

Abstract
TRPV4 is a widely expressed member of the transient receptor potential (TRP) family that facilitates Ca2+ entry into nonexcitable cells. TRPV4 is activated by several stimuli, but it is largely unknown how the activity of this channel is terminated. Here, we show that ubiquitination represents an important mechanism to control the presence of TRPV4 at the plasma membrane. Ubiquitination of TRPV4 is dramatically increased by the HECT (homologous to E6-AP carboxyl terminus)-family ubiquitin ligase AIP4 without inducing degradation of this channel. Instead, AIP4 promotes the endocytosis of TRPV4 and decreases its amount at the plasma membrane. Consequently, the basal activity of TRPV4 is reduced despite an overall increase in TRPV4 levels. This mode of regulation is not limited to TRPV4. TRPC4, another member of the TRP channel family, is also strongly ubiquitinated in the presence of AIP4, leading to the increased intracellular localization of TRPC4 and the reduction of its basal activity. However, ubiquitination of several other TRP channels is not affected by AIP4, demonstrating that AIP4-mediated regulation is a unique property of select TRP channels.
Keywords: endocytosis, transient receptor potential, TRPC4, TRPV4, ubiquitination
Introduction
Transient receptor potential (TRP) proteins constitute a family of structurally related cation channels that are primarily responsible for Ca2+ entry into nonexcitable cells (Montell et al, 2002). Based on sequence similarities, they have been divided into several subfamilies, including the TRPC (canonical), TRPV (vanilloid receptor), TRPM (melastatin), and TRPP (polycystin) subfamilies. TRP proteins differ substantially in selectivity and activation mechanisms, and have been implicated in diverse biological processes. TRPV4, a nonselective cation channel of the TRPV subfamily with a slight preference for calcium ions, can be activated by several stimuli, including hypotonicity, phorbol esters, arachidonic acid, and heat (reviewed by Nilius et al, 2004). TRPV4 knockout mice show a moderate impairment in osmoregulation and antidiuretic hormone release (Liedtke and Friedman, 2003; Mizuno et al, 2003). As TRPV4 is highly expressed in the kidney and paraventricular regions of the central nervous system (Liedtke et al, 2000; Strotmann et al, 2000; Wissenbach et al, 2000; Tian et al, 2004), these findings hint towards a role of TRPV4 as a cellular or systemic osmosensor. All TRP proteins comprise six-transmembrane (TM) segments, a pore region situated between TM5 and TM6, as well as cytosolic N- and C-terminal tails of variable length. The functional TRP channels require homo- or heteromeric assembly of four subunits. TRPV4 preferably forms homooligomers, a process likely mediated by the amino-terminal ankyrin repeats (Hellwig et al, 2005). The naturally occurring splice variants of TRPV4 with deletions in these repeats do not oligomerize, and fail to reach the plasma membrane (Arniges et al, 2006).
The presence of channels and receptors at the cell membrane must be tightly controlled to ensure correct timing and duration of signals relayed by such proteins. Adjusting the rate of endocytosis or exocytosis of functional plasma membrane proteins can exert this control. Defects in the regulatory mechanisms may result in their aberrant activation, and, in consequence, may be deleterious to the cell and the whole organism. For instance, insufficient removal of the epithelial sodium channel (ENaC) from the cell surface is the cause of the Liddle's syndrome, a hereditary form of human hypertension. Under normal conditions, the ENaC channel subunits are efficiently endocytosed and degraded in the lysosome, as a result of their ubiquitination by HECT (homologous to E6-AP carboxyl terminus)-family ubiquitin ligases (Staub et al, 1997, see below). Similarly, many G protein-coupled receptors and receptor tyrosine kinases are internalized and degraded in the lysosome upon activation (Katzmann et al, 2002).
Internalized plasma membrane proteins destined for lysosomal degradation traffic through early and late endosomes. The crucial step seems to be successful targeting into the lumenal vesicles of multivesicular bodies (MVBs; Katzmann et al, 2002). Proteins that are not properly sorted into the MVB vesicles escape degradation and can recycle back to the plasma membrane. Marking proteins with a single ubiquitin moiety on one (monoubiquitination) or more lysine residues (multiubiquitination) is an important signal for both internalization and sorting into the MVB (Hicke, 2001; d'Azzo et al, 2005). Such modification differs from generating chains of four or more ubiquitins on proteins (polyubiquitination), a signal for proteasomal degradation. The attachment of ubiquitin is carried out by the sequential action of three classes of enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and finally by a ubiquitin ligase (E3), which provides substrate specificity (Pickart, 2001). Ubiquitin ligases can be divided into two families, RING (really interesting new gene) and HECT ligases (d'Azzo et al, 2005). RING-type ligases serve as adaptors between ubiquitin-conjugating enzymes and substrate proteins. In contrast, ubiquitin ligases of the HECT family participate directly in the catalytic reaction by accepting the ubiquitin moiety on a conserved cysteine residue before transferring it to the substrate.
Activation of several TRP channels correlates with their increased appearance at the plasma membrane (Bezzerides et al, 2004; Cayouette et al, 2004; Morenilla-Palao et al, 2004; Singh et al, 2004), although the idea of a direct coupling between these two events has been challenged recently (Smyth et al, 2006). These TRP channels reside in vesicles just underneath the plasma membrane and translocate to the cell surface upon stimulation. Supporting this concept, TRPV1 and TRPC3 interact and colocalize with proteins of the exocytic machinery (Morenilla-Palao et al, 2004; Singh et al, 2004). Thus, the delivery of at least some TRP channels to the plasma membrane appears to be a regulated process. However, much less is known about the regulatory mechanisms that curtail the activity of TRP channels at the plasma membrane. In the present study, we identified AIP4, a member of the HECT family of ubiquitin ligases, to regulate the cell-surface expression of TRPV4. AIP4 mediates the ubiquitination of TRPV4 and other TRP channels. However, TRPV4 ubiquitination does not target this protein for degradation, but affects its endocytic trafficking.
Results
Multiubiquitination of TRPV4 channel is increased in the presence of AIP4 ubiquitin ligase
We speculated that TRPV4, similar to several other plasma membrane proteins, is modified by the covalent attachment of one or more ubiquitin moieties. As TRPV4 contains a sequence resembling the PY motif recognized by HECT-family ubiquitin ligases (see below), we further predicted that such a modification is carried out by a member of this family. To test this hypothesis, we performed an in vivo ubiquitination assay in human embryonic kidney (HEK) 293T cells. The cells were transiently transfected with plasmids coding for His-tagged TRPV4 (TRPV4-His), FLAG-tagged ubiquitin (FLAG-ubiquitin), and one of the three different HECT-family ligases: Nedd4, Nedd4-2, or AIP4. TRPV4-His was purified from cell lysates on Ni-NTA beads. Denaturing conditions were applied to minimize protein degradation, deubiquitination, and to prevent non-covalent protein–protein interactions. Western blot analysis with anti-FLAG antibodies revealed a smear in the region above 100 kDa (Figure 1A, lane 2), demonstrating that TRPV4 is ubiquitinated in HEK 293T cells. This basal level of ubiquitination was dramatically increased when TRPV4 was coexpressed with AIP4 (Figure 1A, lane 3). In contrast, there was only a marginal increase of TRPV4 ubiquitination in the presence of Nedd4 and Nedd4-2. This result led us to investigate the AIP4-mediated ubiquitination of TRPV4 in more detail. We found that wild-type AIP4 (AIP4 WT) typically led to the accumulation of ubiquitinated TRPV4 species that migrated on SDS–PAGE predominantly in a region between 100 and 150 kDa (Figure 1B, lane 3). In contrast, overexpression of catalytically inactive dominant-negative AIP4 (AIP4 DN) mutant did not increase, but moderately decreased the level of TRPV4 ubiquitination (Figure 1B, lane 4). These results could be reproduced in the absence of exogenous ubiquitin, using P4D1 α-ubiquitin antibodies to detect the modified TRPV4 (Figure 1C). To further explore the type of TRPV4 ubiquitination, we took advantage of the fact that the P4D1 antibody detects all ubiquitinated proteins, whereas another monoclonal antibody, FK1, recognizes only polyubiquitinated proteins (Haglund et al, 2003). The ubiquitinated TRPV4, both at the basal level and in the presence of overexpressed AIP4, could be detected only by P4D1 antibodies, implying multi- rather than polyubiquitination as the major mode of ubiquitination (Figure 1D). In contrast, increased ubiquitination of c-Jun was detectable by either antibody, revealing the polyubiquitination of c-Jun in the presence of AIP4 or MEKK1 (Gao et al, 2004, see below). Although we cannot exclude that a fraction of TRPV4 is polyubiquitinated in the presence of AIP4, it was not detectable by FK1 antibodies.
Figure 1.

The AIP4 ubiquitin ligase increases multiubiquitination of TRPV4. (A) HEK 293T cells were transfected with plasmids encoding TRPV4-His, FLAG-ubiquitin, and three different ubiquitin ligases of the HECT family, as indicated. The TRPV4-His protein purified on Ni-NTA beads was analyzed by Western blotting for ubiquitin conjugation using α-FLAG antibodies. (B) The in vivo ubiquitination assay was performed as above, using constructs, as indicated, to compare the effects of AIP4 WT and AIP4 DN on the ubiquitination of TRPV4. (C) The cells were transfected as in (B), except that the plasmid encoding FLAG-ubiquitin was omitted. Ubiquitinated TRPV4 was detected with α-ubiquitin P4D1 antibodies. (D) The TRPV4-His protein purified from transfected HEK 293T cells was analyzed by Western blotting using monoclonal antibodies P4D1, which recognize all ubiquitinated proteins, and FK1, which recognize only polyubiquitinated proteins. As control, His-tagged c-Jun was purified from cells overexpressing AIP4 WT or MEKK1 CA. A nonspecific band is marked with an asterisk. (E) HEK 293T cells were transfected with the indicated plasmids and used for an in vivo ubiquitination assay. The ubiquitinated TRPV4 was detected with α-FLAG antibodies. The bars represent the level of TRPV4 ubiquitination in MEKK1 CA-expressing cells (lanes 5–7). For quantifications, the amount of ubiquitinated TRPV4 was normalized to the amount of the non-ubiquitinated form; the signal in lane 8 was treated as background. The depletion of endogenous AIP4 by RNAi (lane 7 in comparison to lane 5) is shown in the bottom panel.
We next investigated whether activation of endogenous AIP4 in HEK 293T cells also leads to increased ubiquitination of TRPV4. The catalytic activity of AIP4 is stimulated by JNK1-mediated phosphorylation (Gao et al, 2004). In agreement with this observation, TRPV4 ubiquitination was increased in the presence of the constitutively active (CA) but not the dominant-negative (DN) mutant of MEKK1, a kinase that activates the JNK pathway (Figure 1E). This increase was inhibited by AIP4 DN or by depletion of endogenous AIP4 by RNAi. The weaker effect of the knock-down approach may result from incomplete depletion of AIP4 and/or the involvement of other ubiquitin ligases activated by MEKK1. The stronger effect of AIP4 DN may be caused by its interaction with TRPV4 (Supplementary Figure 1), preventing the access of active ubiquitin ligases.
The amino-terminus of TRPV4 is sufficient for AIP4-mediated ubiquitination
We next attempted to determine the motifs in TRPV4 that mediate the ubiquitination of this channel by AIP4. TRPV4 contains in its N-terminal tail a sequence similar to the extended PY motif with consensus sequence PPxYxxL found in all three subunits of the ENaC channel (Figure 2A; Kanelis et al, 2001). In TRPV4 however, the conserved proline at position +1 is changed to glycine. To test whether this PY-like motif in TRPV4 still functions as a determinant for AIP4-mediated ubiquitination, we deleted amino acids 411–437 of TRPV4, entailing this motif. The resulting plasmid (TRPV4-V5/His ΔPY) was used in the in vivo ubiquitination assay, using HEK 293T cells. As shown in Figure 2B, TRPV4 lacking the potential PY motif was still efficiently ubiquitinated in the presence of AIP4. Thus, this motif either does not function as a recognition sequence for AIP4-depedent ubiquitination or it is redundant.
Figure 2.
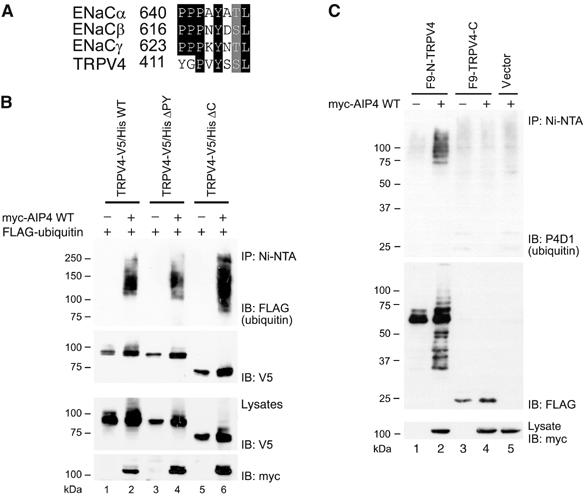
The N-terminus, but not the C-terminus, is required for the ubiquitination of TRPV4. (A) The sequence alignment between extended PY motifs of ENaC channel subunits: α (Swissprot accession number P37088), β (P51168) and γ (P51170) and a similar region found in the N-terminus of murine TRPV4 (Q9EPK8). (B) HEK 293T cells were transfected with plasmids encoding V5/His-tagged WT TRPV4 and TRPV4 mutants lacking either the PY-like motif (ΔPY) or the whole C-terminus (ΔC), along with plasmids encoding myc-AIP4 and FLAG-ubiquitin, as indicated. The cell lysates were incubated with Ni-NTA beads and the purified proteins were resolved on SDS–PAGE. Following Western blotting, V5/His-tagged TRPV4 proteins were detected with α-V5 antibodies and their ubiquitin-conjugated fractions with α-FLAG antibodies. (C) HEK 293T cells were transfected with plasmids encoding FLAG/His9-tagged N-terminus of TRPV4 (F9-N-TRPV4) or similarly tagged C-terminus of TRPV4 (F9-TRPV4-C), along with the plasmid encoding myc-AIP4, as indicated. Cell lysates were treated as in (B). The purified N- and C-termini of TRPV4 were detected with α-FLAG antibodies and the conjugated endogenous ubiquitin with P4D1 α-ubiquitin antibodies.
The N- and C-terminal domains of TRPV4 are located within the cytoplasm, and could both contain the motifs required for AIP4-dependent ubiquitination. To extend our analysis, we deleted the entire C-terminal tail of TRPV4. The expressed truncated protein (TRPV4-V5/His ΔC) was still modified by FLAG-ubiquitin in an AIP4-dependent fashion (Figure 2B). This implied that the C-terminus of TRPV4 is dispensable for AIP4-dependent ubiquitination. To confirm this conclusion, we expressed the N- and C-terminal intracellular domains of TRPV4 in HEK 293T cells. Only the N-terminus, but not the C-terminus, was conjugated to endogenous ubiquitin in the presence of AIP4, suggesting that the N-terminal tail of TRPV4 carries the determinants necessary for the ubiquitination of TRPV4 (Figure 2C).
AIP4 does not accelerate the rate of TRPV4 degradation in HEK 293T cells
Ubiquitination of proteins frequently targets them for accelerated degradation. Therefore, we investigated whether AIP4-mediated ubiquitination shortens the half-life of TRPV4. HEK 293T cells were cotransfected with plasmids coding for TRPV4-V5/His, myc-AIP4, and GFP. Surprisingly, the steady-state level of TRPV4 was reproducibly increased in untreated cells in the presence of AIP4 WT (Figures 3A and 5). This effect required the ubiquitin ligase activity of AIP4, as it was not observed in the presence of DN AIP4. Inhibition of protein synthesis by cycloheximide led to a reduction in TRPV4 levels that was comparable between the AIP4-expressing and control cells, confirming that AIP4 does not increase the degradation rate of TRPV4 (Figure 3B and C). We probed whether inhibition of proteasomal or lysosomal degradation, with MG132 and chloroquine, respectively, leads to the accumulation of ubiquitinated TRPV4. MG132 treatment caused accumulation of a ubiquitinated TRPV4 fraction with a molecular weight >150 kDa, both in the absence and presence of AIP4 WT (Figure 3D). The appearance of higher molecular weight TRPV4 species points towards polyubiquitination of TRPV4 rather than multiubiquitination, appears to occur independently of AIP4, and is probably mediated by a different ubiquitin ligase. However, the fraction of ubiquitinated TRPV4 with a lower molecular weight (100–150 kDa), whose accumulation is AIP4-dependent, was influenced neither by MG132 nor by chloroquine treatment. Taken together, these results demonstrate that AIP4 enhances the ubiquitination, but does not facilitate the degradation of TRPV4 in HEK 293T cells.
Figure 3.
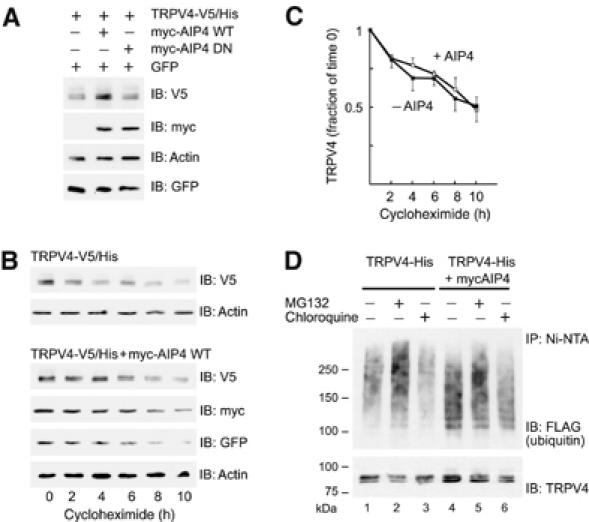
AIP4 does not accelerate the degradation of TRPV4. (A) HEK 293T cells were transfected with plasmids encoding TRPV4-V5/His, GFP, and myc-AIP4 WT or dominant-negative (DN), as indicated. Shown are steady-state levels of TRPV4 in comparison to cotransfected GFP and to actin. (B) The cells transfected as above, with and without myc-AIP4 WT, were treated with 30 μg/ml cycloheximide for 0–10 h. The cell lysates were resolved on SDS–PAGE and the protein turnover was analyzed by Western blotting with α-V5, α-GFP, α-myc, and α-actin antibodies. The amount of cell lysates loaded on gels was adjusted for the initial difference in the TRPV4-V5/His protein levels between myc-AIP4-expressing and control cells. (C) The graph depicts the fraction of the remaining TRPV4-V5/His protein at 2–10 h after cycloheximide treatment for each condition analyzed in (B). Note that, for clarity, the error bars (s.e.m., n=3) point to opposite directions for each curve. (D) HEK 293T cells were transfected with plasmids encoding TRPV4-His and FLAG-ubiquitin, either with or without a plasmid encoding myc-AIP4 WT, as indicated. Cells were treated with 12 μM MG132 or 200 μM chloroquine for 200 min. The lysates were incubated with Ni-NTA beads and the purified proteins analyzed by Western blotting. TRPV4 was detected with α-TRPV4 antibodies and the ubiquitin conjugated to TRPV4 with α-FLAG antibodies.
Figure 5.
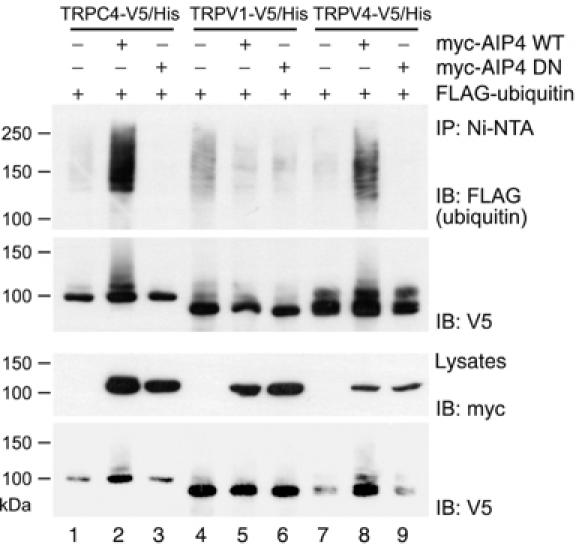
AIP4 ubiquitin ligase is involved in the ubiquitination of both TRPV4 and TRPC4. HEK 293T cells were transfected with plasmids encoding V5/His-tagged TRPC4, TRPV1, or TRPV4 along with plasmids encoding FLAG-ubiquitin and either catalytically active (WT) or inactive (DN) versions of myc-AIP4. The TRP channels were purified from cell lysates on Ni-NTA beads and resolved on SDS–PAGE. Following Western blotting, V5-tagged TRP channels were detected using α-V5 antibodies and the ubiquitin conjugates with α-FLAG antibodies.
TRPV4 is a stable protein
To further address the kinetics of TRPV4 turnover, we examined its stability in Madin–Darby canine kidney (MDCK) cells, which express native TRPV4. As our antisera did not detect the native channel, we created MDCK cell lines stably expressing murine TRPV4, and confirmed that TRPV4 is also ubiquitinated in these cells (Supplementary Figure 2A). Yet, treatment of MDCK cells with cycloheximide revealed only a moderate decrease of TRPV4 levels after 7 h in comparison to actin (Supplementary Figure 2B), corroborating that despite ubiquitination, TRPV4 is not rapidly degraded. To investigate whether TRPV4 is subjected to proteasomal or lysosomal degradation in MDCK cells, we treated MDCK-TRPV4 cells with MG132 and with chloroquine. However, neither treatment had any significant effect on TRPV4 protein concentrations (Supplementary Figure 2B, lanes 4–8), indicating that under normal conditions TRPV4 is not efficiently targeted to either degradation pathway. Subsequently, we analyzed the subcellular localization of stably expressed murine TRPV4 by confocal microscopy using HeLa cells, because a greater selection of marker antibodies is available for human cell lines. The localization of TRPV4, detected by an affinity-purified rabbit α-TRPV4 antibody, resembled that of syntaxin-4, a t-SNARE protein associated with the plasma membrane (Supplementary Figure 2C, panels a–c). These two proteins also partially colocalized, indicating that in HeLa cells TRPV4 is present at the plasma membrane. In contrast, we did not observe any colocalization between TRPV4 and Lamp-1, a marker of late endosomes and lysosomes, even after a prolonged treatment with chloroquine (Supplementary Figure 2C, panels d–f). There was no significant colocalization between TRPV4 and Golgin-97 (marker of Golgi stacks), CI-MPR (marker of late endosomes), and only a low level of colocalization with EEA1, a marker of early endosomes (data not shown). This analysis suggests that TRPV4 in HeLa cells localizes predominantly to the plasma membrane and to a subcortical region, but is not readily detectable in late endosomal or lysosomal compartments.
AIP4 decreases the amount of TRPV4 at the plasma membrane
As ubiquitination of plasma membrane proteins can serve as an internalization signal (Haglund et al, 2003), we examined the amount of TRPV4 at the cell surface in the absence and presence of AIP4. HEK 293T cells expressing TRPV4 with a V5 tag engineered into the first extracellular loop (TRPV4-V5-loop) were incubated with α-V5 antibodies at 4°C for 45 min, washed, and fixed. Cell surface expression of TRPV4-V5-loop was measured by enzyme-linked immunosorbent assay (ELISA), using alkaline phosphatase (AP)-coupled secondary antibodies. Cells not expressing any V5-tagged protein or cells expressing TRPV4 tagged with V5 at the C-terminus (TRPV4-V5-C-term) were used as controls. There was a clear difference in AP activity between cells expressing the two controls and cells expressing the TRPV4-V5-loop (Figure 4A). These results confirmed both the specificity of the α-V5 antibody labeling reaction and the predicted topology of TRPV4. When coexpressed with AIP4, the amount of TRPV4-V5-loop decreased at the plasma membrane, whereas the total amount of protein, analyzed by Western blotting, increased (Figure 4A). Analysis of the results from six independent experiments showed that the ratio of surface fraction to the total amount of TRPV4 was reduced to 31% in AIP4 coexpressing cells compared to control cells (Figure 4B). The ratio of surface TRPV4-V5-loop to the total amount of coexpressed GFP (representing the number of transfected cells) was also significantly decreased to 60.5% (Figure 4B). These results revealed that AIP4 decreases the surface fraction of TRPV4 despite an increase of total TRPV4 protein.
Figure 4.
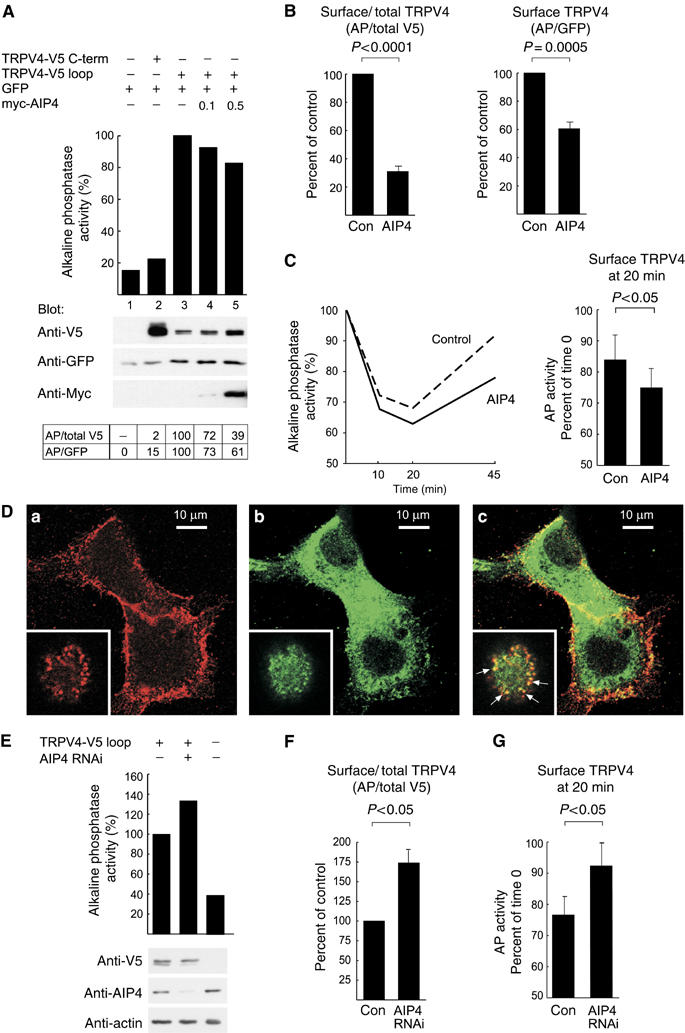
AIP4 decreases the amount of TRPV4 at the plasma membrane. (A) In the representative experiment shown, HEK 293T cells were transfected with plasmids encoding C-terminally V5-tagged TRPV4 (TRPV4-V5 C-term), TRPV4 tagged with V5 in the first extracellular loop (TRPV4-V5 loop), GFP and myc-tagged AIP4 (0.1 or 0.5 μg), as indicated. The transfected cells were incubated with α-V5 antibodies at 4°C, washed, and fixed. The amount of V5 epitope associated with the plasma membrane was measured by ELISA (AP activity). The total amounts of proteins were determined by Western blotting. Results obtained for cells not expressing any V5-tagged protein (lane 1) were treated as background. Results obtained for cells expressing TRPV4-V5 loop in the absence of myc-AIP4 (lane 3) were normalized to 100%. Ratios of AP activity to the total amount of V5 epitope (AP/total V5), and to the total amount of GFP (AP/GFP) are shown in the table. (B) Statistical analysis of six independent experiments, similar to the one presented in (A). The ratios of surface TRPV4 (measured by ELISA) to total TRPV4, as well as coexpressed GFP (analyzed by Western blotting), in the presence of myc-AIP4 (AIP4) relative to control (Con), are depicted. Statistical analysis was performed with one sample t-test; the calculated P-values are indicated. (C) HEK 293T cells expressing TRPV4-V5 loop, with and without myc-AIP4, were incubated with α-V5 antibodies at 4°C, washed, returned to the incubator for 0, 10, 20, or 45 min, and subsequently fixed. The amount of V5 epitope present at the plasma membrane was measured by ELISA (left panel). For both control and myc-AIP4-expressing cells, the AP activity at time 0 was set to 100%. A statistical analysis (means and s.e.m., n=3) of the amount of TRPV4 at the cell surface at the 20-min time point relative to time 0 is shown on the right. The P-value was calculated with a paired t-test. (D) Confocal immunofluorescence analysis of HEK 293T cells expressing TRPV4-V5 loop and myc-AIP4. The cells were incubated with α-V5 antibodies at 4°C, washed, returned to the incubator for 20 min, and subsequently fixed. The bound α-V5 antibody was detected with a Cy3-conjugated donkey α-mouse IgG (a; red). The fixed cells were also stained with α-AIP4 antibodies followed by Alexa488-conjugated donkey α-goat IgG (b; green). The overlay of both pictures is shown in panel c. The colocalization of TRPV4-V5 loop and myc-AIP4 in vesicular structures is best seen at the top section of the cells, as shown in the insets (slightly enlarged relative to main pictures). The arrows in the overlay picture point to several vesicles, where TRPV4-V5 loop and mycAIP4 colocalize. (E) Depletion of endogenous AIP4 in HEK 293T cells by coexpression of the α-AIP4 shRNA construct (AIP4 RNAi) increases the surface localization of TRPV4. A representative experiment is shown. Surface expression of TRPV4-V5 loop was measured by ELISA; total levels of proteins were analyzed by Western blotting. (F) Measurement of the ratio of surface TRPV4 to its total levels in cells depleted of AIP4 (AIP4 RNAi) relative to control cells (Con). Depicted are means and s.e.m. from four experiments. The P-value was calculated with one sample t-test. (G) Depletion of AIP4 results in decreased levels of internalized TRPV4, as measured at 20-min chase time point. Depicted are means and s.e.m. from three experiments performed and analyzed as in (C). A plasmid devoid of the α-AIP4 shRNA sequence was transfected as control.
Decreased surface expression could result either from attenuated trafficking of TRPV4 to the plasma membrane or its accelerated internalization. To distinguish between these two possibilities, we modified the ELISA analysis to measure TRPV4 endocytosis. TRPV4-V5-loop-expressing cells were incubated with α-V5 antibodies at 4°C, washed, and either fixed immediately or returned to the incubator for 10, 20, or 45 min before fixation. Subsequently, the AP activity was determined to follow the endocytosis of TRPV4. TRPV4 was partially removed from the cell surface within the first 20-min chase period, but reappeared after 45 min of the chase (Figure 4C). Significantly more TRPV4 was internalized at the 20-min time point in the presence of AIP4 (Figure 4C). We also analyzed the cells treated in a similar way by confocal immunofluorescence microscopy. This analysis showed that internalized TRPV4-V5-loop protein colocalizes with myc-AIP4 at the cell periphery (Figure 4D), indicating that AIP4 may participate directly in the endocytosis of TRPV4. To analyze whether endogenous AIP4 plays a role in the plasma membrane localization of TRPV4, we determined the turnover of TRPV4 at the surface of HEK 293T cells, in which AIP4 protein was depleted by RNAi. In these cells, the TRPV4-V5 loop protein was more abundant at cell surface as compared to control cells (Figure 4E). Analysis of four independent experiments showed significantly increased ratio of plasma membrane-localized TRPV4 to its total level (Figure 4F). Moreover, the fraction of internalized TRPV4 was significantly smaller in cells depleted of AIP4, measured at the 20-min chase time point (Figure 4G). Thus, AIP4 appears to play a role in TRPV4 trafficking by facilitating its internalization.
AIP4 is involved in the ubiquitination of other TRP-family members
The TRP channel superfamily consists of more than 20 members grouped into six subfamilies based on sequence similarities (Montell et al, 2002). We speculated that the AIP4-dependent ubiquitination is not restricted to TRPV4, but may represent a more general principle of TRP channel regulation. To answer this question, we chose TRPV1 as a second representative of the TRPV subfamily, TRPC4 and TRPC6 of the TRPC subfamily, and PKD2/TRPP2 of the atypical TRPP subfamily. Full-length proteins were tagged with a C-terminal V5/His. The in vivo ubiquitination assay in HEK 293T cells showed that TRPC4 ubiquitination was dramatically increased in the presence of AIP4 WT, but not in the presence of AIP4 DN (Figure 5). In contrast, the ubiquitination of TRPV1 (Figure 5), TRPC6, or TRPP2 (Supplementary Figure 3) was only marginally affected by coexpression of AIP4 WT or AIP4 DN. We also found that coexpression of MEKK1 CA led to a profound increase of TRPC4 ubiquitination; this effect was prevented by simultaneous coexpression of AIP4 DN (data not shown). This cross-talk between JNK signaling and TRP channel trafficking provides a novel insight into the regulation of TRP channel ubiquitination by mitogen-activated protein kinases.
AIP4 alters the subcellular localization of TRPC4
As TRPC4 was heavily ubiquitinated in the presence of AIP4, we examined the basal and receptor-induced entry of divalent cations in HEK 293 cells coexpressing AIP4. As shown by Mn2+-quench experiments in fura 2-loaded HEK 293 cells (Figure 6A), activation of TRPC4 C-terminally fused to the yellow fluorescent protein (TRPC4-YFP) by an endogenously expressed muscarinic receptor resulted in an acceleration of basal Mn2+ entry. When coexpressed with AIP4, the basal Mn2+ entry was significantly attenuated, whereas the receptor-induced activation appeared only slightly reduced (Figure 6B and C). To control for a possible acceleration of TRPC4 degradation in the presence of AIP4, fluorescence intensities of TRPC4-YFP were integrated over single cells and compared to those in cells coexpressing TRPC4-YFP and AIP4. The mean single-cell fluorescence signals of TRPC4-YFP collected in three independent transfection experiments for each setting were about 40% higher in cells that coexpressed AIP4 compared to control cells expressing TRPC4-YFP alone. Most striking was the re-localization of TRPC4-YFP in the presence of AIP4. TRPC4-YFP, normally localized in clusters at the plasma membrane, accumulated in intracellular structures in cells coexpressing AIP4 (Figure 6D). Although the changes in subcellular localization of TRPV4-YFP were less dramatic (data not shown), we observed a similar reduction in the basal activity of cells expressing TRPV4-YFP in the presence of AIP4 (Supplementary Figure 4). AIP4 alone did not significantly alter the resting [Ca2+]i in the cells not expressing TRPV4 (Supplementary Figure 4A).
Figure 6.
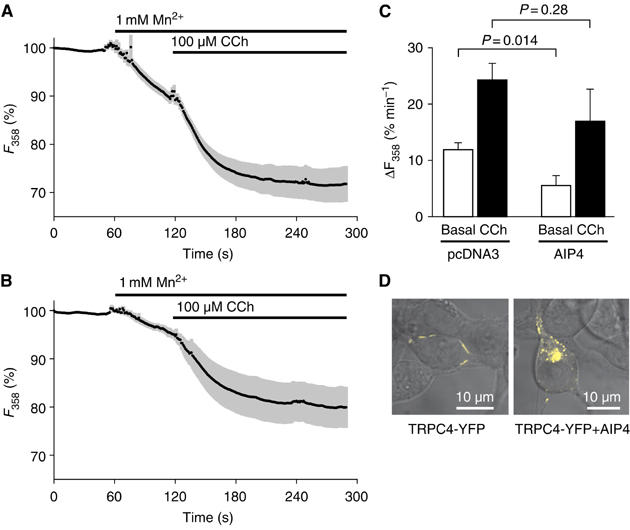
Basal TRPC4 activity and surface expression are reduced upon coexpression with AIP4. (A–C) Mn2+ influx through TRPC4-YFP was detected with the Mn2+-quench technique in fura 2-loaded HEK 293 cells expressing either TRPC4-YFP alone (A; 1.5 μg of cDNA plasmid and 0.5 μg of pcDNA3) or TRPC4-YFP together with AIP4 (B; 0.5 μg AIP4-encoding cDNA plasmid instead of pcDNA3). (A, B) Representative data sets depict means (black symbols)±s.e.m. of n=6 independent transfection experiments containing data from a total number of 267–282 cells. The Ca2+-independent fluorescence intensity was measured by exciting the probe at 358 nm, which corresponds to the isosbestic point of fura 2 in our system. Background intensities and contaminating YFP signals were subtracted before the analysis. (C) Statistical analysis of the data shown in (A, B). The Mn2+-induced loss of 358-nm-excited fura 2 fluorescence (ΔF358) was calculated over a 1-min interval before (Basal) and after (CCh) the addition of 100 μM carbachol and expressed as percentage of the initial intensity. The indicated P-values were calculated by unpaired Student's t-test (n=6). (D) Confocal live cell microscopy shows the typical distribution pattern of TRPC4-YFP without (left panel) and with coexpressed AIP4 (right panel).
Taken together, these results demonstrate that AIP4 does not induce degradation of TRP channels, but rather promotes their accumulation in intracellular compartments.
Discussion
The presented data demonstrate for the first time that trafficking of select TRP channels is regulated by AIP4, a HECT-E3 ubiquitin ligase. AIP4 facilitates the ubiquitination of TRPV4 and TRPC4, increases their steady-state levels, but decreases the amount of both channels at the plasma membrane. The latter effect is not caused by degradation, but results from accelerated internalization.
Ubiquitination is a common post-translational modification of proteins affecting their turnover and/or trafficking. The fate of the modified protein is determined by the type of ubiquitin attachment. Proteins with polyubiquitin chains consisting of at least four ubiquitin molecules conjugated to each other via lysine 48 (polyubiquitination) undergo proteasome-mediated degradation (Pickart, 2001). In contrast, ligation of a single ubiquitin molecule to one (monoubiquitination) or more (multiubiquitination) lysine residues represents a sorting signal for the MVB pathway and targets cell surface proteins for lysosomal degradation (Hicke, 2001; Katzmann et al, 2002). Proteins destined for ubiquitination are recognized by specific E3 ubiquitin ligases. However, the same ubiquitin ligase can mediate polyubiquitination as well as mono-/multiubiquitination (reviewed by d'Azzo et al, 2005), and it is presently unknown what determines the type of ubiquitination. Our results show that TRP channels are new members of the constantly growing group of proteins regulated by ubiquitin attachment. AIP4, a member of the HECT family of ubiquitin ligases, dramatically increases the ubiquitination of TRPV4, as well as TRPC4. The requirement of the catalytic activity of AIP4 for this increase (Figures 1B and 5), the interaction between AIP4 and TRPV4 (Supplementary Figure 1), and their colocalization at the cell periphery (Figure 4D) indicate that AIP4 directly participates in this reaction. Using P4D1 and FK1 α-ubiquitin antibodies, which differ in their ability to recognize monoubiquitinated proteins (Haglund et al, 2003), we determined that TRPV4 is modified by multi- rather than by polyubiquitination. Therefore, we hypothesized that AIP4-driven ubiquitination plays a specific and hitherto unknown role in the endosomal trafficking of TRPV4.
Supporting this concept, coexpression of AIP4 decreases the cell surface fraction of TRPV4 and the basal activity of the channel, although it does not induce degradation of TRPV4. The half-life of TRPV4 is not shortened and its steady-state levels are actually increased in the presence of AIP4. Accumulation of TRPV4 in intracellular compartments is likely less harmful to the cell than its presence at the plasma membrane. Furthermore, our data reveal increased amounts of internalized TRPV4 channel in the presence of AIP4 (Figure 4C and G), suggesting that AIP4-mediated ubiquitination of TRPV4 facilitates its endocytosis. However, a role of AIP4 in slowing the recycling of TRPV4 to the plasma membrane cannot be excluded. We also found that AIP4 colocalizes with internalized TRPV4 in subcortical structures, which is reminiscent of the colocalization of AIP4 and CXCR4 in AP-2-positive structures underneath the plasma membrane (Marchese et al, 2003). Taken together, these results indicate that AIP4 participates in the endocytic trafficking of TRPV4, a concept that is in agreement with the proposed role of monoubiquitination in the internalization process (Haglund et al, 2003). Similarly, TRPC4 is ubiquitinated and localizes to an intracellular compartment in the presence of AIP4.
Internalized and ubiquitinated plasma membrane proteins are usually sorted to the MVBs, leading to their degradation in the lysosome (Katzmann et al, 2002). This process is aided by numerous proteins, some of which directly recognize and bind the ubiquitinated cargo (Babst, 2005). We cannot rule out the possibility that one or more such trans-acting factors, involved in the sorting of TRPV4 to MVBs, are limiting in our experimental conditions. However, our results clearly indicate that internalized TRPV4 is not efficiently degraded, but rather recycles back to the plasma membrane. Three observations support this conclusion. First, exogenously expressed TRPV4 is relatively stable. Second, TRPV4 is mostly confined to the plasma membrane region, and is not detectable in late endosomes or lysosomes even after inhibition of lysosomal degradation. Third, endocytosed TRPV4 efficiently recycles to the plasma membrane (Figure 4C). Thus, TRPV4 likely undergoes repetitive rounds of endocytosis and recycling to the plasma membrane, a process termed constitutive cycling (Royle and Murrell-Lagnado, 2003). AIP4 affects this process leading to an increase of the intracellular TRPV4 pool at the expense of the surface fraction. Our findings extend the current model of recycling that has been observed for several mammalian TRP channels (Bezzerides et al, 2004; Cayouette et al, 2004; Morenilla-Palao et al, 2004; Singh et al, 2004; Smyth et al, 2006). Insertion of TRP channels into the plasma membrane can be stimulated by growth factors (TRPC3, TRPC5), carbachol (TRPC3, TRPC6), PKC (TRPV1), or appears to be unregulated (TRPC1). The rapid reappearance of internalized TRPV4 at the cell surface suggests that the exocytosis of TRPV4 is part of a constitutive cycling process, and is not regulated. Thus, constitutive cycling may represent a common phenomenon for TRP channels, but the regulatory mechanisms involved to determine the concentration of ion channels at the plasma membrane may differ between members of the TRP family.
Substrate recognition by HECT-family ligases typically involves interaction between their WW domains and PPxY (also referred to as PY) motifs in ubiquitinated proteins (Ingham et al, 2004). However, TRPV4 does not contain a consensus PY motif, and the removal of a sequence resembling the extended PY motif found in ENaC subunits did not abrogate the AIP4-mediated ubiquitination (Figure 2). Thus, another motif must be responsible for the increased ubiquitination in the presence of AIP4. Interestingly, several proteins that lack the PY motif are substrates of AIP4, including Notch, CBLC, CXCR4, and endophilin (Qiu et al, 2000; Courbard et al, 2002; Marchese et al, 2003; Angers et al, 2004). Our data add members of the TRP family to this growing group of noncanonical AIP4 substrates. Of note, WW domains from proteins other than HECT-family ubiquitin ligases recognize diverse motifs, sharing only the presence of proline residue(s). Alternatively, AIP4 could be recruited to TRPV4 by an adaptor protein, a hypothesis that has been invoked for CXCR4 (Marchese et al, 2003). Our results suggest that the sequence required for AIP4-mediated TRPV4 ubiquitination resides in the N-terminal, intracellular tail of TRPV4. This region mediates TRPV4 oligomerization and interaction with other proteins, thereby increasing the plasma membrane localization of the channel (Arniges et al, 2006; Cuajungco et al, 2006). Our data adds to the list of trafficking signals encoded by the N-terminal tail of TRPV4 and to the complexity of pathways that regulate TRP channel localization.
In conclusion, our findings uncover a novel mechanism that regulates the concentration of a subset of TRP channels at the cell surface. It will be interesting to probe whether defective endocytosis of TRP channels is associated with human disease known to occur for Nedd4-dependent regulation of ENaC.
Materials and methods
Reagents and plasmids
MG132 (Calbiochem), 4α-phorbol 12,13-didecanoate (4α-PDD), carbachol, cycloheximide, and chloroquine (Sigma-Aldrich) were used at concentrations as indicated. All plasmids containing mouse TRPV4 sequences were generated from pcDNA3-TRPV4 plasmid provided by U Wissenbach (Wissenbach et al, 2000). The following constructs were generated by PCR and standard cloning techniques in pcDNA3 vector (Invitrogen): TRPV4-His, TRPV4-V5 C-term, TRPV4-V5 loop; or in pcDNA6-V5/His vector (Invitrogen): TRPV4-V5/His, TRPV4-V5/His ΔPY (aa 411–437 removed), TRPV4-V5/His ΔC (aa 1–726). The intracellular amino (aa 1–468) and carboxyl tails (aa 713–871) of TRPV4 were tagged at their N-termini with FLAG and His9 (F9) in a derivative of the pCDM8 vector (Invitrogen), creating F9-N-TRPV4 and F9-TRPV4-C plasmids, respectively. All TRPV4 amino-acid numbering is according to the Swissprot entry Q9EPK8. The construction of plasmids encoding autofluorescent TRPC4 and TRPV4 C-terminally fused to YFP has been described (Hofmann et al, 2002; Hellwig et al, 2005). The cDNAs coding for mouse TRPC4 (isoform beta), human TRPV1, TRPC6, and PKD2 were cloned in pcDNA6-V5/His vector (Invitrogen). Plasmids coding for myc-tagged AIP4 and Nedd4-2 were provided by T Pawson (Winberg et al, 2000), T7-tagged Nedd4 by D Rotin (Staub et al, 1996), FLAG-tagged ubiquitin by I Dikic (Soubeyran et al, 2002), His-tagged c-Jun by D Bohmann (Treier et al, 1994), and CA MEKK1 (Δ367) and DN MEKK1 (Δ367 KR) by DJ Templeton (Yan et al, 1994). For the experiments involving knock down of AIP4, the microRNA-adapted shRNA approach was used (Open Biosystems). The shRNA with the human AIP4 mRNA target sequence CCAGTTGGACTCAAGGATTTA was cloned into pCMV-GIN-ZEO vector. The plasmid lacking the shRNA was used as a negative control in the internalization assay (Figure 4G).
Cell cultures and transfections
HEK 293T and HEK 293 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. Transient transfections were carried out using the calcium phosphate method or Fugene 6 reagent (Roche).
Antibodies
The α-TRPV4 polyclonal antibody was generated in rabbits and subsequently purified using the C-terminal peptide CDGHQQGYAPKWRTDDAPL as antigen. The following antibodies were used in this work: α-FLAG M2 (Sigma-Aldrich), α-V5 (Serotec), α-AIP4 D-20, α-GFP (Santa Cruz), α-myc 9E10 (Oncogene), α-T7 (Novagen), α-CI-MPR 2G11 (Affinity Bioreagents), α-LAMP1 H4A3, α-EEA1, α-AIP4 (BD Biosciences), α-ubiquitin P4D1 (Covance), and FK1 (Biomol).
Ubiquitination assays
At 48 h after transfection, HEK 293T cells were washed with PBS and lysed in buffer A (8 M urea, 100 mM NaH2PO4, 10 mM Tris, 1% Triton X-100, pH 8.0; all steps at room temperature). The supernatant obtained after centrifugation at 75 000 g was used for purification on Ni2+-NTA agarose (Qiagen) for 1 h, washed twice with buffer A and twice with buffer B (same as A, except for 0.5% Triton X-100 and pH 6.3). Bound proteins were eluted with buffer C (same as A, except for 0.1% Triton X-100 and pH 4.5).
Protein stability assay
HEK 293T were transfected in 10-cm dishes by the calcium phosphate method and split 24 h later into 35-mm dishes. At 48 h after transfection, the cells were treated with 30 μg/ml cycloheximide in DMEM for 0–10 h and lysed in buffer A (see above).
Immunofluorescence
Cells were fixed in 3.7% paraformaldehyde in PBS, permeabilized with Triton X-100, and blocked in PBS containing 1% horse serum and 0.1% Tween 20. All incubations with antibodies were carried out in the blocking solution. Alexa Fluor 488- and Cy3-labeled antibodies were purchased from Molecular Probes and Jackson ImmunoResearch, respectively. Optical sections (0.49 μm) of cells were taken using a Zeiss LSM 510 confocal microscope. For surface labeling of HEK 293T cells expressing the TRPV4-V5 loop protein, the cells were incubated with α-V5 antibody on ice as for ELISA (see below), washed, returned to the incubator for 20 min and fixed. Confocal live cell imaging of YFP-tagged proteins was performed as described (Schaefer et al, 2000).
ELISA
HEK 293T cells transfected by the calcium phosphate method were split into poly-L-lysine-coated 48-well dishes. The cells were cooled on ice, incubated with α-V5 antibody (1.33 μg/ml) in DMEM containing 10% FBS and 20 mM HEPES at 4°C for 45 min, washed three times and fixed. For kinetic experiments, washed cells were returned to the incubator for a defined period of time before fixing. Fixed cells were subsequently blocked in 2% horse serum in PBS and incubated with AP-coupled α-mouse antibodies (Sigma-Aldrich). Following washings with PBS, the enzymatic reaction was performed in 0.1 M glycine, 1 mM MgCl2, 1 mM ZnCl2, pH 10.4, using 1 mg/ml p-nitrophenyl phosphate (Sigma-Aldrich) as substrate. The absorbance was read at 405 nm. For each experiment, all conditions were tested in triplicate. For the quantification of total amounts of the overexpressed proteins, the cells split in parallel were lysed in buffer A (see above) and the lysates were analyzed by Western blotting. The blots were scanned and the bands were quantified using ImageJ software (NIH, Bethesda).
Calcium measurements and detection of Mn2+ influx
Intracellular calcium concentrations were determined and calibrated as described recently (Lenz et al, 2002). Briefly, transfected HEK cells were loaded for 30 min with fura 2/AM (3 μM; Invitrogen–Molecular Probes, Carlsbad, CA) in HEPES-buffered saline (pH 7.4) containing 135 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5.5 mM glucose, 10 mM HEPES, and 0.2% (w/v) bovine serum albumin. Coverslips were imaged through a Fluar × 10/0.5 objective in a monochromator-equipped (Polychrome II, TILL-Photonics, Martinsried, Germany) inverted microscope (Axiovert 100, Carl Zeiss, Göttingen, Germany). Fluorescence was excited by sequentially applying monochromatic light at 340, 358, 380, and 470 nm, filtered (510 nm long pass filter), and recorded with a cooled 12-bit CCD camera (IMAGO, TILL-Photonics). To detect Mn2+ influx, we applied the Mn2+-quenching technique in fura 2-loaded HEK 293 cells. The Ca2+-independent fluorescence of fura-2 was excited at its isosbestic point of 358 nm and corrected for background signals and for residual fluorescence of YFP excited at 358 nm by a spectral linear unmixing method as described earlier (Lenz et al, 2002).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Legend to Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Materials and Methods
Acknowledgments
We thank B Müller for expert technical assistance, the members of the Walz lab for helpful discussions, M Köttgen for critical reading of the manuscript and insightful comments, as well as Drs I Dikic, T Pawson, D Rotin, U Wissenbach, D Bohmann, and D Templeton for cDNA plasmids. This work was supported by grants of the Deutsche Forschungsgemeinschaft to MS (SFB 366) and GW (Wa 597).
References
- Angers A, Ramjaun AR, McPherson PS (2004) The HECT domain ligase itch ubiquitinates endophilin and localizes to the trans-Golgi network and endosomal system. J Biol Chem 279: 11471–11479 [DOI] [PubMed] [Google Scholar]
- Arniges M, Fernandez-Fernandez JM, Albrecht N, Schaefer M, Valverde MA (2006) Human TRPV4 channel splice variants revealed a key role of ankyrin domains in multimerization and trafficking. J Biol Chem 281: 1580–1586 [DOI] [PubMed] [Google Scholar]
- Babst M (2005) A protein's final ESCRT. Traffic 6: 2–9 [DOI] [PubMed] [Google Scholar]
- Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE (2004) Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 6: 709–720 [DOI] [PubMed] [Google Scholar]
- Cayouette S, Lussier MP, Mathieu EL, Bousquet SM, Boulay G (2004) Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem 279: 7241–7246 [DOI] [PubMed] [Google Scholar]
- Courbard JR, Fiore F, Adelaide J, Borg JP, Birnbaum D, Ollendorff V (2002) Interaction between two ubiquitin-protein isopeptide ligases of different classes, CBLC and AIP4/ITCH. J Biol Chem 277: 45267–45275 [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Grimm C, Oshima K, D'Hoedt D, Nilius B, Mensenkamp AR, Bindels RJ, Plomann M, Heller S (2006) PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J Biol Chem 281: 18753–18762 [DOI] [PubMed] [Google Scholar]
- d'Azzo A, Bongiovanni A, Nastasi T (2005) E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 6: 429–441 [DOI] [PubMed] [Google Scholar]
- Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, Karin M (2004) Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306: 271–275 [DOI] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5: 461–466 [DOI] [PubMed] [Google Scholar]
- Hellwig N, Albrecht N, Harteneck C, Schultz G, Schaefer M (2005) Homo- and heteromeric assembly of TRPV channel subunits. J Cell Sci 118: 917–928 [DOI] [PubMed] [Google Scholar]
- Hicke L (2001) Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2: 195–201 [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99: 7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham RJ, Gish G, Pawson T (2004) The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23: 1972–1984 [DOI] [PubMed] [Google Scholar]
- Kanelis V, Rotin D, Forman-Kay JD (2001) Solution structure of a Nedd4 WW domain–ENaC peptide complex. Nat Struct Biol 8: 407–412 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3: 893–905 [DOI] [PubMed] [Google Scholar]
- Lenz JC, Reusch HP, Albrecht N, Schultz G, Schaefer M (2002) Ca2+-controlled competitive diacylglycerol binding of protein kinase C isoenzymes in living cells. J Cell Biol 159: 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM (2003) Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 100: 13698–13703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell 5: 709–722 [DOI] [PubMed] [Google Scholar]
- Mizuno A, Matsumoto N, Imai M, Suzuki M (2003) Impaired osmotic sensation in mice lacking TRPV4. Am J Physiol Cell Physiol 285: C96–C101 [DOI] [PubMed] [Google Scholar]
- Montell C, Birnbaumer L, Flockerzi V (2002) The TRP channels, a remarkably functional family. Cell 108: 595–598 [DOI] [PubMed] [Google Scholar]
- Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A (2004) Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem 279: 25665–25672 [DOI] [PubMed] [Google Scholar]
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T (2004) TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286: C195–C205 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, Fang D, Hunter T, Liu YC (2000) Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J Biol Chem 275: 35734–35737 [DOI] [PubMed] [Google Scholar]
- Royle SJ, Murrell-Lagnado RD (2003) Constitutive cycling: a general mechanism to regulate cell surface proteins. BioEssays 25: 39–46 [DOI] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G (2000) Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem 275: 17517–17526 [DOI] [PubMed] [Google Scholar]
- Singh BB, Lockwich TP, Bandyopadhyay BC, Liu X, Bollimuntha S, Brazer SC, Combs C, Das S, Leenders AG, Sheng ZH, Knepper MA, Ambudkar SV, Ambudkar IS (2004) VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol Cell 15: 635–646 [DOI] [PubMed] [Google Scholar]
- Smyth JT, Lemonnier L, Vazquez G, Bird GS, Putney JW Jr (2006) Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J Biol Chem 281: 11712–11720 [DOI] [PubMed] [Google Scholar]
- Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I (2002) Cbl–CIN85–endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416: 183–187 [DOI] [PubMed] [Google Scholar]
- Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D (1996) WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J 15: 2371–2380 [PMC free article] [PubMed] [Google Scholar]
- Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 16: 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD (2000) OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2: 695–702 [DOI] [PubMed] [Google Scholar]
- Tian W, Salanova M, Xu H, Lindsley JN, Oyama TT, Anderson S, Bachmann S, Cohen DM (2004) Renal expression of osmotically responsive cation channel TRPV4 is restricted to water-impermeant nephron segments. Am J Physiol Renal Physiol 287: F17–F24 [DOI] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78: 787–798 [DOI] [PubMed] [Google Scholar]
- Winberg G, Matskova L, Chen F, Plant P, Rotin D, Gish G, Ingham R, Ernberg I, Pawson T (2000) Latent membrane protein 2A of Epstein–Barr virus binds WW domain E3 protein-ubiquitin ligases that ubiquitinate B-cell tyrosine kinases. Mol Cell Biol 20: 8526–8535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissenbach U, Bodding M, Freichel M, Flockerzi V (2000) Trp12, a novel Trp related protein from kidney. FEBS Lett 485: 127–134 [DOI] [PubMed] [Google Scholar]
- Yan M, Dai T, Deak JC, Kyriakis JM, Zon LI, Woodgett JR, Templeton DJ (1994) Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature 372: 798–800 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Legend to Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Materials and Methods