


Abstract
Accurate detection of protein families allows assignment of protein function and the analysis of functional diversity in complete genomes. Recently, we presented a novel algorithm called TribeMCL for the detection of protein families that is both accurate and efficient. This method allows family analysis to be carried out on a very large scale. Using TribeMCL, we have generated a resource called Tribes that contains protein family information, comprising annotations, protein sequence alignments and phylogenetic distributions describing 311 257 proteins from 83 completely sequenced genomes. The analysis of at least 60 934 detected protein families reveals that, with the essential families excluded, paralogy levels are similar between prokaryotes, irrespective of genome size. The number of essential families is estimated to be between 366 and 426. We also show that the currently known space of protein families is scale free and discuss the implications of this distribution. In addition, we show that smaller families are often formed by shorter proteins and discuss the reasons for this intriguing pattern. Finally, we analyse the functional diversity of protein families in entire genome sequences. The Tribes protein family resource is accessible at http://www.ebi.ac.uk/research/cgg/tribes/.
INTRODUCTION
The advent of complete genome sequencing has generated an enormous amount of data for computational and molecular biologists. The rapidly increasing quantity of protein sequences, predicted from entire genomes, has necessitated the development of computational genomics techniques, including the assignment of accurate and descriptive functional annotations to these predicted protein sequences (1). One such approach involves the detection of ‘protein families’ from complete genomes (2).
Protein families can be defined as those groups of molecules that share significant sequence similarity and a common evolutionary history (3). It is well known that proteins within protein families preserve their molecular structure and thus can maintain similar or even identical biochemical functions across vast evolutionary distances (4). For this reason, accurate detection of families assists the functional annotation of protein sequences. These annotations might be based on family information and not on individual sequence similarities. This approach is advantageous as annotation of sequences based on individual pair-wise sequence similarities can be rather limiting (5). This approach is dependent both on the quality of the similarity search method and the annotation quality of any similar sequences (6). Protein families are detected on the basis of multiple sequence similarities and are hence less prone to errors originating from individual false assignments. Furthermore, the function of a family can be obtained by a consensus annotation from all family members whose functions may be characterised, and not on individual annotations.
Protein families are generated using a technique called ‘sequence clustering’. Sequence clustering involves the detection of all pair-wise sequence similarities within a given set of protein sequences (7). Proteins are then assigned into clusters (families) based on their sharing of significant sequence similarity patterns. When sequence clustering is performed accurately, proteins within a family may be considered as sharing a common evolutionary history and possibly similar or identical functions (8).
Many excellent methods exist aiming for accurate protein clustering (9–15). However, the rapid and expanding growth of protein sequence data also requires that such methods be both automatic and rapid. These methods must be able to work effectively with the many hundreds of thousands of sequences already available, and the millions of sequences that should be available within the next few years.
Recently, we presented a novel method called TribeMCL for the rapid and accurate detection of protein families from complete genome sequences (8). This method uses a Markov cluster (MCL) algorithm (16) that overcomes many of the problems presented by protein domains, fragment peptides and sequence similarity errors. The method has been rigorously tested and validated on a number of databases, including SwissProt, InterPro, SCOP and the draft human genome, and has been shown to be both robust and reliable, with an accuracy of at least 87% (8). One clear advantage of the TribeMCL method is its speed. Using TribeMCL, it is possible to cluster hundreds of thousands of sequences in a few hours, rather than days or weeks. The method has already been used for protein family analysis in the draft human genome (17).
To this end, we have produced an exhaustive set of protein family assignments for 83 complete genomes using the TribeMCL method. In addition to these 83 genomes, we also add protein sequences from the SwissProt database (18) (see Materials and Methods). These entries are added in order to aid the annotation of families, as functional annotations for SwissProt are generally more reliable than those obtained from complete genome sequences. Family assignments are stored in a publicly available database called Tribes. This database provides a convenient method to analyse protein function for individual protein families and also to analyse the distribution of protein families across genomes. The fully automatic and rapid nature of this system allows us to keep this database up to date and consistent, through the constant addition of complete genome sequences as they become available.
The Tribes resource is a unique snapshot of protein sequence space in complete genomes and hence allows many interesting questions to be addressed. We have used Tribes to analyse protein function and evolution in complete genomes (19), as well as the history of protein family discovery (20). Herein, we describe the properties of protein family space, including the phylogenetic and functional distribution of protein families and the relationship between numbers of genes and families in entire genomes. We also investigate the distribution of family sizes in the database and the effect of sequence length and sequence similarity on the detection of protein families. To our knowledge this is the first time that an analysis of this kind has been carried out on such a large scale across entire genome sequences.
MATERIALS AND METHODS
Predicted protein sequences from completely sequenced genomes were obtained from the COGENT database (21). This database stores protein sequences and publication ranking order for all completely sequenced genome sequences. Sequences in COGENT were obtained from their original sequencing centres (where possible). A total of 311 257 sequences were extracted from the COGENT database (release 83) and combined with a further 108 158 protein sequences from the SwissProt database.
All sequences were then compared against each other using BLASTp 2.0 with an E-value threshold of 1 × 10–10. Query sequences were filtered for low complexity regions using the CAST algorithm (22). This analysis was performed in parallel on a 400 node Compaq Alpha DS10 cluster, and took ∼8 h to complete. The results from this analysis (over 30 million similarities) were loaded into a similarity table in the Tribes MySQL database. This similarity table was then processed to correct asymmetric BLAST hits and scores. The resulting symmetric similarity table was converted into a Markov matrix and clustered into protein families using the TribeMCL algorithm (8). This process took ∼6 h to complete.
For each detected protein family, annotations are obtained from the COGENT MySQL database, and a consensus annotation for that family is generated from all members using the recursive longest common substring algorithm RLCS (23). It should be stressed, however, that the consensus annotation may not be a reliable way of identifying the functions of the individual members of a family, because of the variability of annotations for different genomes. Instead, Tribes provides the family information that can be further studied by other means, such as multiple sequence alignments and dendrograms.
Finally, all families are loaded into the core Tribes family database table, which can be queried at http://www.ebi.ac.uk/research/cgg/tribes/ via a simple user interface or, alternatively, using SQL statements.
RESULTS AND DISCUSSION
Basic statistics
We have used the TribeMCL algorithm to cluster 311 257 proteins from 83 publicly available genomes and the SwissProt database into protein families. The SwissProt database was used in order to allow higher quality consensus annotation of families that contain SwissProt members. The TribeMCL algorithm allows clustering to be performed at multiple granularities by altering the inflation parameter (8). This feature allows us to narrow (or broaden) our definition of protein families and is reflected by increasing (or decreasing) sequence similarity levels within these families. Multiple inflation values were used for this clustering (1.1, 2.0 and 3.0). With inflation value 3 (the narrowest or tightest clustering), 82 692 families were obtained. Other broader inflation values produced similar results, resulting in 75 635 and 60 934 families for inflation values of 2.0 and 1.1, respectively.
Distribution of families across domains of life
Very few detected families are universally present in all three domains of life, illustrating the functional diversity of the different species, reflected in their genome content. Only 756 (1.2%) of all 60 934 (at inflation value 1.1) families have members from the Archaea, Bacteria and Eukarya, compared to 622 COGs (12). Bacterial diversity appears to be highest, with 48% of all families being bacterial specific (Fig. 1), although this figure is almost certainly skewed by over-representation of Bacteria in the 83 genomes used. Eukaryotic genomes are the second highest at 34%, while only 13% of families are archaeal specific. Interestingly, Archaea share 10 times more families with Bacteria (2%) than with Eukarya (0.2%). This is consistent with previous reports of greater similarity between Archaea and Bacteria rather then Eukarya (24). Finally, a total of 48 families appear to be present in all 83 genomes (at inflation value 1.1). These highly conserved, universal protein families represent important protein families involved in metabolism, transcription and translation.
Figure 1.
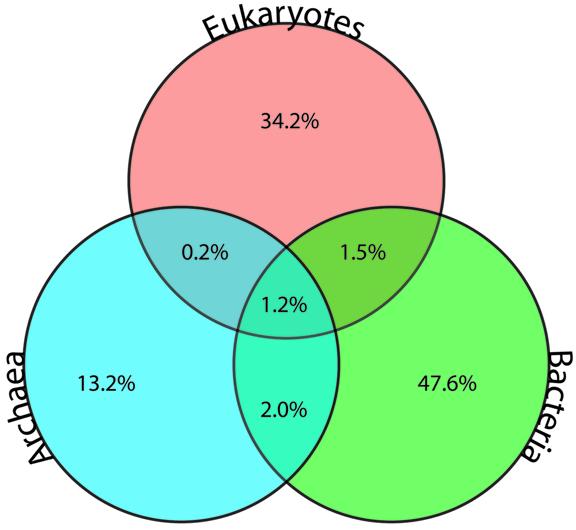
Phylogenetic distribution of protein families in the Tribes database. The numbers show relative abundance of protein families unique to each domain as well as shared ones across the three domains of life.
Relationship between genome size and number of families
As expected, the number of families in each genome strongly correlates with the number of genes (Fig. 2). This is especially true for both Bacteria and Archaea and, when Eukarya are excluded, this relationship is linear. Eukarya, with larger genomes, deviate strongly from the linear dependency and are discussed further below.
Figure 2.

Correspondence between number of genes and number of Tribes families in available genomes. Colours show correspodence to the domains of life, and eukaryotic genomes are named.
The linear dependency is clear for prokaryotes at all inflation values tested (R2 > 0.94), and can be described by equation 1:
f = P × g + Fini1
where f is the number of protein families in an organism, g is the number of genes and P and Fini are two constants, representing the paralogy constant (slope of the line) and the minimal number of families, respectively.
The minimal number of families most likely represents the indispensable core set of families in the prokaryotic genome. Depending on the inflation value used, this value varies between 366 and 426. It is remarkable that this estimate agrees closely with estimates for the number of genes in the minimal genome (25) and the number of gene families in the reconstruction of the last universal common ancestor (26). The paralogy levels represent the ratio of genes to families in the species under consideration. The values obtained (with the essential families excluded) indicate that paralogy levels are similar between prokaryotes irrespective of genome size. This paralogy constant varies between 0.42 and 0.63, depending on the inflation value used. This result indicates that for all observed archaeal and bacterial species there are on average two genes per protein family, which suggests that the number of duplicated genes across all species is remarkably similar.
The linear dependency between the number of genes and families found for prokaryotic genomes does not apply to the set of currently available eukaryotic genomes, as they deviate strongly from a straight line (Fig. 2). Eukarya in this case have higher paralogy levels (i.e. more genes per family) than prokaryotes. This contrasting character reflects radically different evolutionary strategies and genome organisation in these groups of organisms. Moreover, it is possible that improvements in gene prediction and the consideration of alternative splicing for a number of genes in Eukarya may further protract the above deviation.
Power law distribution
We have examined the distribution of family sizes contained within the Tribes database. Interestingly, the current distribution of protein family size follows a power law (Fig. 3) for all inflation values, suggesting that the currently known space of protein sequence similarities represents a scale-free network. This scale-free behaviour of family sizes has been previously reported for individual genomes (27) and has several implications when associated with family size distributions from multiple genomes.
Figure 3.

The power law distribution of the Tribes family sizes. Counts of families for each family size are shown.
First, the power law distribution suggests that the TribeMCL family detection method is robust, as it is not biased towards any specific family size. A preference for a particular family size would create a deviation from the power law graph (Fig. 3). Absence of this type of deviation serves as additional support for the robustness of the method. In addition, a similar pattern can be obtained from the ProtoMap clustering project, for which the cluster size distribution is available (not shown).
The second conclusion from this graph addresses the quality of gene prediction. Our dataset includes ‘hypothetical’ proteins coming from genome projects that may represent potential gene prediction errors. Erroneously predicted genes are expected to appear as singletons (i.e. families of size one) on the similarity graph. The fact that families of size one are also placed on the power law line suggests that erroneous gene prediction might not be a major contributor to the number of predicted genes.
Protein lengths and cluster sizes
We have also examined the length distribution of proteins in families of various sizes (Fig. 4). Interestingly, we have observed that, in general, families with fewer members are composed of shorter proteins than families with more members. As family size increases, there is a corresponding increase in the average protein length within families. For example, the peak protein length for singletons is ∼100 residues, for families of size three the peak is ∼125 residues and for families comprising seven or more members the peak is >300 residues. There are several possible explanations for this phenomenon. One possibility is that the majority of these proteins result from erroneous gene prediction, however, this is not consistent with the gradual increase in protein length with increasing family size. The possibility that this size irregularity is caused by erroneous gene prediction also contradicts the clear power law distribution of protein family sizes, although this effect may indeed be present on a smaller scale. Also, because families are constructed from pair-wise similarity scores (see Materials and Methods), longer proteins have a greater chance of finding a sequence similarity (28) and are thus found in larger families. Another possibility is that some of the shorter proteins emerged de novo. This possibility is consistent with their short length and singularity in sequence space.
Figure 4.

Distribution of protein lengths in the Tribes families of various sizes. Note that smaller families are composed of shorter proteins (see text for discussion).
The proportion of singletons is highest in Archaea (Table 1), intermediate in Eukarya and smallest in Bacteria. This distribution most probably results from the differential taxonomic sampling of genomes sequenced from the three domains of life. This result can also be partially influenced by erroneous gene predictions.
Table 1. Fraction of singletons (proteins forming families with a single member) in various domains of life/data sources.
| Singletons | Total | Fraction (%) of singletons | |
|---|---|---|---|
| Archaea | 5785 | 36 194 | 16.0 |
| Bacteria | 18 639 | 203 535 | 9.2 |
| Eukarya | 13 015 | 101 146 | 12.9 |
| SwissProt | 3694 | 108 158 | 3.4 |
| Total | 41 133 | 449 033 | 9.2 |
Functional diversity of families
Using information from the GeneQuiz (29) automated genome annotation system, it is possible to perform detailed functional analysis of protein sequences from complete genomes. Currently GeneQuiz annotations are available for 60 out of the 83 genomes used in this analysis. One advantage of GeneQuiz is that it automatically assigns proteins to one of 14 functional classes (e.g. ‘cell envelope’ or ‘energy metabolism’). GeneQuiz also automatically determines the annotation category for a given protein using sequence similarity searches. Proteins are sorted into four annotation categories, according to their similarity to other proteins of known structure, function (but no structure), sequence (but no function) or just themselves (no similarity to any protein).
Using GeneQuiz functional classes and annotation categories for the available 60 genomes, we have detected the most likely class and annotation category for each Tribes family that contains proteins from one of the 60 genomes annotated with GeneQuiz. In total, it was possible to annotate 48 096 families in this manner. Annotation was performed using a consensus approach, i.e. for any given family we scan GeneQuiz annotations for all members of that family (where available) and transfer the most common annotation to the family. This annotation transfer appears to be very robust, as consensus annotations were on average present in over 94% of all family members.
Analysis of annotation categories indicates that for all families, approximately 6% have significant similarity to a known three-dimensional structure, 25% have clear similarity to well-characterised proteins and the rest are either uncharacterised or poorly characterised (Fig. 5a). When each domain is analysed separately, it becomes apparent that the annotation quality is significantly higher for Eukarya, intermediate for Bacteria, while Archaea represent the domain with the lowest amount of annotation. For archaeal specific families only 13% have a clear functional assignment or a three-dimensional structure. For Bacteria this rises to 27%, while for Eukarya this value is highest at 38% (Fig. 5a).
Figure 5.

Annotation categories for protein families in the three domains of life. The analysis was performed with GeneQuiz (29). (Top) Families are divided according to annotation quality (current knowledge) and (bottom) according to functional class membership.
Classification of characterised families also sheds light on the functional diversity of the three separate domains (Fig. 5b). When all families from all domains are considered, there is a relatively even distribution of functional class assignments. The lowest fraction of families is represented by three functional classes (‘biosynthesis of amino acids’, ‘biosynthesis of cofactors’ and ‘fatty acid and phospholipid metabolism’) (each corresponding to 3% of families), while the highest fractions correspond to another three functional classes (‘cell envelope’, ‘regulatory functions’ and ‘transport and binding proteins’) (at 13%). Broken down for families specific to each of the three domains, this distribution changes dramatically, reflecting the functional properties of the corresponding domains: for instance, in Archaea, the largest proportion of characterised families correspond to energy metabolism and transcription, in contrast to ‘cell envelope’ in Bacteria and ‘regulatory functions’ in Eukarya.
Conclusion
We have presented Tribes, an automatically generated and easy to update protein family resource, containing sequences from most of the publicly available entire genomes. Our results suggest that this approach is scalable and could readily deal with an ever-increasing data avalanche from genome projects, to support research in computational and experimental genomics.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
REFERENCES
- 1.Eisenberg D., Marcotte,E.M., Xenarios,I. and Yeates,T.O. (2000) Protein function in the post-genomic era. Nature, 405, 823–826. [DOI] [PubMed] [Google Scholar]
- 2.Tatusov R.L., Koonin,E.V. and Lipman,D.J. (1997) A genomic perspective on protein families. Science, 278, 631–637. [DOI] [PubMed] [Google Scholar]
- 3.Doolittle R.F. (1981) Similar amino acid sequences: chance or common ancestry? Science, 214, 149–159. [DOI] [PubMed] [Google Scholar]
- 4.Chothia C. and Lesk,A.M. (1986) The relationship between the divergence of sequence and structure in proteins. EMBO J., 5, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devos D. and Valencia,A. (2000) Practical limits of function prediction. Proteins, 41, 98–107. [PubMed] [Google Scholar]
- 6.Andrade M.A., Brown,N.P., Leroy,C., Hoersch,S., de Daruvar,A., Reich,C., Franchini,A., Tamames,J., Valencia,A., Ouzounis,C. et al. (1999) Automated genome sequence analysis and annotation. Bioinformatics, 15, 391–412. [DOI] [PubMed] [Google Scholar]
- 7.Liu J. and Rost,B. (2003) Domains, motifs and clusters in the protein universe. Curr. Opin. Chem. Biol., 7, 5–11. [DOI] [PubMed] [Google Scholar]
- 8.Enright A.J., Van Dongen,S. and Ouzounis,C.A. (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res., 30, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enright A.J. and Ouzounis,C.A. (2000) GeneRAGE: a robust algorithm for sequence clustering and domain detection. Bioinformatics, 16, 451–457. [DOI] [PubMed] [Google Scholar]
- 10.Krause A., Haas,S.A., Coward,E. and Vingron,M. (2002) SYSTERS, GeneNest, SpliceNest: exploring sequence space from genome to protein. Nucleic Acids Res., 30, 299–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kriventseva E.V., Fleischmann,W., Zdobnov,E.M. and Apweiler,R. (2001) CluSTr: a database of clusters of SWISS-PROT+TrEMBL proteins. Nucleic Acids Res., 29, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatusov R.L., Natale,D.A., Garkavtsev,I.V., Tatusova,T.A., Shankavaram,U.T., Rao,B.S., Kiryutin,B., Galperin,M.Y., Fedorova,N.D. and Koonin,E.V. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res., 29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yona G., Linial,N. and Linial,M. (2000) ProtoMap: automatic classification of protein sequences and hierarchy of protein families. Nucleic Acids Res., 28, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bateman A., Birney,E., Cerruti,L., Durbin,R., Etwiller,L., Eddy,S.R., Griffiths-Jones,S., Howe,K.L., Marshall,M. and Sonnhammer,E.L. (2002) The Pfam protein families database. Nucleic Acids Res., 30, 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaffer A.A., Wolf,Y.I., Ponting,C.P., Koonin,E.V., Aravind,L. and Altschul,S.F. (1999) IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics, 15, 1000–1011. [DOI] [PubMed] [Google Scholar]
- 16.Van Dongen S. (2000) Graph clustering by flow simulation. PhD thesis, Utrecht University, The Netherlands.
- 17.Birney E., Bateman,A., Clamp,M.E. and Hubbard,T.J. (2001) Mining the draft human genome. Nature, 409, 827–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bairoch A. and Apweiler,R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunin V. and Ouzounis,C.A. (2003) GeneTrace: reconstruction of gene content of ancestral species. Bioinformatics, 31, 1412–1416. [DOI] [PubMed] [Google Scholar]
- 20.Kunin V., Cases,I., Enright,A.J., de Lorenzo,V. and Ouzounis,C.A. (2003) Myriads of protein families and still counting. Genome Biol., 4, i401.401–i401.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janssen P.J., Enright,A.J., Audit,B., Cases,I., Goldovsky,L., Harte,N., Kunin,V. and Ouzounis,C.A. (2003) COmplete GENome Tracking (COGENT): a flexible data environment for computational genomics. Bioinformatics, 31, 1451–1452. [DOI] [PubMed] [Google Scholar]
- 22.Promponas V.J., Enright,A.J., Tsoka,S., Kreil,D.P., Leroy,C., Hamodrakas,S., Sander,C. and Ouzounis,C.A. (2000) CAST: an iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics, 16, 915–922. [DOI] [PubMed] [Google Scholar]
- 23.Enright A.J. (2002) Computational analysis of protein function within complete genomes, PhD thesis, University of Cambridge.
- 24.Andrade M.A., Ouzounis,C., Sander,C., Tamames,J. and Valencia,A. (1999) Functional classes in the three domains of life. J. Mol. Evol., 49, 551–557. [DOI] [PubMed] [Google Scholar]
- 25.Hutchison C.A., Peterson,S.N., Gill,S.R., Cline,R.T., White,O., Fraser,C.M., Smith,H.O. and Venter,J.C. (1999) Global transposon mutagenesis and a minimal Mycoplasma genome. Science, 286, 2165–2169. [DOI] [PubMed] [Google Scholar]
- 26.Kyrpides N., Overbeek,R. and Ouzounis,C. (1999) Universal protein families and the functional content of the last universal common ancestor. J. Mol. Evol., 49, 413–423. [DOI] [PubMed] [Google Scholar]
- 27.Harrison P.M. and Gerstein,M. (2002) Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J. Mol. Biol., 318, 1155–1174. [DOI] [PubMed] [Google Scholar]
- 28.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoersch S., Leroy,C., Brown,N.P., Andrade,M.A. and Sander,C. (2000) The GeneQuiz web server: protein functional analysis through the Web. Trends Biochem. Sci., 25, 33–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.