Google Scholar]
- 2.Storch K. J., Wagner D. A., Young V. R. Methionine kinetics in adult men: effects of dietary betaine on L-[2H3-methyl-1-13C]methionine. Am. J. Clin. Nutr. 1991;54:386–394. doi: 10.1093/ajcn/54.2.386. [DOI] [PubMed] [Google Scholar]
- 3.Lu S. C., Alvarez L., Huang Z. Z., Chen L., An W., Corrales F. J., Avila M. A., Kanel G., Mato J. M. Methionine adenosyltransferase 1 A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barak A. J., Beckenjauer H. C., Tuma D. J. Betaine, ethanol, and the liver: a review. Alcohol. 1996;13:395–398. doi: 10.1016/0741-8329(96)00030-4. [DOI] [PubMed] [Google Scholar]
- 5.Lu S. C., Huang Z. Z., Yang H. P., Mato J. M., Avila M. A., Tsukamoto H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in the alcoholic rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G178–G185. doi: 10.1152/ajpgi.2000.279.1.G178. [DOI] [PubMed] [Google Scholar]
- 6.Mato J. M., Corrales F. J., Lu S. C., Avila M. A. S-adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 7.Finkelstein J. D., Martin J. J. Methionine metabolism in mammals. J. Biol. Chem. 1986;261:1582–1587. [PubMed] [Google Scholar]
- 8.Bose N., Greenspan P., Momany C. Expression of recombinant human betaine: homocysteine S-methyltransferase for x-ray crystallographic studies and further characterization of interaction with S-adenosylmethionine. Protein Expr. Purif. 2002;25:73–80. doi: 10.1006/prep.2001.1611. [DOI] [PubMed] [Google Scholar]
- 9.Forestier M., Bänninger R., Reichen J., Solioz M. Betaine homocysteinemethyltransferase: gene cloning and expression analysis in rat liver cirrhosis. Biochim. Biophys. Acta. 2003;1638:29–34. doi: 10.1016/s0925-4439(03)00037-1. [DOI] [PubMed] [Google Scholar]
- 10.Hultberg B., Berglund M., Andersson A., Frank A. Elevated plasma homocysteine in alcoholics. Alcohol. Clin. Exp. Res. 1993;17:687–689. doi: 10.1111/j.1530-0277.1993.tb00820.x. [DOI] [PubMed] [Google Scholar]
- 11.Avila M. A., Berasain C., Torres L., Martin-Duce A., Corrales F. J., Yang H., Prieto J., Lu S. C., Caballeria J., Rodes J., et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J. Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 12.Refsum H., Ueland P. M., Nygard O., Vollset S. E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 13.Ji C., Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 14.Yang H. P., Magilnick N., Ou X. P., Lu S. C. Tumor necrosis factor α induces coordinated activation of rat GSH synthetic enzymes via NF-κB and AP-1. Biochem. J. 2005;391:399–408. doi: 10.1042/BJ20050795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrow T. A. Purification, kinetic properties, and cDNA cloning of mammalian betaine–homocysteine methyltransferase. J. Biol. Chem. 1996;271:22831–22838. doi: 10.1074/jbc.271.37.22831. [DOI] [PubMed] [Google Scholar]
- 16.Yang H. P., Sadda M. R., Li M., Zeng Y., Chen L. X., Bae W. J., Ou X. P., Runnegar M. T., Mato J. M., Lu S. C. S-adenosylmethionine and its metabolite induce apoptosis in HepG2 cells: role of protein phosphatase 1 and Bcl-xS. Hepatology. 2004;40:221–231. doi: 10.1002/hep.20274. [DOI] [PubMed] [Google Scholar]
- 17.Yang H. P., Huang Z. Z., Wang J. H., Lu S. C. The role of c-Myb and Sp1 in the up-regulation of methionine adenosyltransferase 2A gene expression in human hepatocellular carcinoma. FASEB J. 2001;15:1507–1516. doi: 10.1096/fj.01-0040com. [DOI] [PubMed] [Google Scholar]
- 18.Vandesomeple J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT–PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:34.1–34.11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giulietti A., Overbergh L., Valckx D., Decallonne B., Bouillon R., Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- 20.Nasuhara Y., Adcock I. M., Catley M., Barnes P. J., Newton R. Differential IκB kinase activation and IκBα degradation by interleukin-1B and tumor necrosis factor-α in human U937 monocytic cells. J. Biol. Chem. 1999;274:19965–19972. doi: 10.1074/jbc.274.28.19965. [DOI] [PubMed] [Google Scholar]
- 21.Ji C., Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J. Gastrol. 2004;10:1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finkelstein J. D. Pathways and regulation of homocysteine metabolism in mammals. Semin. Thromb. Hemost. 2000;26:219–225. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 23.Barak A. J., Beckenjauer H. C., Junnilla M., Tuma D. J. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol. Clin. Exp. Res. 1993;17:552–555. doi: 10.1111/j.1530-0277.1993.tb00798.x. [DOI] [PubMed] [Google Scholar]
- 24.Wu S. E., Huskey W. P., Borchardt R. T., Schowen R. E. L. Chiral instability at sulfur of S-adenosylmethionine. Biochemistry. 1983;22:2828–2832. doi: 10.1021/bi00281a009. [DOI] [PubMed] [Google Scholar]
- 25.Latasa M. U., Boukaba A., Garcìa-Trevijano E. R., Torres L., Rodríguez J., Caballería L., Lu S. C., López-Rodas G., Franco L., Mato J. M., Avila M. A. Hepatocyte growth factor induces MAT2A expression and histone acetylation in rat hepatocytes. Role in liver regeneration. FASEB J. 2001;15:1248–1250. doi: 10.1096/fj.00-0556fjev1. [DOI] [PubMed] [Google Scholar]
- 26.Veal N., Hsieh C. L., Xiong S., Lu S. C., Tsukamoto H. Inhibition of lipopolysaccharide-stimulated TNFα promoter activity by S-adenosylmethionine and 5′-methylthioadenosine. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G352–G362. doi: 10.1152/ajpgi.00316.2003. [DOI] [PubMed] [Google Scholar]
- 27.Dante R., Anaud M., Niveleau A. Effects of 5′deoxy-5′-methylthioadenosine on the metabolism of S-adenosylmethionine. Biochem. Biophys. Res. Commun. 1983;114:214–221. doi: 10.1016/0006-291x(83)91615-7. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W., Kone B. C. NF-κB inhibits transcription of the H+-K+-ATPase α2-subunit gene: role of histone deacetylases. Am. J. Physiol. Renal Physiol. 2002;283:F904–F911. doi: 10.1152/ajprenal.00156.2002. [DOI] [PubMed] [Google Scholar]
- 29.Ashburner B. P., Westerheide S. D., Baldwin A. S., Jr The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Z., Chiao P., Zhang X., Zhang X. H., Lazar M. A., Seto E., Young H. A., Ye J. Coactivators and corepressors of NF-κB in IκBα gene promoter. J. Biol. Chem. 2005;280:21091–21098. doi: 10.1074/jbc.M500754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Z. Z., Chen C. J., Zeng Z. H., Yang HP., Oh J., Chen L. X., Lu S. C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001;15:19–21. doi: 10.1096/fj.00-0445fje. [DOI] [PubMed] [Google Scholar]
- 32.Castro C. C., Breksa A. P., III, Salisbury E. M., Garrow T. A. Betaine–homocysteine S-methyltransferase transcription is inhibited by S-adenosylmethionine. In: Milstien S., Kapatos G., Levine R., Shane B., editors. Chemistry and Biology of Pteridines and Folates: Proceedings of the 12th International Symposium on Pteridines and Folates. Norwell, MA: Kluwer Publishers; 2001. pp. 1–8. [Google Scholar]
- 33.Stramentinoli G. Pharmacologic aspects of S-adenosylmethionine. Am. J. Med. 1987;83(Suppl. 5A):35–42. doi: 10.1016/0002-9343(87)90849-7. [DOI] [PubMed] [Google Scholar]
- 34.Friedel H. A., Goa K. L., Benfield P. S-adenosyl-L-methionine. A review of its pharmacological properties and therapeutic potential in liver dysfunction and affective disorders in relation to its physiological role in cell metabolism. Drugs. 1989;38:389–416. doi: 10.2165/00003495-198938030-00004. [DOI] [PubMed] [Google Scholar]
- 35.Finkelstein J. D., Kyle W. E., Harris B. J., Martin J. J. Methionine metabolism in mammals: concentration of metabolites in rat tissues. J. Nutr. 1982;112:1011–1018. doi: 10.1093/jn/112.5.1011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.