

Abstract
α-RIMs (RIM1α and RIM2α) are multidomain active zone proteins of presynaptic terminals. α-RIMs bind to Rab3 on synaptic vesicles and to Munc13 on the active zone via their N-terminal region, and interact with other synaptic proteins via their central and C-terminal regions. Although RIM1α has been well characterized, nothing is known about the function of RIM2α. We now show that RIM1α and RIM2α are expressed in overlapping but distinct patterns throughout the brain. To examine and compare their functions, we generated knockout mice lacking RIM2α, and crossed them with previously produced RIM1α knockout mice. We found that deletion of either RIM1α or RIM2α is not lethal, but ablation of both α-RIMs causes postnatal death. This lethality is not due to a loss of synapse structure or a developmental change, but to a defect in neurotransmitter release. Synapses without α-RIMs still contain active zones and release neurotransmitters, but are unable to mediate normal Ca2+-triggered release. Our data thus demonstrate that α-RIMs are not essential for synapse formation or synaptic exocytosis, but are required for normal Ca2+-triggering of exocytosis.
Keywords: active zone, neurotransmitter release, RIM, synapse, synaptic plasticity
Introduction
In presynaptic nerve terminals, exocytosis of transmitter-filled synaptic vesicles at the plasma membrane is under tight spatial and temporal control. Synaptic vesicle exocytosis is restricted to a specialized area of the plasma membrane, the active zone. The protein network that constitutes the active zone organizes the docking and priming of synaptic vesicles. In addition, the active zone mediates use-dependent changes in release during short- and long-term forms of presynaptic plasticity (reviewed in Dresbach et al, 2001; Rosenmund et al, 2003; Kaeser and Südhof, 2005).
The active zone is composed of a network of proteins that includes RIMs (for Rab3-interacting molecule; see Wang et al, 1997, 2000) as central components (Koushika et al, 2001; Castillo et al, 2002; Schoch et al, 2002). RIMs are multidomain proteins that are expressed in variable forms: two α-RIMs (RIM1α and 2α) that include a full complement of RIM domains (an N-terminal region that comprises a Rab3-binding sequence and a Munc13-binding zinc-finger; a central PDZ-domain; and two C-terminal C2-domains that however do not bind Ca2+), a single β-RIM (RIM2β) that contains all of the domains of α-RIMs except for the N-terminal Rab3- and Munc13-binding sequences;, and three γ-RIMs (RIM2γ, 3γ, and 4γ) that are composed of only the C-terminal C2-domain preceded by a short N-terminal flanking sequence (Wang et al, 2000). RIMs are encoded by four genes, of which the RIM1, RIM3, and RIM4 genes express only a single isoform (RIM1α, 3γ, and 4γ, respectively), whereas the RIM2 gene expresses three isoforms (RIM2α, 2β, and 2γ; Wang and Südhof, 2003). Further variation is introduced into α- and β-RIMs (but not γ-RIMs) by extensive alternative splicing (Wang et al, 2000; Johnson et al, 2003; Wang and Südhof, 2003).
In addition to interacting with Munc13 and Rab3, α-RIMs bind to multiple other synaptic proteins: ELKS via the central PDZ-domain (Ohtsuka et al, 2002; Wang et al, 2002), RIM-BPs via an SH3-domain-binding sequence between the two C2-domains (Wang et al, 2000), and α-liprins and synaptotagmin 1 via the C-terminal C2-domain (Coppola et al, 2001; Schoch et al, 2002). Furthermore, in vitro interactions with several proteins have been described, including cAMP–GEFII (guanine nucleotide-exchange factor II) (Ozaki et al, 2000), SNAP-25 (Coppola et al, 2001), N-type Ca2+ channels (Coppola et al, 2001), and 14-3-3 adaptor proteins (Sun et al, 2003; Simsek-Duran et al, 2004). RIMs are connected indirectly with the active zone proteins Piccolo and Bassoon via ELKS (Takao-Rikitsu et al, 2004) and with receptor tyrosine phosphatases via liprins (Serra-Pages et al, 1998). Of these interactions, only RIM1α and 2α bind to Munc13 and Rab3, whereas γ-RIMs bind only to α-liprins and synaptotagmin 1. The binding of the N-terminal region of α-RIMs to Rab3 on synaptic vesicles and Munc13s is particularly interesting because a relatively short sequence (<150 residues) contains two nested subdomains, an α-helical region that binds to Rab3 (Wang et al, 2001) and a zinc-finger that binds to Munc13 (Betz et al, 2001; Dulubova et al, 2005). This binding is mutually compatible with each other, resulting in a trimeric complex in which the α-RIM/Munc13 dimer on the active zone is coupled to the synaptic vesicle protein Rab3 (Dulubova et al, 2005). Finally, RIMs are substrates for cAMP-dependent protein kinase (PKA) that phosphorylates RIM1α and RIM2α/β at two sites (Lonart et al, 2003).
Analysis of RIM1α knockout (KO) mice showed that RIM1α plays a key regulatory role in synaptic vesicle exocytosis at the active zone, from vesicle priming to short- and long-term synaptic plasticity (Castillo et al, 2002; Schoch et al, 2002; Calakos et al, 2004). RIM1α-deficient synapses did not exhibit major changes in ultrastructure, suggesting that it is essential only for regulating exocytosis, and not for building an active zone architecture (Schoch et al, 2002). Although important, loss of this function does not impair mouse survival, as RIM1α KO mice have a normal apparent life expectancy (Schoch et al, 2002). The importance of RIM1α function nevertheless is apparent from the severe behavioral abnormalities observed in these mice, which include impairments in spatial learning and in fear conditioning as well as an increase in locomotor responses to novelty (Powell et al, 2004).
The currently available data confirm that RIM1α is an active zone protein with a central role in regulating neurotransmitter release, and suggest that the other RIM isoforms may also be involved in the regulation of synaptic vesicle exocytosis. However, so far, only RIM1α has been analyzed. Although the various RIM isoforms are coexpressed in brain, their relative expression patterns are unknown, and it is unclear how much potential redundancy may exist among RIM isoforms. Such redundancy could exist, for example, between RIM1α and RIM2α because both of these RIM isoforms bind to Munc13 and to Rab3 (Dulubova et al, 2005), although they are the only isoforms that do so. Therefore, major questions remain unanswered: (1) in which cell types are the various RIM isoforms expressed? (2) Are RIM1α and RIM2α functionally redundant? (3) How do the two α-RIMs relate to each other? (4) Does the deletion of both α-RIMs lead to ultrastructural changes? To examine the role of the α-RIMs in synaptic transmission, we generated single and double KO mice (DKO) lacking either or both α-RIMs. Our data demonstrate that the RIM-α-isoforms are essential for survival and exhibit partially overlapping functions in the regulation of synaptic transmission, but are not required for building a normal synapse.
Results
Differential expression of RIM1 and RIM2 isoforms
To examine whether RIM1α, RIM2α, RIM2β, and RIM2γ are differentially expressed in brain, we performed in situ hybridizations on brain sections from adult rats (Figure 1A, left panels). Two oligonucleotides were used for each RIM isoform to ensure that the same labeling patterns were obtained (data not shown). This labeling was abolished when excess unlabeled oligonucleotides were added to the hybridization mix (Figure 1A, right panels).
Figure 1.
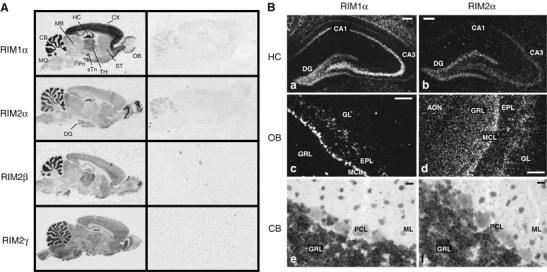
In situ hybridization of RIM mRNAs in the rat brain. (A) Film images showing the distribution of RIM mRNAs in the adult rat brain (CB, cerebellum; CX, cerebral cortex; DG, dentate gyrus; HC, hippocampus; MB, midbrain; OB, olfactory bulb; Pn, pontine nucleus; sTn, subthalamic nucleus; ST, striatum; TH, thalamus; MO medulla oblongata). (B) Dark-field images of emulsion-dipped sections from rat hippocampus, olfactory bulb, and cerebellum (AON, anterior olfactory nucleus; DG, dentate gyrus; EPL, external plexiform layer; GL, glomerular layer; GRL, granule cell layer; MCL, mitral cell layer; ML, molecular layer; PCL, Purkinje cell layer; scale bar B, a–d=100 μm, B, e–f 20 μm).
Autoradiographs of hybridized rat brain sections revealed differential but overlapping expression patterns of RIM mRNAs. In each case, regions rich in glial cells (e.g., white matter of cerebral cortex and cerebellum) were unlabeled, indicating a neuron-specific expression of RIM isoforms (Figure 1A, left panel). RIM1α mRNA is present throughout the brain, with the highest levels in the cortex, cerebellum, hippocampus and thalamus. RIM2α mRNA is concentrated in the cerebellum, the olfactory bulb and the dentate gyrus of the hippocampus. However, comparison of the signals obtained with the negative control indicates that RIM2α is also ubiquitously expressed in brain, albeit at low levels. RIM2β and RIM2γ mRNAs are present in a pattern similar to RIM1α, but both are probably expressed at lower levels based on the hybridization signal and exhibit regional differences (e.g., RIM2β is expressed more in the thalamus, and RIM2γ more in the brainstem; Figure 1A). These results show that the various RIM1- and RIM2-isoforms exhibit a differential, but highly overlapping expression pattern in rat brain.
To determine the cellular distribution of RIM mRNA species, we analyzed emulsion-dipped sections. In the hippocampus, RIM1α can be detected at high levels in the dentate gyrus and the CA3 region, and at lower levels in the CA1 region (Figure 1B). In contrast, strong labeling for RIM2α was largely restricted to the dentate gyrus, with lower levels observed in the CA3 region. In the olfactory bulb, significant RIM1α expression was detected in the mitral cell layer (MCL) and in some cells of the external plexiform layer (EPL), whereas the cells of the granule cell layer (GRL) and the glomerular layer (GL) were devoid of a RIM1α signal (Figure 1B). In contrast, RIM2α showed strong expression in the GL, the MCL, and the GRL. Within the cerebellum, RIM1α and RIM2α expression is highly concentrated in the GRL (Figure 1B). Clearly, most neurons express multiple isoforms of RIMs with distinct relative expression levels.
Characterization of RIM2α KO mice
We generated mutant mice in which the fifth exon of the RIM2α gene (which encodes part of the zinc-finger; Wang and Südhof, 2003) is flanked by loxP sites (Figure 2A). Mice containing the targeted floxed RIM2α gene synthesized RIM2α at approximately wild-type levels (data not shown). We then crossed the floxed mice to transgenic mice that express cre recombinase in the male germ line to produce KO mice in which the floxed exon was deleted (Figure 2A) (O'Gorman et al, 1997). The conditional KO mice may prove to be useful in the analysis of the function of the α-RIMs in specific brain regions. However, in the present study, we focused on the constitutive RIM2α KO mice in order to establish the baseline function of the α-RIM isoforms.
Figure 2.
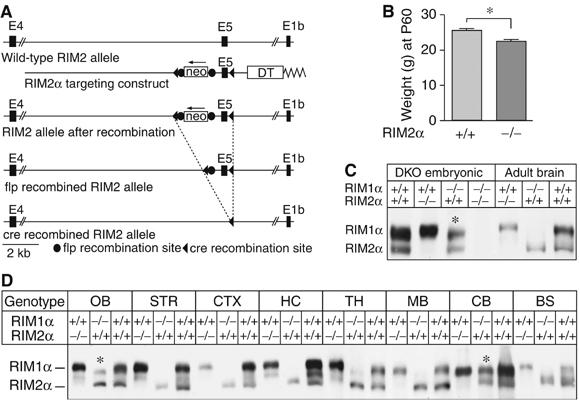
Generation of RIM2α KO mice. (A) Structures of the RIM2 wild-type gene (wild-type allele), of the targeting vector used for homologous recombination (targeting construct), and of the mutant RIM2 alleles after homologous recombination before and after further recombination of flp and cre recombinases. In the targeting vector, exon 5 is flanked by loxP sites (black triangles) and the neomycin resistance gene cassette (neo) that is flanked by flp recombination sites (black circles). A diphtheria toxin gene (DT) is included for negative selection. (B) Weights of male RIM2α KO and littermate control mice (N=36, *P<0.001). (C) Immunoblots of E18.5 embryonic and adult wild-type, RIM1α KO, RIM2α KO and embryonic DKO whole brain homogenates. (D) α-RIM immunoblots of proteins from different brain regions from adult RIM1α KO, RIM2α KO, and wild-type control mice; blots were probed with an antibody that recognizes both RIM1α and RIM2α (abbreviations of brain areas: OB, olfactory bulb; STR, striatum; CTX, cortex; HC, hippocampus; TH, thalamus; MB, midbrain, BS, brain stem; CB, cerebellum; *, bands of alternatively spliced RIM2α isoforms of higher molecular weight).
Homozygous RIM2α KO mice were viable and fertile. However, systematic analysis of the offspring from heterozygous matings revealed that homozygous KO mice were slightly smaller than littermate wild-type controls (males, N=36) (Figure 2B), and less frequent than would be expected based on Mendelian inheritance (24.1% wild type, 56.3% heterozygous, and 19.6% homozygous mutant mice in offspring at >1 month of age; N=591; P<0.01). Immunoblotting confirmed that the KO mice lacked RIM2α (Figure 2C and D and Supplementary Figure 2). Similar to the RIM1α KO mice (Schoch et al, 2002), RIM2α mutant mice exhibited a deficit in maternal behavior, as they did not take care of their litters even after multiple pregnancies. Brains of single RIM2α KO mice showed a normal cell density, cytoarchitecture, connectivity, distribution/density of synapses, and ultrastructural morphology as assessed by H&E staining, NeuN immunohistochemistry, and electron microscopy (Supplementary Figure 1). Taken together, these data demonstrate that although RIM2α KO mice are viable and exhibit no apparent developmental abnormalities, the behavior and survival of these mice are slightly impaired.
To search for compensatory changes in the expression of RIM1α as the only other α-RIM isoform in RIM2α KO mice, we compared the relative expression of RIM1α and RIM2α in various brain regions in wild-type, RIM1α KO, and RIM2α KO mice (Figure 2D and Supplementary Figure 2). Antibodies to the zinc-finger region of RIM1α and RIM2α often crossreact because of their structural similarities (Schoch et al, 2002). Moreover, these antibodies detect multiple bands even when one of the two α-RIM isoforms is deleted because of the extensive alternative splicing of RIMs (Wang and Südhof, 2003). Consistent with the in situ hybridization data, immunoblotting of different brain regions in single RIM1α and RIM2α KO mice showed that RIM1α is the more abundant isoform, whereas RIM2α is found at low levels in most rostral brain regions examined, but at high levels in the cerebellum and olfactory bulb (Figure 2D). However, we detected no region-specific compensatory changes in RIM1α protein levels in RIM2α KO mice, or conversely in RIM2α protein levels in RIM1α KO mice (Figure 2D and Supplementary Figure 2). Interestingly, alternative splicing of RIM2α seems to be regulated in a region-specific manner. Although in most brain regions, a smaller isoform is detected, in cerebellum and olfactory bulb an additional larger protein could be observed (asterisks in Figure 2D and Supplementary Figure 2).
In RIM1α KO mice, synaptic transmission in the hippocampus is severely impaired (Castillo et al, 2002; Schoch et al, 2002; Calakos et al, 2004). To test whether RIM2α performs a similar fundamental role in synaptic transmission in the hippocampus, we recorded excitatory and inhibitory synaptic responses in acute hippocampal slices (Supplementary Figure 3). We first probed excitatory synaptic transmission in the CA1 region of the hippocampus, and compared synaptic responses of wild-type and KO mice in four paradigms that measure different types of short-term synaptic plasticity: paired-pulse facilitation (Supplementary Figure 3A), post-tetanic potentiation (Supplementary Figure 3B), and use-dependent depression (Supplementary Figure 3C). In addition, we examined the synaptic release probability by analyzing the progressive block of NMDA-dependent synaptic responses by the irreversible NMDA receptor antagonist MK801 (Supplementary Figure 3D). In all of these measurements, we observed no significant difference between wild-type and RIM2α KO mice. Next, we tested whether RIM2α KO mice exhibited a change in mossy fiber LTP in excitatory synapses of the CA3 region, which is abolished in RIM1α KO mice (Castillo et al, 2002). Again, we found no change in RIM2α KO mice (Supplementary Figure 3E). Finally, we examined inhibitory synaptic transmission at CA1 pyramidal cells, but failed to detect significant changes in paired-pulse-ratio (Supplementary Figure 3F). Thus, RIM2α-deficient synapses, different from RIM1α-deficient synapses, do not exhibit a major impairment of synaptic transmission in the hippocampus.
Impaired survival of RIM1α/2α DKO mice
To assess the redundant or divergent functions of the two α-RIMs, we generated RIM1α/2α DKO mice. Analysis of the offspring from systematic breedings of double heterozygous RIM1α/2α KO mice revealed that the survival of RIM1α or RIM2α homozygous mutant mice exhibited no significant decrease as long as two RIM1α or RIM2α wild-type alleles were present (Figure 3A). However, in >100 crossings, no surviving mouse that was homozygous mutant for both RIM1α and RIM2α was observed, demonstrating that RIM1α and RIM2α are redundant in terms of survival. Moreover, even single homozygous RIM1α or RIM2α KO mice exhibited significantly impaired survival when one of the other two α-RIM alleles was also deleted (i.e., in the RIM1α−/−2α+/− or RIM1α+/−2α−/− genotypes, P<0.001; see Figure 3A).
Figure 3.
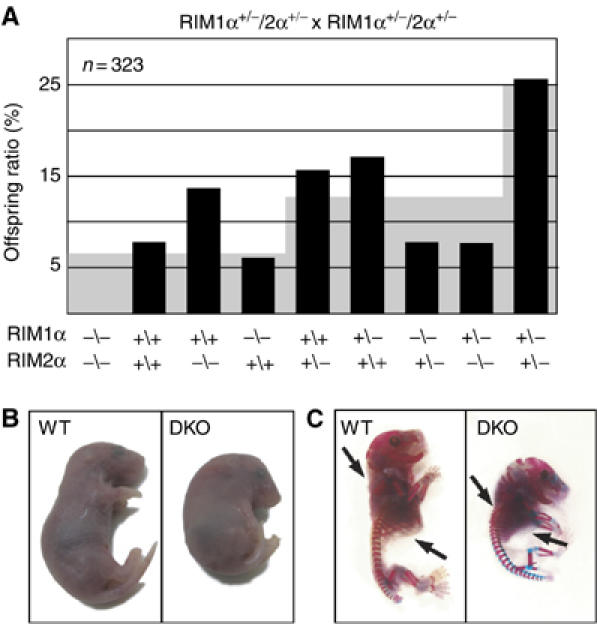
(A) Survival analysis of the offspring from matings of double heterozygous RIM1α/2α mutant mice. The black bars plot the observed frequency of the indicated genotypes as percentage of the total, whereas the gray background indicates the expected frequency based on Mendelian inheritance (N=323). (B, C) Images of E18.5 RIM1α/2α DKO mutant and control littermate mice overall morphology (B) and skeleton (C, bones are stained in blue and cartilage in pink; black arrows point to ribcage and cervical vertebrae).
RIM1α/2α DKO mice died immediately after birth because they could not breathe. The mutant mice did not respond to tactile stimuli, and exhibited an abnormal body posture similar to synaptobrevin 2 KO mice (Figure 3; Schoch et al, 2001), suggesting that they were paralyzed. Double KO mice could be recovered alive, however, at E18.5 as embryos by hysterectomy; at this age, DKO mice were present in a normal Mendelian ratio, suggesting that the deletion of both α-RIMs does not impair survival in utero (data not shown). Thus, for all subsequent studies on DKO mice, we analyzed embryos at E18.5. Although whole-body staining for bone and cartilage revealed no major developmental defects in the DKO mice, their vertebrae in the KO skeleton seemed more compact at the cervical level, and the ribcage appeared enlarged (Figure 3C), possibly because of permanent paralysis of the embryo during development.
Brain composition and synapse structure of RIM1α/2α double KO mice
Histochemistry and immunocytochemistry of brain sections revealed that RIM1α/2α DKO mice displayed no obvious impairments in central nervous system development (Figure 4A and data not shown). However, H&E- and NeuN-stained vibratome sections of paraffin-embedded spinal cords showed an increased number of ventral horn motoneurons at all cervical levels in RIM1α/2α DKO mice as compared with control littermates (Figure 4B). A morphometric analysis of NeuN-immunopositive motoneurons revealed a significant increase (**P<0.005) in motoneuron density in E18.5 DKO spinal chords (Figure 4C). No sign of neurodegeneration was detectable in the spinal cord by TUNEL or Fluoro JadeB staining (data not shown). The increase in motoneurons may be due to decreased synaptic transmission at the neuromuscular junction (NMJ) (see below) (Harris and McCaig, 1984; Oppenheim, 1991).
Figure 4.
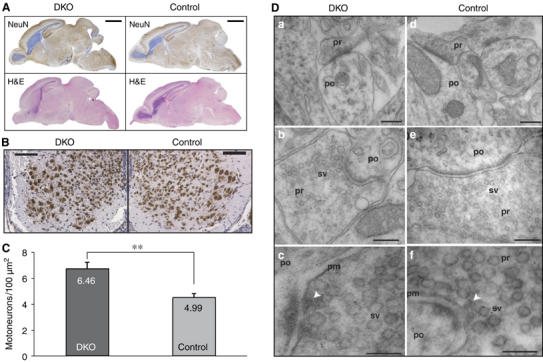
Morphology of RIM1α/2α DKO brains and spinal cord. (A) H&E- and NeuN-stained sagittal section of brains from E18.5 mice of the indicated genotype (scale bar=2 mm). (B) NeuN-stained coronal sections of spinal cord from E18.5 mice of the indicated genotype (scale bar=100 μm). (C) Morphometric analysis of NeuN-immunopositive spinal motoneurons (**P<0.005). (D) Electron micrographs of synapses in the spinal cord. The arrows in (c, f) point to presynaptic dense projections in active zones. Abbreviations: pr, presynaptic; po, postsynaptic; sv, synaptic vesicles; pm, presynaptic plasma membrane. Scale bars, a=300 nm, d=370 nm, b=300 nm, e=240 nm, c=100 nm, f=130 nm.
To test whether deletion of α-RIMs causes a major change in the protein composition of the brain, we quantified the levels of neuronal proteins in adult littermate wild-type, heterozygous, and homozygous mutant RIM2α KO mice (Figure 5A and C and Supplementary Table I) and in E18.5 embryos that were homozygous for the RIM1α KO allele and wild-type, heterozygous, or homozygous mutant for the RIM2α KO allele (Figure 5B and D and Supplementary Table II). We detected no significant changes in any protein in adult RIM2α KO mice compared with controls; in particular, no significant decrease in Munc13-1 in RIM1α/2α DKO embryos compared with RIM1α KO embryos was observed. To rule out region-specific changes in Munc13-1 level, we investigated different brain areas, but detected no obvious alterations (Figure 5E). These results were unexpected because previous studies showed that Munc13-1 is decreased ∼50% in RIM1α KO mice (Schoch et al, 2002), which prompted the hypothesis that the remaining Munc13-1 is stabilized by RIM2α. As shown here, however, additional deletion of RIM2α does not cause a major further decrease in Munc13-1 levels beyond that observed in RIM1α KO mice. A recent report described a role for RIM1α in the regulation of presynaptic recruitment of Munc13-1 and ubMunc13-2 (Andrews-Zwilling et al, 2006). Therefore, we examined if deletion of both α-RIMs results in a redistribution of Munc13-1 from the membrane-bound to the soluble fraction (Supplementary Figure 4B and data not shown). Soluble and membrane-bound protein fractions from wild-type and RIM1α/2α DKO embryonic brains were separated by ultrathurrax homogenization in buffer or in the presence of detergents of different strength and high-speed centrifugation. In agreement with Andrews-Zwilling et al (2006) we detected an increase of Munc13-1 in the soluble fraction in the brains of DKO mice. However, the majority of Munc13 was still found in the insoluble fraction with buffer or nondenaturing detergents but was solubilized with SDS. Thus, consistent with the fact that only some of the Munc13 isoforms contain the N-terminal C2-domain that binds to α-RIMs (Brose et al, 1995; Betz et al, 2001; Dulubova et al, 2005), only approximately half of the synaptic Munc13-1 depends on α-RIMs, and α-RIMs are not absolutely essential for Munc13 function.
Figure 5.
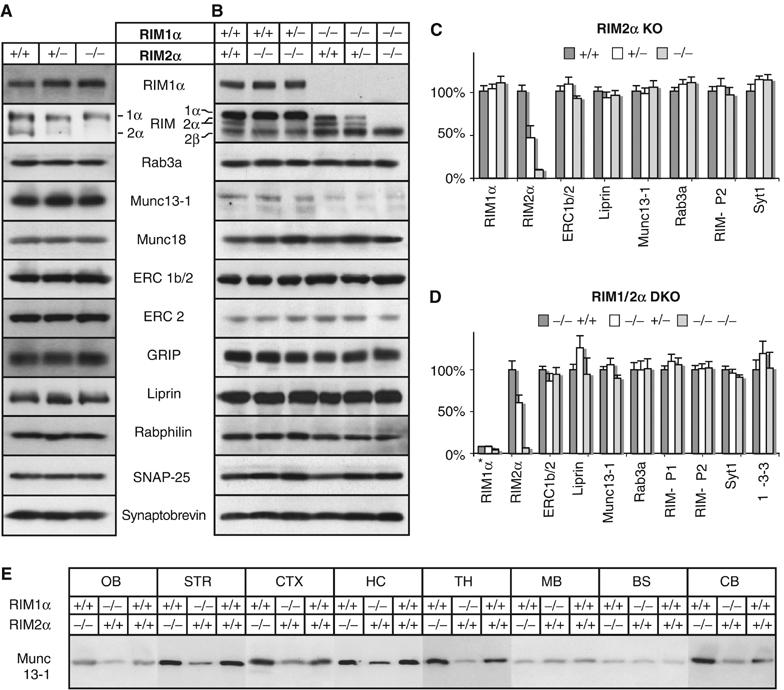
Analysis of synaptic protein levels in brains from RIM2α KO mice and RIM1α/2α DKO embryos. Brain homogenates of the indicated genotypes were analyzed by immunoblotting using antibodies to the indicated proteins (A, B), the blots were quantified and all data normalized to control level (C, D, RIM1α level in (D) normalized to wild type). (A, C) RIM 2α-KO (adult), (B, D) RIM1α/RIM2α-DKO (E18.5). (E) Proteins from different brain areas of adult RIM1α KO, RIM2α KO, and wild-type mice were analyzed with an antibody against Munc13-1 (abbreviations of brain areas: OB, olfactory bulb; STR, striatum; CTX, cortex; HC, hippocampus; TH, thalamus; MB, midbrain; BS, brain stem; CB, cerebellum).
To investigate whether the deletion of both α-RIMs alters synapse assembly, we analyzed the ultrastructure of RIM1α/2α DKO synapses in the spinal cord by quantitative electron microscopy. We observed typical synaptic structures in RIM1α/2α DKO neurons (Figure 4D), and detected no obvious changes in synapse density (DKO=35.7±24.1 per 103 μm2; control [RIM1α−/−, RIM2α+/+]=25.1±21.3 per 103 μm2; N=3 for both genotypes; 50 synapses for both genotypes), presynaptic bouton area (DKO=0.381±0.222 μm2; control=0.371±0.274 μm2; N=3 and 100 synapses), density of synaptic vesicles (DKO=196±136 synaptic vesicles per μm2 bouton area; control=202±150 synaptic vesicles per μm2 bouton area; N=3 and 50 synapses), active zone length (DKO=397±177 nm; control=354±165 nm; N=3 and 50 synapses), or number of docked vesicles (DKO=7.8±4.2 per μm active zone; control=8.3±5.4 per μm active zone; N=3 and 50 synapses). Statistical analysis was performed with Student's t-tests (two-sided, unpaired data with unequal variances). Thus, deletion of α-RIMs does not abolish the formation of synapses with docked vesicles and an ultrastructurally clearly visible active zone.
Structure of the NMJ in RIM1α/2α double KO mice
We next examined the morphology of the NMJ in the phrenic nerve/diaphragm muscle preparation, and performed immunostainings of whole-mount muscles at E18.5 with antibodies to presynaptic proteins (synaptotagmin 2, SV2B, synaptophysin, Rab3A, liprin-α, and ELKS1) and with Texas Red-conjugated α-bungarotoxin that labels postsynaptic acetylcholine receptors (Figure 6A and Supplementary Figure 4A and data not shown). In wild-type mice, the left and right phrenic nerves form synapses on the diaphragm in a discrete spatially restricted endplate band within the central region of each hemidiaphragm. Strikingly, in RIM1α/2α DKO mice, synapses as visualized by synaptotagmin 2 and acetylcholine receptor staining were only poorly aligned along the midline of the hemidiaphragm but more randomly distributed across a broader region of the muscle (Figure 6A). Similar results were obtained for other presynaptic markers, and in control as well as in RIM1α/2α DKO diaphragms, all axon terminals were always juxtaposed to acetylcholine receptors clusters. Furthermore, immunostainings of the whole-mount muscles with antibodies against the RIM-binding proteins liprin, ELKS, and Rab3 revealed a synaptic localization even in the absence of α-RIMs (Figure 6A and Supplementary Figure 4A and data not shown).
Figure 6.
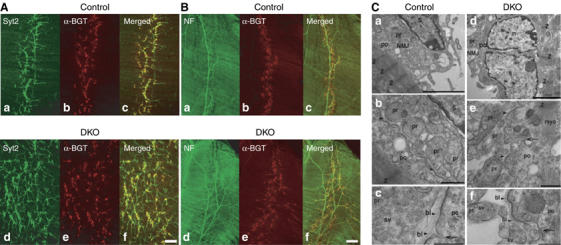
Morphology of diaphragm NMJs in RIM1α/2α DKO mice. (A, B) Confocal micrographs of E18.5 RIM1α/2α DKO mutant and control littermate whole-mount diaphragm muscles labeled with Texas Red-conjugated α-bungarotoxin (α-BGT) and antibodies to synaptotagmin-2 (Syt2) (A, a–d) or to neurofilament (NF) (B, a–d). (C) Electron micrographs of NMJs in RIM1α/2α DKO mice and control littermates demonstrate normal ultrastructure of NMJs in control and DKO mice. (a, d) Low-magnification electron micrographs of the NMJ demonstrating normal overall architecture of the NMJ in control and DKO mice. (b, c) and (e, f) Normal ultrastructural appearance of the NMJs at higher magnifications. Arrows point to postsynaptic invaginations that are not yet fully developed at E18 in both control and DKO mice. Arrowheads point to basal lamina material in the synaptic cleft of the NMJ. Abbreviations: NMJ, neuromuscular junction; S, Schwann cell; n, nucleus of a postsynaptic muscle fiber; Z, Z stripes in sarcomeres of postsynaptic muscle fibers; myo, myofibrils; pr, presynaptic; po, postsynaptic; bl, basal lamina of neuromuscular junction between the presynaptic terminals and the postsynaptic plasma membrane; sv, synaptic vesicles. Scale bars, A=100 μm, B=200 μm, C, a, d=3 μm, b, e=700 nm, c, f=400 nm.
The observed pattern of synaptic contacts in the mutant NMJs suggests an increase in innervation or an expansion of innervation. To confirm this observation, we stained whole-mount diaphragm muscles with antibodies to the neurofilament protein or to syntaxin 1, both of which label axons. Costaining with Texas Red-conjugated α-bungarotoxin showed that in control mice, synapses were distributed regularly (Figure 6B). In RIM1α/2α DKO mice, however, phrenic nerves exhibited abnormally extensive branching throughout the muscle, and their terminal arborizations covered a much broader surface of the muscle (Figure 6B).
To examine synapse ultrastructure of the NMJ, we performed electron microscopy at E18.5. Both in control and RIM1α/2α DKO mice, typical but immature synapses were observed with clusters of normal-sized small synaptic vesicles that were occasionally docked at the active zone, and with continuously aligned pre- and postsynaptic membranes that were separated by a well-developed basal lamina (Figure 6C). Thus, similar to central synapses (Figure 4), on a qualitative level no obvious ultrastructural changes were detectable in NMJs.
Electrophysiological properties of the NMJ in RIM1α/2α DKO mice
To examine synaptic transmission in the NMJ, we performed electrophysiological recordings in the phrenic nerve/diaphragm preparation at E18.5. Because of the impossibility of obtaining sufficient numbers of matched littermate wild-type control mice for these experiments, we used in these experiments control embryos that were from the same litter as the tested RIM1α/2α DKO embryos and that contained at least one wild-type RIM1α allele, that is, embryos that could have up to three mutant α-RIM alleles.
Recordings of spontaneous miniature endplate potentials (mEPPs) in normal Ringer solution revealed that the mEPP frequency and amplitudes were not significantly different between control samples and RIM1α/2α DKO mice (frequency: control, 0.71 Hz±0.12 (N=3 embryos, 13 muscle fibers); DKO, 0.65 Hz±0.07 (N=4 embryos, 25 muscle fibers); amplitudes: control, 1.7 mV±0.16 (N=3 embryos, 13 muscle fibers); DKO, 1.9 mV±0.16 (N=4 embryos, 25 muscle fibers)). The very low frequency of mEPPs at E18.5, a reflection of the immaturity of the NMJ at this stage in development, may have precluded observing a difference between control and RIM1α/2α DKO mice. We therefore increased the mEPP frequency by depolarizing the nerve terminals with 40 mM K+ in the presence of extracellular Ca2+ concentrations ranging from 0 to 4 mM (Figure 7). Quantitation of the mEPP frequency uncovered a significant difference between control and RIM1α/2α DKO mice at all examined extracellular Ca2+ concentrations. At 0 and 1 mM Ca2+, the mEPP frequencies were low in control and RIM1α/2α double-deficient NMJs. However, in RIM1α/2α DKO NMJs, the mEPP frequency was reduced by half as compared with control NMJs (P<0.0005). At 2 and 4 mM Ca2+, the mEPP frequency increased >3-fold in control NMJs but remained almost unchanged in RIM1α/2α-deficient NMJs (Figure 7B) (P<5 × 10−8).
Figure 7.
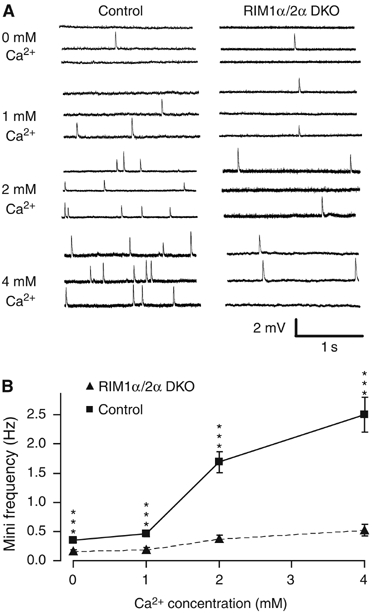
Spontaneous miniature endplate potentials (mEPPs) at the diaphragm NMJ from control and RIM1α/2α DKO mice recorded in 40 mM KCl in 0–4 mM extracellular Ca2+. (A) Representative traces from muscles from control (left panel) and double-mutant mice (right panel). (B) Average mEPP frequencies as a function of extracellular Ca2+ concentration (data shown are means±s.e.m.; n (number of cells) for mutants/controls are 34/27 (0 μM Ca2+), 27/26 (1 μM Ca2+), 30/27 (2 μM Ca2+), and 26/22 (4 μM Ca2+), respectively, ***P<0.0005, Student's t-test).
The mEPP frequency data suggest that synaptic vesicle exocytosis is not abolished in RIM1α/2α DKO mice, but that Ca2+ triggering of exocytosis may be impaired. To confirm this conclusion, we examined evoked EPPs elicited by a single stimulation or at 10 Hz (Figure 8). Deletion of both α-RIMs did not abolish evoked responses, but severely impaired them. The EPP amplitudes were almost 10-fold smaller in RIM1α/2α DKO than in control mice (control: 23±3 mV (N=3 embryos, 16 muscle fibers); DKO: 2.7±0.5 mV (N=4 embryos, 19 muscle fibers); P<0.001; failures were excluded from this analysis), and the failure rates were dramatically increased—in three control embryos, there was only one failure in response to a total of 309 stimuli (0.32±0.15%, N=3 mice, 25 muscle fibers), whereas in four double mutants analyzed, there were 115 failures in response to a total of 518 stimuli (22.2±2.39%, N=4 mice, 39 muscle fibers; Figure 8C). These measurements likely underestimate the degree of impairment as the controls are not wild-type mice but mice that contain a partial lack of α-RIMs. Thus, α-RIMs are not essential for exocytosis as such, but are required for normal Ca2+-triggering of exocytosis.
Figure 8.
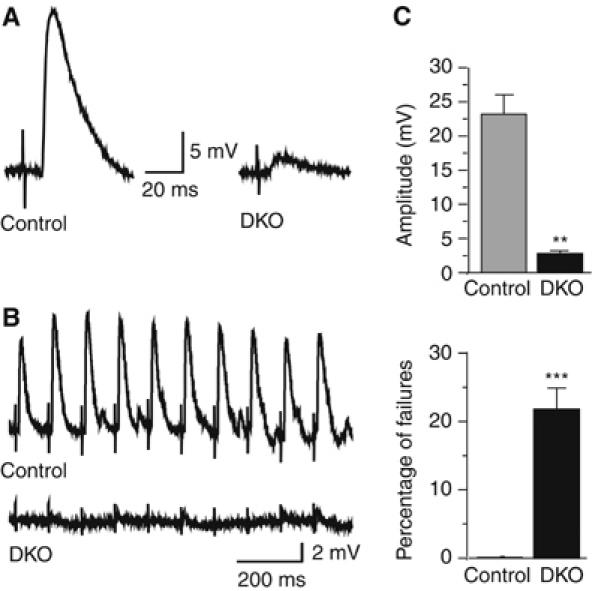
Impaired evoked neurotransmitter release at the NMJ of RIM1α/2α DKO mice. (A, B) Representative traces of EPPs evoked at low frequency (A) or at 10 Hz (B) in muscles from control (left trace in (A), top trace in (B)) or RIM1α/2α DKO embryos (right trace in (A); bottom trace in (B)). (C) Decreased amplitudes (upper panel) and increased failure rates (lower panel) of evoked EPPs in α-RIM-deficient NMJs (for amplitudes (excludes failures), controls, n=3 embryos, 16 cells; DKO, n=4 embryos, 19 cells; for failure rates, controls=1 failure in 309 stimuli, N=3 mice, 25 cells; DKO=115 failures in 518 stimuli, N=4 mice, 39 cells, **P<0.001, ***P<0.0005, Student's t test).
Insulin secretion in RIM1α and RIM2α knockout mice
Recent reports suggested that RIM2α plays a central role in regulating insulin secretion (Iezzi et al, 2000; Kashima et al, 2001). Therefore, we examined the glucose metabolism in the single RIM1α and RIM2α KO mice with two standardized tests: body fat measurement by NMR and a glucose tolerance test. Deletion of either RIM1α or RIM2α had no significant effect on body fat levels in mice (Supplementary Figure 5). Intraperitoneal glucose tolerance tests were performed on the same mice after an overnight fast, and also did not reveal any significant alterations in the KO mice (Supplementary Figure 5).
Discussion
RIMs comprise a family of active zone proteins, with two full-length α-isoforms (RIM1α and RIM2α) that interact with a host of other active zone proteins in addition to synaptic vesicle proteins, and β- and γ-isoforms that exhibit more restricted interaction patterns. However, to date, only RIM1α has been analyzed functionally (Castillo et al, 2002; Schoch et al, 2002; Lonart et al, 2003; Calakos et al, 2004; Powell et al, 2004). The currently available data demonstrate that RIM1α performs a central role in regulating neurotransmitter release, but suggest that other RIM isoforms may also be involved in the regulation of synaptic vesicle exocytosis. Although the various RIM isoforms are coexpressed in brain, their relative expression patterns were unknown, and it was unclear whether potential redundancy among RIM isoforms exists. Such redundancy could be present, for example, between RIM1α and RIM2α because these two RIM isoforms are the only RIMs that bind to Munc13 and Rab3 (Dulubova et al, 2005).
In the present study, we have examined the expression patterns of various RIM isoforms, generated RIM2α KO mice to explore its functions, and tested the possibility that RIM1α and RIM2α are functionally redundant. Moreover, we have probed the extent of their functions in release by comparing the effect of a double RIM1α and 2α deletion on synapse structure and neurotransmitter release. Our data demonstrate that α-RIMs are expressed in differential but largely overlapping expression patterns, they are functionally redundant, and they are selectively required to maintain Ca2+-triggered neurotransmitter release, not synapse structure. Among others, our data demonstrate that the linkage of Munc13s to α-RIMs in the active zone, and of Rab3 on synaptic vesicles to α-RIMs, is not essential for synaptic vesicle docking, priming, or exocytosis because in the RIM1α/2α double-deficient neurons, these linkages are abolished whereas exocytosis continues. Our data demonstrate, however, that this linkage is required for Ca2+-triggered release because the release is decreased approximately 10-fold in the double-deficient neurons.
Distribution of RIMs
To analyze if RIM isoforms are coexpressed and could thus be redundant, we performed in situ hybridizations and immunoblotting. Our results show that RIM1α, 2α, 2β, and 2γ are ubiquitously present throughout the brain in overlapping, but non-identical expression patterns (Figure 1). RIM1α is the most abundant isoform that appears to be expressed uniformly. RIM2α is also present in all brain areas, but at lower levels; we observed high levels of RIM2α in a small subset of neurons, such as olfactory bulb granule cells, hippocampal dentate gyrus granule cells, and cerebellar granule cells. RIM2β and RIM2γ are also expressed ubiquitously in the brain, again with high levels in a subset of neurons, for example the thalamus for RIM2β and the medulla oblongata for RIM2γ. Immunoblotting of protein homogenates from single RIM1α and RIM2α KO mice verified the differential distribution and divergent expression levels of RIM1α and RIM2α in the brain (Figure 2D). These findings suggest that in some neurons, all RIM isoforms appear to be present, most conspicuously cerebellar granule cells, whereas in other neurons, one isoform predominates, for example, hippocampal CA1 neurons that primarily express RIM1α, or olfactory bulb granule cells that primarily express RIM2α (Figure 1B).
Essential functions of RIM2α
Whereas deletion of RIM1α causes large changes in the amount and regulation of neurotransmitter release (Schoch et al, 2002; Calakos et al, 2004), we found that the single deletion of RIM2α, the only other α-RIM, has milder phenotypic consequences compared with the deletion of RIM1α. RIM2α-deficient mice are impaired, but their release properties in hippocampal synapses appear to be normal (Supplementary Figure 3). It is puzzling that RIM1α but not RIM2α KO mice exhibit a loss of mossy fiber LTP in the CA3 region of the hippocampus (Castillo et al, 2002; Supplementary Figure 3E), because RIM1α and RIM2α are both highly expressed in the dentate granule cell neurons that supply the mossy fiber synapses, and both are phosphorylated by PKA at a site equivalent to serine 413 in RIM1α (Lonart et al, 2003). Furthermore, it has been postulated that α-RIMs play a role in regulating insulin release from pancreatic β-cells (Iezzi et al, 2000; Kashima et al, 2001). However, we were unable to detect any changes in body fat content or in blood glucose levels in RIM1α and 2α KO mice (Supplementary Figure 5).
Redundant functions of RIM1α and 2α
Although deletion of either RIM1α or RIM2α does not significantly impair survival of mice, deletion of both α-RIMs causes complete lethality at birth, whereas the absence of only three of the four α-RIM alleles—in either combination—induces an ∼40% lethality (Figure 3A). We detected no obvious developmental abnormalities, changes in synapse structure, or alterations in brain composition in the RIM1α/2α DKO mice (Figures 4 and 5), suggesting that these mice perished because of a functional and not a developmental defect.
Because of the severe loss of motor control in RIM1α/2α DKO mice, we studied the NMJ as a model system to understand the consequences of deleting both α-RIMs. As indirect indicators of defective synaptic transmission at the neuromuscular junction, we found an increased density of motoneurons in the ventral horn of the spinal cord, increased branching of presynaptic motor nerves, and increased numbers of irregularly distributed motor synapses on skeletal muscle fibers. These effects are considered to be consequences of an impaired function at the neuromuscular junction (Harris and McCaig, 1984; Dahm and Landmesser, 1991; Oppenheim, 1991). Direct measurement of synaptic transmission at the neuromuscular junction revealed profound defects. We found that spontaneous release was unchanged in frequency, but evoked synaptic transmission was strongly reduced (Figures 7 and 8). The miniature frequency could not be enhanced in the double mutants by increasing the Ca2+ concentration under depolarizing conditions (Figure 7), whereas the amplitudes and failure rate of evoked release were massively decreased and increased, respectively (Figure 8). This phenotype demonstrates that deletion of α-RIMs does not abolish spontaneous or Ca2+-dependent exocytosis, but severely and specifically impairs Ca2+-triggering of exocytosis. Interestingly, a recent study at the NMJ of Caenorhabditis elegans has shown that Unc-10/RIM localization is restricted to dense projections and that the number of membrane-contacting vesicles is reduced in unc10-deficient animals (Weimer et al, 2006). It was therefore hypothesized that the Rab3–unc10/RIM–unc13/Munc13 tripartite complex localizes primed synaptic vesicles in close proximity of calcium channel containing dense projections. Consistent with a loss of synaptic transmission, the pattern of synapses in the NMJs of DKO mice exhibited morphological changes that are typically observed upon complete block of synaptic transmission in NMJs (Figure 6; Misgeld et al, 2002; Brandon et al, 2003).
Munc13-deficient NMJs also display an impairment of evoked transmitter release similar to NMJs lacking α-RIMs. But different from NMJs lacking α-RIMs, Munc13-deficient NMJs exhibit an increased frequency of spontaneous transmitter release (Varoqueaux et al, 2005). Moreover, NMJs deficient for the SNARE protein SNAP-25 also exhibit increased spontaneous transmitter release but lack evoked release (Washbourne et al, 2002). Based on the observation of decreased evoked and increased spontaneous release in Munc13 and SNAP-25 KO mice, it was hypothesized that Munc13-mediated priming via SNARE proteins is essential for the majority of synaptic vesicles at the NMJ, whereas a small subpopulation of vesicles undergo spontaneous Munc13-independent priming (Varoqueaux et al, 2005). The increase in mini-frequency observed in Munc13 and SNAP-25 KO mice was then explained by an increase in the number of synapses in mutant NMJs. However, NMJs lacking α-RIMs exhibited the same morphological changes (increase in number of motoneurons, in branching of presynaptic nerve fibers, and in neuromuscular synapse formation) and the same loss of evoked release as Munc13-deficient synapses, but the mini-frequency is not increased. These results thus suggest that the increase in mini-frequency in Munc13- and SNAP-25-deficient synapses is not due to an increased number of synaptic contacts in the NMJ, but could be a homeostatic response to the decrease in neurotransmitter release. Thus, α-RIMs may act as downstream effectors of Munc13s, presumably via the N-terminal interaction between these two proteins (Betz et al, 2001; Dulubova et al, 2005).
Our study indicates that in some synapses as at the NMJ, loss of either RIM1α or RIM2α can be compensated by the respective other, whereas in hippocampal excitatory and inhibitory neurons, RIM1α can compensate for RIM2α but not vice versa. A similar result has been obtained in the analysis of Munc13-1/-2-deficient neurons, which show a complex pattern of redundancy (Varoqueaux et al, 2002). In GABAergic hippocampal neurons, both isoforms are redundant; however, in glutamatergic neurons, the presence of Munc13-1 is required. Furthermore, Munc13-1- or Munc13-2-equipped synapses exhibit diverging short-term plasticity characteristics in spite of their high structural homology (Rosenmund et al, 2002). Nevertheless, the molecular mechanisms underlying this phenotype are still unclear. As RIM1α and RIM2α are also structurally highly homologous, interact with the same set of proteins. and are both PKA substrates, a detailed gain-of-function and loss-of-function analysis in RIM1α/2α-deficient neurons can now address the molecular basis that determines their individual and redundant functions. However, our study clearly demonstrates a severe defect in synaptic transmission that can at least partially explain the dramatic lethal phenotype that we observe in the absence of α-RIMs.
Materials and methods
Generation and characterization of RIM2α KO mice
The targeting vector (Figure 2A) was constructed and employed to generate mutant embryonic R1 stem cells (Nagy et al, 1993) using standard procedures (Rosahl et al, 1995) (see Supplementary data for a detailed description). Mice were generated by blastocyst injections, and the floxed fifth exon was removed by cre-mediated recombination in the germ line (O'Gorman et al, 1997). Mice were bred using standard mouse husbandry, and genotyped by PCR. RIM1α mutant alleles were genotyped as described (Schoch et al, 2002).
In situ hybridizations were performed on cryostat sections from the brains of male Wistar rats (4-week-old; see Supplementary data for detailed procedures) with 35S-labeled oligonucleotides. Antisense oligonucleotides used corresponded to bp 952–907 and 1381–1337 of RIM1α (GenBank acc. no. NM_052829.1); bp 536–491 and 745–701 of RIM2α (GenBank acc. no. NM_053945.1); bp 111–67 and 194–150 of RIM2β (GenBank acc. no. AF548738.1); bp 311–267 and 337–293 of RIM2γ (GenBank acc. no. NM_145881.1). Oligonucleotide sequences (45-mers, 50–60% GC, low hairpin and dimer formation probability) were selected by using Oligo 6.7 (Cascade, CO, USA). In control experiments, hybridizations were performed with a 1000-fold excess of the respective unlabeled oligonucleotide. The hybridization signal was visualized either directly on Kodak BioMax film (Eastman Kodak, New Haven, CT, USA) or after dipping in the photographic emulsion (LM1; Amersham Biosciences) and development in Kodak D-19 developer (Eastman Kodak). For analysis with bright-field optics, sections were counterstained with H&E.
Protein quantifications were performed on brain homogenates from four 2-month-old RIM2α littermate KO and wild-type mice, and four E18.5 RIM1/2α DKO mice using littermate controls. Quantitations were performed by immunoblotting using 125I-labeled secondary antibodies and PhosphoImager (Fuji) detection with annexin, VCP, and α-tubulin as internal standards (Rosahl et al, 1995; Schoch et al, 2002). PhosphoImager data were then quantified with the image data analyzer software AIDA 3.20.116 (raytest GmbH, Germany).
Microscopy
Whole-body staining of eviscerated E18.5 embryos was performed after immersion fixation in absolute ethanol for 4 days and acetone for 3 days (see Supplementary data). For light and electron microscopy, brain sections from 2-month-old and E18.5 mice were examined using standard procedures (Rosahl et al, 1995) (see Supplementary data).
Hippocampal electrophysiology
Vibratome-cut transverse hippocampal slices (400-μm thick) from 3- to 5-week-old mice were used for field recordings using standard procedures (Castillo et al, 2002; Schoch et al, 2002).
NMJ electrophysiology
NMJ recordings were carried out at room temperature using intracellular sharp glass microelectrodes filled with 3 M KCl on acutely isolated phrenic nerve/diaphragm preparations at E18.5. Muscles were dissected in oxygenated normal mouse Ringer's solution (136.8 mM NaCl, 5 mM KCl, 12 mM NaHCO3, 1 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, and 11 mM glucose; pH 7.4) (Liley, 1956), pinned to Sylgard coated dishes, and continuously perfused with oxygenated Ringer's solution. Evoked EPPs were elicited by suprathreshold stimulation (5 V, 1 ms) of the phrenic nerve via a suction electrode. Data were collected with an AxonClamp 2B amplifier, digitized, and analyzed using PClamp (v. 9.0) (Axon Instruments Inc.) and MiniAnalysis (Synaptosoft Inc.).
Glucose tolerance test and body fat measurement
Mice at 10–12 weeks of age were fasted overnight with free access to water. Glucose was injected into the peritoneal cavity at 2 mg/g of body weight. Blood samples were collected from the tail vein immediately before and 15, 30, 60, 90, and 120 min after glucose injection. Plasma was prepared by low-speed centrifugation (5000 g for 10 min) and used for glucose measurement using Sigma Diagnostics glucose (Trinder) reagent. Body fat content in mice was measured with a Bruker Minispec mq7.5 NMR analyzer (Bruker Optics, TX, USA).
Miscellaneous
All chemicals were of the highest available purity and were purchased from standard sources. SDS–PAGE and immunoblotting were performed following standard procedures (Laemmli, 1970; Towbin et al, 1979) using the antibodies described (Schoch et al, 2002).
Statistical analysis
Data are presented as means±s.e.m. Statistical analysis of quantitative Western blots was performed using one sample t-test (GraphPad Prism) to determine the significance of the difference from mean to control level (normalized to 100% for each blot). Differences were considered significant at P<0.05. EM: Statistical analysis was performed with Student's t-tests (two-sided, unpaired data with unequal variances); data represent means and s.d.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Materials
Acknowledgments
We thank Dr A Ho, Dr M Khvotchev, Dr S Chandra, Dr A Becker, and Dr H Beck for helpful scientific discussions, support, and advice, S Bauerkämper, I Kornblum, A Roth, and E Borowicz for excellent technical assistance, and N Hamlin and J Mitchell for animal care. This work was supported by the German Research Foundation (Emmy Noether Fellowship to SS) and a grant to FS (SFB 530 TPC11), by the Swiss National Science Foundation (Advanced Postdoctoral Fellowship to PSK) and local funding (BONFOR to SS). The PEC laboratory is funded by the NIH and the PEW Biomedical Program.
References
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N (2006) Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem 281: 19720–19731 [DOI] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N (2001) Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron 30: 183–196 [DOI] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D'Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23: 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Hofmann K, Hata Y, Südhof TC (1995) Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem 270: 25273–25280 [DOI] [PubMed] [Google Scholar]
- Calakos N, Schoch S, Südhof TC, Malenka RC (2004) Multiple roles for the active zone protein RIM1alpha in late stages of neurotransmitter release. Neuron 42: 889–896 [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC (2002) RIM1alpha is required for presynaptic long-term potentiation. Nature 415: 327–330 [DOI] [PubMed] [Google Scholar]
- Coppola T, Magnin-Luthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R (2001) Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J Biol Chem 276: 32756–32762 [DOI] [PubMed] [Google Scholar]
- Dahm LM, Landmesser LT (1991) The regulation of synaptogenesis during normal development and following activity blockade. J Neurosci 11: 238–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresbach T, Qualmann B, Kessels MM, Garner CC, Gundelfinger ED (2001) The presynaptic cytomatrix of brain synapses. Cell Mol Life Sci 58: 94–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Südhof TC, Rizo J (2005) A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J 24: 2839–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AJ, McCaig CD (1984) Motoneuron death and motor unit size during embryonic development of the rat. J Neurosci 4: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi M, Regazzi R, Wollheim CB (2000) The Rab3-interacting molecule RIM is expressed in pancreatic beta-cells and is implicated in insulin exocytosis. FEBS Lett 474: 66–70 [DOI] [PubMed] [Google Scholar]
- Johnson S, Halford S, Morris AG, Patel RJ, Wilkie SE, Hardcastle AJ, Moore AT, Zhang K, Hunt DM (2003) Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7). Genomics 81: 304–314 [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Südhof TC (2005) RIM function in short- and long-term synaptic plasticity. Biochem Soc Trans 33: 1345–1349 [DOI] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S (2001) Critical role of cAMP–GEFII–Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem 276: 46046–46053 [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001) A post-docking role for active zone protein Rim. Nat Neurosci 4: 997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- Liley AW (1956) An investigation of spontaneous activity at the neuromuscular junction of the rat. J Physiol 132: 650–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ (2003) Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115: 49–60 [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635–648 [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC (1993) Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA 90: 8424–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y (1997) Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci USA 94: 14602–14607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y (2002) Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J Cell Biol 158: 577–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453–501 [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S (2000) cAMP–GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2: 805–811 [DOI] [PubMed] [Google Scholar]
- Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Südhof TC, Nestler EJ (2004) The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. Neuron 42: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC (1995) Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375: 488–493 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Rettig J, Brose N (2003) Molecular mechanisms of active zone function. Curr Opin Neurobiol 13: 509–519 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS (2002) Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 33: 411–424 [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC (2002) RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415: 321–326 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294: 1117–1122 [DOI] [PubMed] [Google Scholar]
- Serra-Pages C, Medley QG, Tang M, Hart A, Streuli M (1998) Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem 273: 15611–15620 [DOI] [PubMed] [Google Scholar]
- Simsek-Duran F, Linden DJ, Lonart G (2004) Adapter protein 14-3-3 is required for a presynaptic form of LTP in the cerebellum. Nat Neurosci 7: 1296–1298 [DOI] [PubMed] [Google Scholar]
- Sun L, Bittner MA, Holz RW (2003) Rim, a component of the presynaptic active zone and modulator of exocytosis, binds 14-3-3 through its N terminus. J Biol Chem 278: 38301–38309 [DOI] [PubMed] [Google Scholar]
- Takao-Rikitsu E, Mochida S, Inoue E, Deguchi-Tawarada M, Inoue M, Ohtsuka T, Takai Y (2004) Physical and functional interaction of the active zone proteins, CAST, RIM1, and Bassoon, in neurotransmitter release. J Cell Biol 164: 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA 99: 9037–9042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sons MS, Plomp JJ, Brose N (2005) Aberrant morphology and residual transmitter release at the munc13-deficient mouse neuromuscular synapse. Mol Cell Biol 25: 5973–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hu B, Zimmermann B, Kilimann MW (2001) Rim1 and rabphilin-3 bind Rab3-GTP by composite determinants partially related through N-terminal alpha-helix motifs. J Biol Chem 276: 32480–32488 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu X, Biederer T, Südhof TC (2002) A family of RIM-binding proteins regulated by alternative splicing: implications for the genesis of synaptic active zones. Proc Natl Acad Sci USA 99: 14464–14469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC (1997) Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 388: 593–598 [DOI] [PubMed] [Google Scholar]
- Wang Y, Südhof TC (2003) Genomic definition of RIM proteins: evolutionary amplification of a family of synaptic regulatory proteins(small star, filled). Genomics 81: 126–137 [DOI] [PubMed] [Google Scholar]
- Wang Y, Sugita S, Südhof TC (2000) The RIM/NIM family of neuronal C2 domain proteins. Interactions with Rab3 and a new class of Src homology 3 domain proteins. J Biol Chem 275: 20033–20044 [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19–26 [DOI] [PubMed] [Google Scholar]
- Weimer RM, Gracheva EO, Meyrignac O, Miller KG, Richmond JE, Bessereau JL (2006) UNC-13 and UNC-10/rim localize synaptic vesicles to specific membrane domains. J Neurosci 26: 8040–8047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Materials