

Abstract
Sphingosine 1-phosphate (S1P), produced by sphingosine kinase (SPHK), acts both by intracellular and extracellular modes. We evaluated the role of SPHK1 and S1P in osteoclastogenesis using bone marrow-derived macrophage (BMM) single and BMM/osteoblast coculture systems. In BMM single cultures, the osteoclastogenic factor receptor activator of NF-κB ligand (RANKL) upregulated SPHK1 and increased S1P production and secretion. SPHK1 siRNA enhanced and SPHK1 overexpression attenuated osteoclastogenesis via modulation of p38 and ERK activities, and NFATc1 and c-Fos levels. Extracellular S1P had no effect in these cultures. These data suggest that intracellular S1P produced in response to RANKL forms a negative feedback loop in BMM single cultures. In contrast, S1P addition to BMM/osteoblast cocultures greatly increased osteoclastogenesis by increasing RANKL in osteoblasts via cyclooxygenase-2 and PGE2 regulation. S1P also stimulated osteoblast migration and survival. The RANKL elevation and chemotactic effects were also observed with T cells. These results indicate that secreted S1P attracts and acts on osteoblasts and T cells to augment osteoclastogenesis. Taken together, S1P plays an important role in osteoclastogenesis regulation and in communication between osteoclasts and osteoblasts or T cells.
Keywords: osteoclast differentiation, osteoclast–osteoblast coupling, RANKL, sphingosine kinase, sphingosine 1-phosphate
Introduction
Bone is a tissue continually regenerated through a remodeling process. In response to biochemical or mechanical signal, the basic multicellular unit (BMU) is organized at discrete sites in bone to perform remodeling. Once the BMU is formed, osteoclasts are activated to resorb old bone. Osteoblasts are recruited to the resorption area and new bone matrix is produced to fill the resorption pits. Subsequent mineralization of the matrix completes the remodeling process. Therefore, the coordinated action of osteoclasts and osteoblasts is important for proper bone remodeling, and an imbalance in the activity of osteoclasts and osteoblasts leads to metabolic bone disorders. However, molecular mechanisms for the intricate interplay between the bone resorbing and forming cells have not been well characterized. Other cells present in bone such as vascular endothelial cells, bone stromal cells, and immune cells may also communicate with osteoclasts and osteoblasts, influencing bone metabolism under both physiological and pathological conditions.
Osteoclasts are specialized polykaryons that differentiate from monocyte–macrophage lineage precursors of hematopoietic cells. Receptor activator of NF-κB ligand (RANKL), a tumor necrosis factor (TNF) family member, plays an essential role in the commitment of precursors to osteoclastic differentiation (Suda et al, 1999; Boyle et al, 2003; Teitelbaum and Ross, 2003). It is also important for activation and survival of differentiated osteoclasts. Macrophage-colony stimulating factor (M-CSF) supports proliferation and survival of osteoclast precursors during differentiation. Early studies with coculture system of bone marrow osteoclast precursor cells and osteoblasts revealed that direct cell-to-cell interaction was needed for osteoclast generation. The cell surface molecular interaction for osteoclastic differentiation was later found to be between RANKL expressed on osteoblasts and RANK present on osteoclast precursors (Suda et al, 1999). Osteoprotegerin (OPG) is a soluble decoy receptor that binds RANKL, but unlike RANK, has no intracellular signaling capacity (Boyle et al, 2003). Therefore, the RANKL–RANK–OPG axis is critical to determine osteoclastogenesis and bone homeostasis.
RANKL binding to its cognate receptor RANK leads to recruitment of adaptor proteins TRAFs (TNF receptor-associated factors) (Lee and Kim, 2003; Boyle et al, 2003; Teitelbaum and Ross, 2003). Many in vitro and in vivo studies have provided evidence that TRAF6 may be the most critical TRAF protein in RANK signaling in osteoclasts (Lomaga et al, 1999; Naito et al, 1999; Kobayashi et al, 2001). The downstream signaling events ensuing TRAF6 recruitment to RANK include activation of kinases such as PI3K/Akt, IKKs, and MAPKs (Wong et al, 1999). The activation of signaling pathways mediated by these kinases leads to cytoskeletal organization necessary for migration and bone resorption activities, and induction of an array of genes required for differentiation and survival (Boyle et al, 2003; Lee and Kim, 2003; Teitelbaum and Ross, 2003). Recent studies have demonstrated that NFATc1 is the transcription factor profoundly increased by RANKL and required for osteoclastogenesis (Takayanagi et al, 2002). In turn, NFATc1 induction by RANKL is dependent on c-Fos and positive feedback by NFATc1 itself (Takayanagi et al, 2002; Matsuo et al, 2004).
Sphingosine kinase (SPHK) is a lipid kinase that phosphorylates sphingosine to generate sphingosine 1-phosphate (S1P). Two mammalian isoforms, SPHK1 and SPHK2, have been identified. S1P has been implicated in a variety of cellular processes, including cell differentiation, apoptosis, proliferation, and motility (Spiegel and Milstien, 2003; Futerman and Hannun, 2004). The diversity in cell responses elicited by S1P may be attained in part by its dual characteristics of being both an intracellular second messenger and an extracellular signal. In addition, the presence of multiple cell surface receptors may contribute to the complexity of S1P effects. Five mammalian S1P receptors have been identified so far—S1P1/endothelial differentiation gene 1 (EDG1), S1P2/EDG5, S1P3/EDG3, S1P4/EDG6, and S1P5/EDG8. Upon engagement with extracellular S1P, each of these receptors couples to a specific set of heterotrimeric G proteins to trigger plethora of intracellular signaling pathways (Spiegel and Milstien, 2003). Although the direct targets of intracellular S1P are not clear, the amount of intracellular S1P relative to ceramide and sphingosine has been suggested to be important in the regulation of cell cycle, apoptosis, and calcium homeostasis (Spiegel and Milstien, 2003). FTY-720, a structural analog of sphingosine and a novel immunosuppressant, can be converted to phosphate ester form by SPHK and phospho-FTY-720 binds with higher affinity than S1P to all S1P receptors except S1P2 (Brinkmann et al, 2002; Yopp et al, 2003). In contrast to S1P-mimicking effects, functional antagonism against certain S1P-stimulated responses, such as T-cell chemotaxis and angiogenesis, have also been demonstrated for FTY-720 (Graeler and Goetzl, 2002; LaMontagne et al, 2006). Although the antagonistic function of FTY-720 has been suggested to be due to internalization and partial degradation of S1P receptors (Kaneider et al, 2004), the precise mechanism of action of FTY-720 as well as that of S1P is far from comprehensive understanding.
In this study, we investigated the possibility of involvement of SPHK1 and S1P in osteoclast differentiation. We found that SPHK1 activated by RANKL plays dichotomous function for osteoclast differentiation: intracellular S1P attenuates osteoclast differentiation program, whereas S1P secreted from stimulated osteoclast precursor cells acts on osteoblasts to augment osteoclastogenesis by increasing RANKL expression. Secreted S1P also chemoattracts osteoblasts and enhances their survival. Our studies identify the SPHK1–S1P system as a novel player in osteoclastogenesis regulation and in communication between osteoclasts and osteoblasts in bone metabolism.
Results
SPHK1 expression and activity increase during RANKL-induced osteoclastogenesis
We analyzed expression levels of SPHK during osteoclast differentiation. Bone marrow-derived macrophages (BMMs) were cultured in the presence of RANKL plus M-CSF (RANKL/M-CSF) for 4 days to generate osteoclasts. Both mRNA and protein levels of SPHK1 and SPHK2 increased during the osteoclastogenesis from BMMs (Figure 1A). The increased gene expression of SPHK1 and SPHK2 was also detected in microarray experiments (data not shown). We next examined whether the activity of SPHK1 is regulated by osteoclastogenic stimuli. SPHK1 activity was found to be stimulated by RANKL/M-CSF (Figure 1B) when an in vitro assay was performed in the presence of Triton X-100, which inhibits SPHK2 activity (Liu et al, 2000). RANKL alone could also stimulate SPHK1 activity; however, a synergy was observed when M-CSF was also present (Figure 1C). The intracellular level of S1P was also elevated in BMMs stimulated with RANKL (Figure 1D). Consistent with the increased SPHK levels during differentiation, the intracellular S1P levels were higher in differentiating prefusion osteoclasts (pOc) than in BMMs (Figure 1E). It has been reported that TRAF2 interacts with and thereby activates SPHK1 in response to TNF-α (Xia et al, 2002). As TRAF6 is a crucial adaptor molecule for RANK signaling, we next determined whether TRAF6 associates with SPHK1. Both in HEK293 cells transfected with TRAF6 and SPHK1 and in untransfected pOc, the complex of TRAF6 and SPHK1 was detected (Figure 1F). The complex formation was weakly stimulated by RANKL (Figure 1F). These results indicate that SPHK1 is upregulated and its activity is stimulated by RANKL in BMMs undergoing osteoclastic differentiation.
Figure 1.
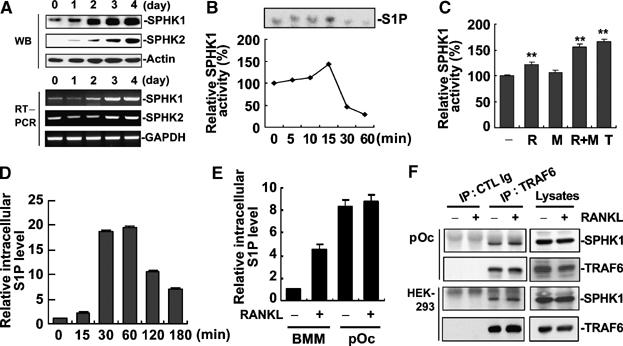
SPHK1 expression and activity is stimulated by RANKL during osteoclastogenesis from BMMs. (A) BMMs were cultured in the presence of RANKL (100 ng/ml) and M-CSF (30 ng/ml). Cells were harvested at the indicated days and protein (upper three panels) and mRNA (lower three panels) levels of SPHK1 and SPHK2 were examined by Western blotting and RT–PCR. (B) BMMs were stimulated with RANKL (100 ng/ml) and M-CSF (30 ng/ml) and SPHK1 activity was measured from 80 μg of cell lysates. (C) BMMs were stimulated with RANKL (100 ng/ml), M-CSF (30 ng/ml), RANKL plus M-CSF, or TNF-α (10 ng/ml) for 15 min and SPHK1 activity in 80 μg cell lysates was measured. **P< 0.005 versus control. (D) BMMs were labeled with 32Pi and stimulated with RANKL (1 μg/ml). Intracellular S1P levels were detected from 1 × 106 cells as described in Materials and methods. (E) 32Pi-labeled BMMs or pOc were stimulated with RANKL (1 μg/ml) for 15 min. Intracellular S1P levels from 1 × 106 cells were measured. (F) HEK293 cells were transfected with RANK, Flag-TRAF6, and HA-SPHK1. The transfected HEK293 cells and untransfected pOc were starved for 3 h and stimulated with RANKL (1 μg/ml) for 15 min. Cell lysates were immunoprecipitated with anti-TRAF6 or control Ig, and the precipitates were immunoblotted with anti-SPHK1 followed by reprobing with anti-TRAF6.
SPHK1 negatively regulates osteoclast differentiation in BMM single culture
Unlike SPHK1, the role of SPHK2 in S1P-mediated cellular responses is elusive and the intracellular location of SPHK2 has not been conclusive (Igarashi et al, 2003; Maceyka et al, 2005). For this reason, we focused on the role of SHPK1 for osteoclastogenesis. First, we investigated whether downregulation of SPHK1 by siRNA would reduce osteoclastogenesis. Infection of BMMs with SPHK1 siRNA retroviruses resulted in a prominent decrease in the SPHK1 mRNA and protein levels (Figure 2A). Infected BMMs were induced to differentiate to osteoclasts by culturing with RANKL/M-CSF. During differentiation, BMMs convert to tartrate-resistant acid phosphatase-positive (TRAP+) cells and these TRAP+ mononuclear cells fuse to generate TRAP+ multinuclear cells (MNCs; osteoclasts). In contrast to our expectation, the formation of TRAP+ MNCs was not reduced, rather modestly increased by SPHK1 siRNA compared with the control luciferase siRNA (Figure 2B and C). The number of generated TRAP+ mononuclear cells was also higher in cells infected with SPHK1 siRNA than in the control cells (data not shown). In experiments with RAW264.7 cells, a model cell line for osteoclastogenesis, using oligocassette siRNA for SPHK1, similar results were observed (Supplementary Figure 1). We next evaluated effects of increased SPHK1 expression. BMMs were infected with retroviruses harboring HA-tagged SPHK1 and expression of HA-SPHK1 was confirmed (Figure 2D). The overexpression of SPHK1 weakly reduced the generation of mature osteoclasts (Figure 2E and F). These data indicate that SPHK1 activity negatively regulates osteoclastogenesis in this BMM culture system.
Figure 2.
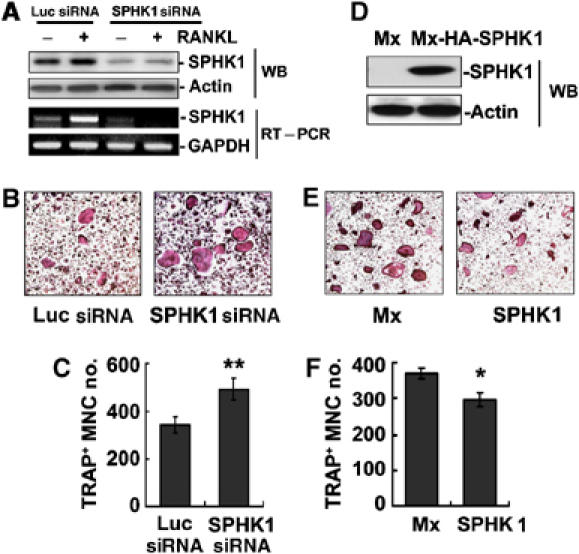
SPHK1 inhibits RANKL-induced osteoclastogenesis in BMM single culture. (A) BMMs were infected with retroviruses encoding SPHK1 siRNA or luciferase (Luc) siRNA. SPHK1 protein and mRNA levels were evaluated by Western blotting and RT–PCR. (B, C) Infected BMMs were cultured for 4 days in the presence of RANKL (100 ng/ml) and M-CSF (30 ng/ml). Cells were TRAP-stained and the number of TRAP+ MNC was counted. **P<0.005 versus Luc siRNA control. (D) BMMs were infected with retroviruses harboring HA-SPHK1 or the control viruses. The expression of SPHK1 was tested by Western blotting with anti-HA. (E, F). Infected BMMs were induced to differentiate to osteoclasts. The number of TRAP+ MNC was scored. *P<0.01 versus control.
SPHK1 reciprocally regulates RANKL activation of p38 and ERK MAPKs in BMMs
To gain insights into the mechanism by which SPHK1 siRNA increases and SPHK1 overexpression decreases osteoclastogenesis, we examined the effects of the SPHK1 modulations on RANK signaling. In BMMs with reduced SPHK1 expression by siRNA, RANKL stimulation of p38 MAPK was enhanced compared with that in the control luciferase siRNA infected cells (Figure 3A and B). On the other hand, ERK activation by RANKL was suppressed by SPHK1 siRNA retrovirus infection (Figure 3A and B). Overexpression of SPHK1 resulted in the opposite effect; reduced p38 and augmented ERK activation by RANKL (Figure 3C and D). Therefore, the RANKL signaling to p38 and ERK MAPKs was reciprocally modulated by the regulation of SPHK1 expression in BMMs.
Figure 3.
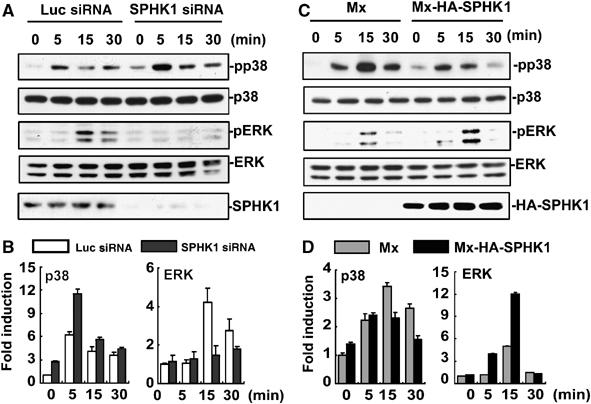
SPHK1 reciprocally modulates RANKL-induced activation of p38 and ERK. (A) BMMs infected with SPHK1 or luciferase siRNA retroviruses were stimulated with RANKL (1 μg/ml). The phosphorylated and total levels of p38 and ERK were examined by Western blotting. SPHK1 protein levels were also examined. (B) Quantification data for the phosphorylated p38 or ERK normalized to the level of total p38 or ERK are presented. (C) BMMs infected with HA-SPHK1 or the control retroviruses were stimulated with RANKL and cell extracts were Western blotted as above. (D) The levels of phosphorylated p38 or ERK were normalized to those of total p38 or ERK.
SPHK1 inhibits NFATc1 and c-Fos induction by RANKL in BMMs
Recent studies have indicated the importance of NFATc1 transcription factor for differentiation of osteoclasts (Ishida et al, 2002; Takayanagi et al, 2002). Thus, we investigated whether the effect of SPHK1 on RANKL-induced osteoclastogenesis might be related to NFATc1. SPHK1 downregulation in BMMs by siRNA infection increased the induction of NFATc1 by RANKL (Figure 4A, top panel). On the contrary, overexpression of SPHK1 suppressed NATc1 induction (Figure 4B, top panel). c-Fos is required for NFATc1 induction by RANKL (Takayanagi et al, 2002; Matsuo et al, 2004). SPHK1 siRNA enhanced RANKL-stimulated increase in c-Fos expression (Figure 4A, middle panel), whereas overexpression of SPHK1 attenuated c-Fos induction (Figure 4B, middle panel). As both the p38 activation (Figure 3B) and NFATc1 and c-Fos induction (Figure 4B) were suppressed by SPHK1 overexpression, the potential link between p38 and these transcription factors was tested. BMMs were incubated with RANKL in the presence of the p38-specific inhibitor SB203580. SB203580 treatment led to a strong inhibition in NFATc1 and c-Fos induction (Figure 4C). Therefore, p38-dependent regulation of c-Fos and NFATc1 mediates the suppressive role of SPHK1 in osteoclast differentiation from this culture system involving only BMMs.
Figure 4.
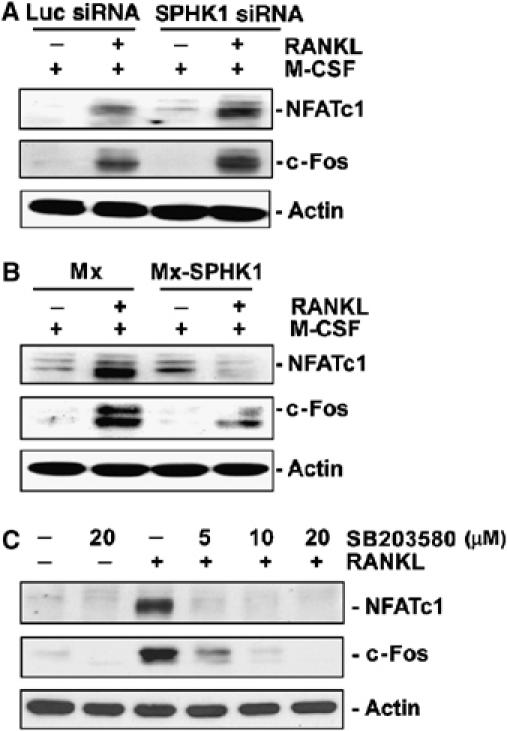
SPHK1 suppresses NFATc1 and c-Fos induction by RANKL. (A) BMMs were infected with SPHK1 or Luc siRNA retroviruses. Cells were cultured for 24 h with RANKL (100 ng/ml) and M-CSF (30 ng/ml) and protein levels of NFATc1 and c-Fos were examined. (B) BMMs were infected with HA-SPHK1 or the control retroviruses and analyzed as in (A). (C) BMMs were stimulated with RANKL (100 ng/ml) in the presence of SB203580 for 24 h. The NFATc1 and c-Fos protein levels were detected.
Extracellular S1P does not affect osteoclast differentiation in BMM single culture
S1P generated by SPHK1 can function as both an extracellular ligand and an intracellular second messenger. To examine the potential role of extracellular S1P in osteoclastogenesis, we first determined whether RANKL increases S1P secretion from BMMs as a result of SPHK activation. As shown in Figure 5A, extracellular S1P level increased by RANKL treatment. Next, we explored the possibility of autocrine function of S1P. For this, the presence of S1P receptors in BMMs and osteoclasts was analyzed. BMMs and differentiating osteoclasts expressed all known S1P receptors except S1P5 (Figure 5B). S1P5 was detected in T cells, indicating that no detection of this receptor in BMMs was not due to an inadequate assay condition (Supplementary Figure 2). As the overexpression of SPHK1 reduced osteoclastogenesis by RANKL (Figure 2F), whereas RANKL stimulated extracellular S1P production (Figure 5A), we next tested whether addition of exogenous S1P would negatively regulate osteoclast differentiation through cell surface S1P receptors. S1P (1 nM–1 μM) did not have any effect on RANKL-induced osteoclastogenesis from BMMs (Figure 5C). Neither S1P addition to the culture with lower RANKL concentrations nor S1P itself in the absence of RANKL could cause any difference in osteoclast differentiation (data not shown). In addition, NFATc1 induction by RANKL was not modulated by exogenous S1P (data not shown). The lack of effects of exogenous S1P was not due to malfunction of the receptors as S1P treatment caused increased migration of BMMs (Figure 5D) and p38 activation (Supplementary Figure 3). Moreover, FTY-720, a potential functional antagonist of S1P receptors, had no effect (Figure 5E). These data point that the apparent inhibition of osteoclastic differentiation of BMMs by increased SPHK1 activity (Figure 2) is not owing to the extracellular action of S1P secreted in response to RANKL. Rather, RANKL stimulation of SPHK1 exerts a moderate osteoclastogenesis inhibitory function through the action of intracellular S1P in BMMs.
Figure 5.
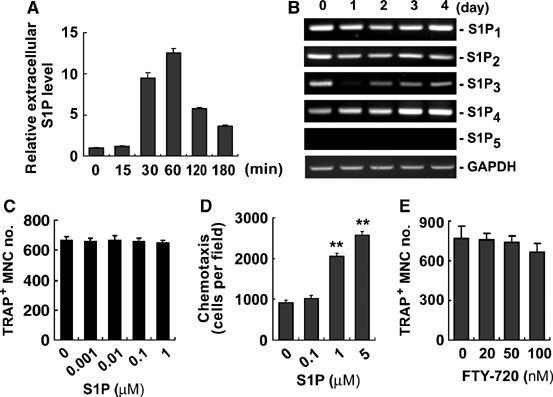
Extracellular S1P has no effect on osteoclastogenesis in BMM single culture. (A) BMMs were labeled with 32Pi and treated with RANKL (1 μg/ml). S1P levels in the culture supernatants from 1 × 106 cells were determined. (B) BMMs were cultured in the presence of RANKL (100 ng/ml) and M-CSF (30 ng/ml). S1P receptor mRNA levels were analyzed. (C) BMMs were cultured for 5 days in medium supplemented with 1% FBS, RANKL (100 ng/ml), M-CSF (30 ng/ml), and S1P. The number of TRAP+ MNCs was determined. (D) Migration of BMMs towards S1P was examined using transwell plates. **P<0.005 versus control. (E) BMMs were cultured for 4 days in the presence of FTY-720, RANKL (100 ng/ml), and M-CSF (30 ng/ml). The number of TRAP+ MNCs was scored.
S1P increases chemotaxis and survival of osteoblasts and activated T cells
Osteoblasts are recruited to resorption sites to fill the eroded cavity with new bone. For osteoblast attraction, factors released by osteoclasts may play an important role. S1P produced by osteoclast precursors during differentiation may function in a paracrine manner to osteoblasts. To explore this possibility, we examined the chemotactic effects of S1P on primary mouse osteoblasts. S1P increased osteoblast chemotaxis at the range of 0.01–1 μM (Figure 6A). The chemotactic effect was abolished by FTY-720 (Figure 6A). In addition, the chemotaxis of osteoblasts was higher toward the conditioned medium (CM) from differentiating BMMs transduced to overexpress SPHK1 (SPHK1-CM) as compared with that from control cells (Figure 6B). Consistently, the CM from differentiating BMMs infected with SPHK1 siRNA retroviruses showed lower chemotactic activity for osteoblasts (Figure 6C). The elevated and reduced S1P production by SPHK1 overexpression and SPHK1 siRNA, respectively, was confirmed (Supplementary Figure 4). S1P also enhanced osteoblast survival in serum-deprived condition (Figure 6D). In support of the responses to S1P, mouse osteoblasts expressed S1P receptors S1P1, S1P2, and S1P3 (Figure 6E). Osteoclast-derived factors may also have effects on T cells in inflammatory bone erosion diseases such as rheumatoid arthritis (RA). We examined whether S1P from osteoclastic cells has chemotactic effects on T cells. The SPHK1-CM increased T-cell chemotaxis (Figure 6F), which was suppressed by FTY-720 (Figure 6G).
Figure 6.
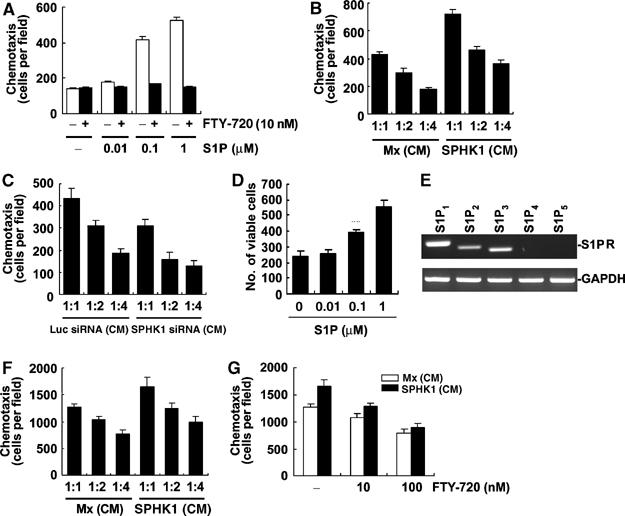
Extracellular S1P stimulates osteoblast chemotaxis and survival. (A) Mouse calvarial osteoblasts were treated with 10 nM FTY-720 or vehicle, and allowed to migrate for 24 h towards 0.01–1 μM S1P using transwell plates. (B) Osteoblasts were allowed to migrate for 24 h towards the indicated CM diluted at the indicated ratio. (C) Osteoblasts were allowed to migrate for 24 h towards the indicated CM. (D) Osteoblasts were incubated with S1P in serum-free media for 72 h. The number of surviving cells was counted after hematoxylin staining. (E) Osteoblasts were collected and S1P receptor mRNA was detected by RT–PCR. (F) T cells were allowed to migrate for 3 h towards the indicated CM. (G) T cells were allowed to migrate for 3 h in the presence of FTY-720 towards the indicated CM diluted at 1:1 ratio.
S1P potentiates osteoclast differentiation in coculture of BMMs and osteoblasts by increasing RANKL expression
Osteoclasts are generated when BMMs and osteoblasts are cocultured in the presence of 1α,25-dihydroxyvitamin D3 (VtD3) and/or prostagladin E2. As S1P released during osteoclastic differentiation can act in a paracrine manner on osteoblasts, we examined the effects of S1P on osteoclastogenesis in the coculture system. Addition of S1P (1 μM) greatly increased VtD3-induced osteoclast formation in the coculture (Figure 7A). This stimulatory effect of S1P on osteoclastogenesis was also observed when the coculture cells were treated with a low concentration of PGE2 (10−8 M), but not with higher concentrations of PGE2 (Figure 7B). In cocultures treated with PGE2 (10−8 M) and VtD3 (10−10 M), S1P also enhanced osteoclast generation (Figure 7C). The lack of S1P effect in the presence of high-dose PGE2 suggests the possibility that the osteoclastogenesis-stimulatory effect of S1P is associated with S1P-dependent PGE2 elevation, which will increase RANKL expression (Li et al, 2002a) and subsequently stimulate osteoclast differentiation. In fact, a prominent increase in RANKL expression was observed in S1P-treated osteoblasts (Figure 7D). When both S1P and VtD3 were present, RANKL expression was further increased, whereas OPG level was decreased, resulting in a greater rise in the ratio of RANKL to OPG. Moreover, osteoblasts treated with SPHK1-CM exhibited higher RANKL expression than those treated with the control CM (Figure 7E). Similar RANKL-elevating effect by S1P and SPHK1-CM was observed in activated T cells (Figure 7F and G). To further characterize the relationship of S1P with osteoclastogenesis, we included FTY-720 in cocultures of BMMs and calvarial osteoblasts. As shown in Figure 7H, FTY-720 dramatically inhibited osteoclastogenesis at nanomolar concentrations. In accordance with the anti-osteoclastogenic effects, FTY-720 significantly suppressed the S1P- and VtD3-stimulated RANKL expression in osteoblasts (Figure 7I). These results indicate that S1P induces RANKL expression in osteoblasts, and thereby facilitates osteoclastogenesis in cocultures of BMMs and osteoblasts. It is interesting that FTY-720 reduced the osteoclastogenesis and RANKL induction responses by VtD3 alone (Figure 7H and I). These findings suggest that S1P receptors may also be involved in the signaling by VtD3 alone.
Figure 7.
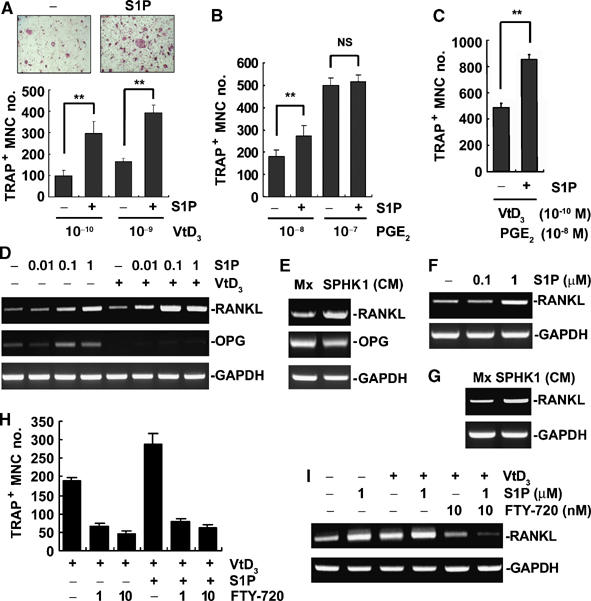
S1P potentiates osteoclast differentiation in coculture of BMMs and osteoblasts. (A) Coculture cells were treated with VtD3 and S1P (1 μM) for 6 days and TRAP+ MNCs generated were scored. **P<0.005 compared with S1P-untreated controls. (B) Cocultures were treated with PGE2 and S1P (1 μM) for 6 days. TRAP+ MNCs were counted. **P<0.005 versus S1P-untreated controls. NS, not significant. (C) Cocultures were carried out in the presence of PGE2 (10−8 M), VtD3 (10−10 M), and S1P (1 μM). TRAP+ MNC formation was assessed. **P<0.005 versus S1P-untreated controls. (D) Primary osteoblasts were treated with S1P and VtD3 (10−8 M) for 24 h. RANKL and OPG mRNA levels were analyzed by RT–PCR. (E) Osteoblasts were incubated for 24 h with indicated CM and RANKL mRNA levels were examined by RT–PCR. (F) T cells were activated with ionomycin and PMA and treated with S1P for 24 h. RANKL mRNA levels were determined by RT–PCR. (G) T cells were activated and treated with SPHK1-CM for 24 h. RT–PCR was performed to detect RANKL mRNA. (H) Coculture was carried out in the presence of FTY-720, VtD3 (10−9 M), and S1P (1 μM). TRAP+ MNCs were scored. (I) Osteoblasts were incubated with VtD3 (10−9 M), S1P, and FTY-720 for 24 h. RANKL mRNA was analyzed by RT–PCR.
S1P increases RANKL expression in osteoblasts by inducing PGE2 secretion through COX2 regulation
We next investigated whether PGE2 is involved in the S1P-dependent regulation of RANKL expression. First, we determined whether S1P stimulates PGE2 production. EIA assays revealed that S1P alone or in combination with VtD3 increased PGE2 production in osteoblasts (Figure 8A). As cyclooxygenase-2 (COX2) and microsomal prostaglandin E synthase-1 (mPGES1) are key enzymes involved in PGE2 production, we next examined the levels of these enzymes. Figure 8B shows that S1P increased protein levels of COX2 in osteoblasts. S1P effect on COX2 mRNA levels was inconsistent (data not shown). S1P had only minimal effect on mPGES1 protein level, whereas VtD3 increased it (Figure 8B). The CM from pOc that overexpressed SPHK1 also increased COX2 compared with the control CM (Figure 8C). The SPHK1 CM had no effect on mPGES1 level (Figure 8C). Whether the catalytic activity itself is stimulated by S1P was also tested by treating the cells with S1P for only 10 min during which no protein level change occurred (Supplementary Figure 5). S1P increased the enzymatic activity of both COX2 and mPGES1, whereas VtD3 alone did not have any effect (Figure 8D and E). Treatment with both S1P and VtD3 showed some synergy (Figure 8D and E). The COX2 inhibitor NS398 attenuated the induction of RANKL mRNA expression (Figure 8F) and the stimulation of osteoclastogenesis by S1P (Figure 8G). Taken together, S1P stimulates RANKL expression in osteoblasts via PGE2 production by upregulating the COX2 protein level and the catalytic activity of COX2 and mPGES1.
Figure 8.
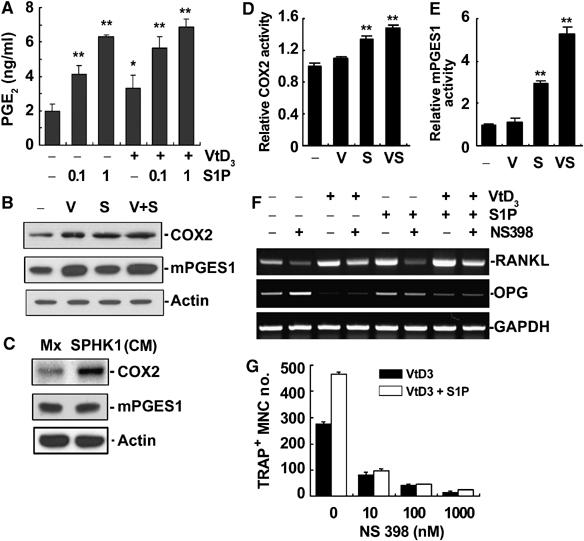
S1P increased RANKL expression by inducing PGE2 secretion through COX2 regulation. (A) Calvarial osteoblasts were treated for 24 h with S1P and VtD3 (10−8 M). PGE2 levels in the culture supernatants were determined by EIA. *P<0.01; **P<0.005 versus control. (B) Osteoblasts were treated with S1P (1 μM) and/or VtD3 (10−8 M) for 24 h. COX2 and mPGES1 protein levels were determined. (C) Osteoblasts were incubated for 24 h with the indicated CM. COX2 and mPGES1 proteins were detected. (D, E) Osteoblasts were stimulated with S1P (1 μM) and/or VtD3 (10−8 M) for 10 min. The COX2 and mPGES1 activities were determined as described in Materials and methods. (F) Osteoblasts were treated with S1P (1 μM), VtD3 (10−8 M), and NS398 (1 μM). RANKL and OPG mRNA levels were examined by RT–PCR. (G) Cocultures of BMMs and osteoblasts were carried out in the presence of VtD3 (10−9 M), S1P (1 μM), and NS398. TRAP+ MNCs were scored.
S1P regulates COX2 expression via ERK and p38 pathways in osteoblasts
S1P binding to its cell surface receptors has been reported to regulate the activity of MAPKs via G-protein-mediated signaling pathways (Spiegel and Milstien, 2003). Therefore, we sought to investigate the potential participation of MAPKs in the upregulation of COX2 and RANKL by S1P in osteoblasts. First, whether S1P can activate ERK and p38 MAPKs in osteoblasts was examined. S1P stimulated ERK and p38, and less effectively JNK, activation in a dose-dependent manner in the range of 0.1–10 μM (Figure 9A and data not shown). Next, the effect of PD98059, SB203580, and SP600125, specific inhibitors of the ERK, p38, and JNK pathways, respectively, on the S1P-stimulated induction of RANKL was assessed. As shown in Figure 9B, all the inhibitors suppressed RANKL induction by S1P. These inhibitors also blocked upregulation of COX2 by S1P in osteoblasts (Figure 9C). Effects of the inhibitors on COX2 mRNA were inconsistent (data not shown). In line with the suppressive effect on COX2, the inhibitors suppressed PGE2 production in osteoblasts (Figure 9D). Interestingly, S1P most strongly stimulated ERK and PD98059 was most potent in the inhibition of PGE2 production. Taken together, S1P activates ERK and p38, and less prominently JNK, to upregulate COX2 level, which leads to increased PGE2 production and subsequent RANKL induction.
Figure 9.
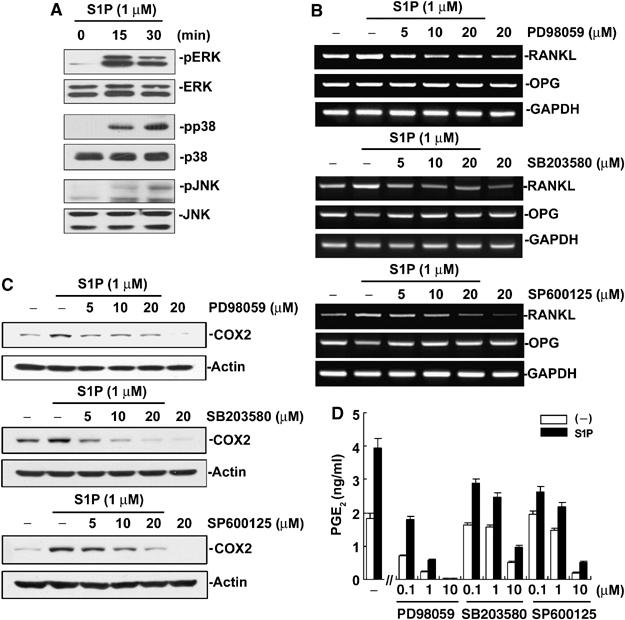
S1P regulates COX2 expression via MAPK pathways in osteoblasts. (A) Primary osteoblasts were stimulated with S1P. The phosphorylated and total levels of ERK, p38, and JNK were examined by Western blotting. (B) Osteoblasts were stimulated with S1P in the presence of PD98059, SB203580, and SP600125 for 24 h. The RANKL and OPG mRNA levels were examined by RT–PCR. (C) Cells were stimulated as in (B) and COX2 protein levels were examined. (D) Osteoblasts were treated with S1P (1 μM) and indicated inhibitor for 24 h. PGE2 in the culture supernatant was determined by EIA.
Discussion
In the present study, we demonstrate that SPHK1 has a dichotomous role in osteoclast differentiation, depending on the microenvironment where osteoclast precursors are present. SPHK1 has a negative role in osteoclastogenesis when a single population of osteoclast precursor cells (BMMs) is induced to differentiate by the soluble osteoclastogenic factor, RANKL. On the other hand, in coculture conditions, where cell surface RANKL on osteoblasts supports osteoclastic differentiation of BMMs, SPHK1 and its product, S1P, function positively in osteoclastogenesis. RANKL stimulates SPHK1 activity and S1P production in the precursor and differentiating BMMs during osteoclastogenesis. However, gene manipulation experiments show that suppression of SPHK1 expression by siRNA introduction enhances, whereas overexpression by SPHK1 retroviral transduction decreases osteoclast formation when BMMs are challenged with RANKL in the absence of osteoblasts. These observations suggest that SPHK1 activation and subsequent S1P production in response to the RANKL differentiation signal comprises a negative feedback loop in osteoclastogenesis. This BMM-autonomous negative feedback is likely to be mediated by intracellular S1P as exogenous addition of S1P does not have any effect on BMM osteoclastogenesis.
RANKL triggers the activation of MAPK family kinases in osteoclast precursors (Lee and Kim, 2003). Evidence for the requirement of p38 MAPKs for osteoclastogenesis has been accumulated by studies with pharmacological inhibitors and dominant-negative forms of kinases (Matsumoto et al, 2000; Lee et al, 2002; Li et al, 2002b; Huang et al, 2006). In contrast, conflicting results have been reported on the role of ERKs in osteoclast differentiation. The ERK pathway inhibitor PD98059 displayed no, suppressive, or augmenting effects on osteoclastogenesis depending on the cell types of osteoclast precursors, culture conditions, and concentrations of the inhibitor (Matsumoto et al, 2000; Hotokezaka et al, 2002; Lee et al, 2002). The contribution of ERKs to osteoclastogenesis may depend on the context of balance with other MAPK activities. In line with this notion, the balance in the p38 and ERK activation appears critical for the function of SPHK1 in osteoclast differentiation. The negative effect of SPHK1 overexpression and the positive effect of SPHK1 knockdown by siRNA on BMM osteoclastogenesis well correlate with the changes in the extents of ERK and p38 activation by RANKL; the increased p38 and decreased ERK activity parallel with enhanced osteoclastogenesis. Whether S1P modulates these MAPKs directly or whether S1P targets other upstream components in the signaling pathways to these MAPKs is an intriguing question to be addressed. Our study also indicates that the influence of SPHK1 on osteoclastogenesis is associated with RANKL regulation of NFATc1 and c-Fos, key transcription factors required for osteoclast differentiation. The induction of NFATc1 and c-Fos by RANKL in BMMs was dependent on p38 activity. Thus, p38 is an important mediator for the modulation of osteoclastogenesis by SPHK1.
In contrast to BMM single cultures, SPHK1 activity positively regulates osteoclastogenesis in cocultures of BMMs and osteoblasts. S1P significantly increases osteoclast formation in coculture treated with VtD3. This effect of S1P appears to be due to stimulation of PGE2 synthesis, which leads to an increase in RANKL expression in osteoblasts (Li et al, 2002a). As RANKL stimulation of BMMs results in S1P production and secretion, the induction of RANKL by S1P in osteoblasts constitutes a positive feedback loop for osteoclastogenesis in the coculture system. Thus, SPHK1 activity can generate both a negative and a positive feedback, depending on the culture system. In normal bone remodeling, osteoclast precursors and differentiating osteoclasts are likely to be in the vicinity of preosteoblasts, osteoblasts, and marrow stromal cells. Therefore, it is more likely that SPHK1 activity functions positively in osteoclast development under this condition. Under conditions where osteoblasts forming new bone matrix are not around, the negative feedback mechanism shown in BMM single culture may have a role in holding massive osteoclastogenesis in check to prevent unbalanced excessive bone resorption.
S1P has been shown to play important roles in cell migration for vascular and cardiac development and lymphocyte trafficking (Spiegel and Milstien, 2003). It also functions as an autocrine chemotactic factor to mast cells (Jolly et al, 2004). S1P produced by RANKL stimulation during osteoclastogenesis may attract, in both autocrine and paracrine manners, osteoclast precursors and other types of cells in bone. In fact, S1P exhibited chemotactic effect on the migration of BMM osteoclast precursors and osteoblasts. Attraction of osteoclast precursors will facilitate cell fusion with differentiating cells to generate multinucleated mature osteoclasts. However, this stimulatory effect on osteoclastogenesis may be compromised by the intrinsic action of intracellular S1P that negatively regulates osteoclast differentiation. Paracrine action of S1P to osteoblasts will attract the bone-forming cells to the vicinity of osteoclasts. In our study, S1P and the CM of BMMs transduced to overexpress SPHK1 stimulated migration and survival of osteoblasts. This paracrine mode of S1P function must be beneficial to coupling between bone resorption and new bone formation in normal bone remodeling.
COX2 catalyzes the rate-limiting step in prostaglandin biosynthesis (Dixon, 2004). The regulation of COX2 levels has been described at both transcriptional and post-transcriptional levels (Dixon et al, 2003; Ramsay et al, 2003). Several transcription factors including AP-1 (Guo et al, 2001) and CREB (Park et al, 2005) have been shown to be involved in the transcriptional induction of COX2. COX2 expression is also regulated by control of mRNA stability and translation efficiency, which is mediated through sequences in the 3′-untranslated region (Mukhopadhyay et al, 2003). In addition, protein degradation has been implicated in COX2 regulation (Zaric and Ruegg, 2005). These diverse aspects lend regulation mechanisms for COX2 complex, and the relative contribution of each element of COX2 regulation appears different in different cell types in response to different stimuli. MAPKs of the ERK, JNK, and p38 families have been implicated in both transcriptional and post-transcriptional control of COX2 expression (Xu et al, 2000; Rousseau et al, 2002, Park et al, 2005; Wu et al, 2006). However, the type of MAPKs involved and the regulation aspect targeted have been differentially described for different experimental conditions. We for the first time demonstrate that S1P increases COX2 in osteoblasts. Our study is also the first to show the involvement of p38 in S1P-induced COX2 upregulation in any cell type. The participation of ERK in S1P-stimulated COX2 increase has been documented in other cell types (Hsieh et al, 2006). The identification of precise targets of ERKs and p38 MAPKs in the COX2 increase by S1P in osteoblasts requires further investigation.
Our data indicate that S1P-stimulated COX2 induction and PGE2 production increase RANKL in osteoblasts, which consequently stimulates osteoclast formation. COX2, PGE2, and RANKL may comprise an important axis in pathogenic conditions involving bone erosion. In ovariectomized animal models of osteoporosis, increased COX2 activity, PGE2 production, and RANKL induction have been suggested to participate in osteoclast generation and bone resorption (Kanematsu et al, 2000). Recently, in synoviocytes from RA patients, S1P was shown to stimulate COX2 expression and PGE2 production (Kitano et al, 2006). RANKL is expressed by synovial fibroblasts and activated T cells from RA tissues (Gravallese et al, 2000; Takayanagi et al, 2000). Although direct evidence is lacking, the S1P–COX2–PGE2 axis is likely to participate in RANKL induction in RA synovium. In RA synovium, SPHK activation and S1P production during osteoclast differentiation will provoke more RANKL expression through COX2–PGE2 production in surrounding cells, such as preosteoblasts, synovial fibroblasts, and T cells. This will compose a vicious cycle of osteoclast generation and bone resorption, exacerbating the disease condition. S1P can also function to recruit activated T cells and other immune cells to the site of osteoclastogenesis in pathologic conditions. We observed that the CM from SPHK1-overexpressing pOc-stimulated T-cell migration. Furthermore, extracellular S1P enhanced RANKL expression in activated T cells. Therefore, S1P produced during osteoclastogenesis under inflammatory conditions, such as RA and periodontal diseases, may play a detrimental role through combined effects on T-cell chemotaxis and RANKL expression.
Based on our data presented here, a schematic diagram of osteoclastogenesis modulation by the SPHK1-S1P signaling is shown in Figure 10. When RANKL binds to RANK on BMMs, osteoclastic differentiation is triggered by activation of downstream signals such as p38, c-Fos, and NFATc1. S1P is also generated upon the activation of SPHK1 in response to RANKL. Intracellular production of S1P comprises a negative feedback loop in osteoclastogenesis owing to its inhibitory effect on p38 activation in BMMs. S1P exported to extracellular environment can bind to S1P receptors in osteoblasts. Binding of S1P to S1P receptors on osteoblasts leads to an increase in COX2 level through a mechanism dependent on ERK and p38. COX2 induction stimulates PGE2 production, which induces RANKL expression. The increased RANKL molecules in osteoblasts in turn bind to more RANK on BMMs and facilitate osteoclastic differentiation. Extracellular S1P also stimulates osteoblast migration and survival, contributing to the coupling between osteoclast and osteoblast activity.
Figure 10.
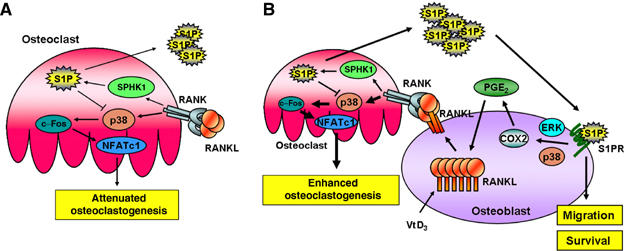
Schematic diagrams for osteoclastogenesis modulation by SPHK1 and S1P. (A) When RANKL binds RANK on BMMs, osteoclastogenesis is started by the activation of downstream signals such as p38, c-Fos, and NFATc1. S1P is also produced by the activation of SPHK1 in response to RANKL. Intracellular S1P constitutes a negative feedback loop in osteoclastogenesis by suppressing p38. (B) S1P produced in BMMs in response to RANKL is exported to extracellular environment and binds the S1P receptor in osteoblasts. The S1P–S1P receptor interaction stimulates MAPKs to upregulate COX2 and PGE2 in osteoblasts. PGE2 then induces RANKL expression, which augments osteoclastogenesis. In addition, extracellular S1P stimulates migration and survival of osteoblasts. In this situation, SPHK1 and S1P comprise a positive feedback loop for osteoclastogenesis.
In conclusion, our studies reveal that SPHK1 and S1P play important roles in osteoclastogenesis regulation. To our knowledge, this is the first study showing involvement of SPHK1 and S1P in osteoclast differentiation. Our results may provide a basis for exploring the therapeutic potential of S1P receptor antagonists for inflammatory disease-associated bone destruction.
Materials and methods
Reagents
See Supplementary data.
Plasmid preparation
The full-length human SPHK1 cDNA was reverse-transcribed amplified from HeLa cell mRNA and cloned into pSRα-HA vector. To generate retroviral vectors harboring HA-tagged SPHK1 (pMX-HA-SPHK1), DNA fragments encoding HA-SPHK1 were sub-cloned into the pMX retroviral vector. For construction of retroviral vectors for siRNA, targeting oligonucleotides were annealed and ligated into the pSuper-retro vector (Oligoengine, Seattle, WA) using BamHI and HindIII sites. The sequences of used oligonucleotides are provided in Supplementary Table 1.
BMM and osteoclast cultures
BMM culture and osteoclast generation were performed as described (Huang et al, 2006; Supplementary data).
Coculture
Mouse calvarial osteoblasts were isolated from 2-day-old ICR mice. The calvarias were digested in HBSS containing 0.02% type I collagenase (GIBCO BRL), 0.05% trypsin, and 0.53 mM EDTA for 20 min at 37°C with vigorous shaking. The digestion procedure was repeated six times, and the cells isolated by the last three digestions were combined. The isolated cells were seeded at 5 × 105 cells per 10-cm culture dish and grown to confluence. The cells of passage 2 were used for coculture. 1 × 104 osteoblasts and 1 × 105 BMMs were seeded in each well of 96-well plates in α-MEM/10% FBS and cultured for 6 days.
Retroviral gene transfer and conditioned medium
Generation of retroviruses and BMM infection were performed as described (Huang et al, 2006; Supplementary data). The CM used in Figures 6, 7 and 8 was collected from pOc generated by culturing BMMs infected with indicated retroviruses for 2–3 days with 100 ng/ml RANKL and 30 ng/ml M-CSF.
Sphingosine kinase activity assay
Sphingosine kinase activity was measured as previously described (Olivera and Spiegel, 1998). Cells were scraped in SPHK assay buffer (20 mM Tris (pH 7.4), 20% glycerol, 1 mM mercaptoethanol, 1 mM EDTA, 1 mM sodium orthovanadate, 40 mM β-glycero-phosphate, 15 mM NaF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, 1 mM phenylmethylsulfonyl fluoride, and 0.5 mM 4-deoxypyridoxine) and disrupted by freeze–thawing. An 80 μg portion of cell extracts in 185 μl volume was mixed with 5 μl of [γ-32P]ATP (5 μCi) containing MgCl2 (0.2 M) and 10 μl of 1 mM sphingosine (dissolved in 5% Triton X-100) and then incubated for 30 min at 37°C. The reaction was terminated with 10 μl of 1 N HCl. A 400 μl portion of chloroform/methanol/HCl (100:200:1, v/v) mixture was added and mixed. Then, 120 μl of chloroform and 120 μl of 2 M KCl were added and phases separated by centrifugation. The organic phase was dried and resolved by thin-layer chromatography (TLC) on silica gel G60 with SPHK1-butanol/methanol/acetic acid/water (80:20:10:20, v/v). The radioactive spots corresponding to S1P were detected using phosphoimager, BAS-1500 (Fujifilm Co. Ltd).
S1P level determination
Cells were seeded at 1 × 106 cells/well in six-well plates and incubated for 4 h with phosphate-free DMEM and metabolically labeled with [32P]orthophosphate for 4 h. Cells were washed with phosphate-free DMEM and treated with 1 μg/ml RANKL in 1 ml/well volume for indicated times. To determine extracellular S1P, the whole culture medium was collected and centrifuged. One milliliter of the supernatant was mixed with 1 ml chloroform, 1 ml methanol, and 9 μl HCl. The organic phase was dried under vacuum, and resolved by TLC as described above. To determine intracellular S1P levels, cells were scraped in 400 μl methanol and 25 μl HCl was added. Lipids were extracted by the addition of 400 μl chloroform, 400 μl KCl, and 40 μl 3 M NaCl. The aqueous phase was mixed with 50 μl HCl and then with 400 μl chloroform, and lipid phase was extracted. The lipid phase extracts were combined, dried under vacuum, and resolved by TLC as above.
Western blotting, co-immunoprecipitation, and RT–PCR analyses
Western blot, co-immunoprecipitation, and RT–PCR experiments were conducted as previously described (Shin et al, 2002; Ha et al, 2003). For Western blotting shown in Figure 1A, boiling sample buffer was used to lyse the cells. For co-immunoprecipitation shown in Figure 1F, a light chain-specific secondary antibody was used and experimental details are described in Supplementary data. The sequences of PCR primers are provided in Supplementary data. The number of amplification cycles was determined to be in a linear range of amplification. Twenty-two cycles were run for GAPDH and 30–35 cycles for the others.
Migration assay
Migration assay was conducted using a 24-well Transwell plate (Corning) possessing 8-μm pores. Cells were serum-starved for 3 h and 5 × 105 osteoblasts or 1 × 106 T cells were applied to the upper well and incubated with various doses of S1P or CM in the lower well. After the indicated time, the side of membranes facing the upper well was wiped of cells and stained with hematoxylin. The number of migrated cells was counted.
T-cell preparation
Total spleen cells were separated from 6-week-old ICR mouse and processed with CD4+ T-cell isolation kits using magnetic cell separation (MACS) (Miltenyi Biotech) as per the manufacturer's instructions. To activate T cells, 750 ng/ml ionomycin and 10 ng/ml PMA were added and cultured for 24 h.
PGE2 production assay
Osteoblasts were incubated with S1P and VtD3 for 24 h. PGE2 secreted into culture supernatants was measured by enzyme-linked immunosorbent assay using a commercial kit (Amersharm).
COX2 and mPGES1 activity assay
Osteoblasts were incubated in the absence or presence of 10 nM NS398 (COX2 inhibitor) or 1 μM MKK886 (mPGES1 inhibitor) for 30 min. The medium was changed with one supplemented with 30 μM arachidonic acid and cells were stimulated with S1P and/or VtD3 for 10 min at 37°C. The released PGE2 was measured as above. The PGE2 level difference between inhibitor-treated and untreated samples was considered as the corresponding enzyme activity.
Statistical analysis
All experiments except ones involving 32Pi-labeling were performed 3–4 times. Each quantitative experiment was performed in 3–5 replicates, except 32Pi-labeling experiments performed in duplicate. The data are expressed as means±s.d. from the representative experiments. Statistical significance of differences between experimental and control groups was evaluated by Student's t test.
Supplementary Material
Supplementary Figures and Tables
Acknowledgments
We thank Dr T Kitamura for kindly providing pMX retroviral vector. This work was supported by the 21C Frontier Functional Proteomics Project Grant (FPR05C2-280) and the Basic Research Program (R01-2005-000-10073-0) from the Korea Science & Engineering Foundation.
References
- Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423: 337–342 [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277: 21453–21457 [DOI] [PubMed] [Google Scholar]
- Dixon DA (2004) Dysregulated post-transcriptional control of COX-2 gene expression in cancer. Curr Pharm Des 10: 635–646 [DOI] [PubMed] [Google Scholar]
- Dixon DA, Balch GC, Kedersha N, Anderson P, Zimmerman GA, Beauchamp RD, Prescott SM (2003) Regulation of cyclooxygenase-2 expression by the translational silencer TIA-1. J Exp Med 198: 475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Hannun YA (2004) The complex life of simple sphingolipids. EMBO R 5: 777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeler M, Goetzl EJ (2002) Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. FASEB J 16: 1874–1878 [DOI] [PubMed] [Google Scholar]
- Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR (2000) Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum 43: 250–258 [DOI] [PubMed] [Google Scholar]
- Guo YS, Hellmich MR, Wen XD, Townsend CM Jr (2001) Activator protein-1 transcription factor mediates bombesin-stimulated cyclooxygenase-2 expression in intestinal epithelial cells. J Biol Chem 276: 22941–22947 [DOI] [PubMed] [Google Scholar]
- Huang H, Ryu J, Ha J, Chang EJ, Kim HJ, Kim HM, Kitamura T, Lee ZH, Kim HH (2006) Osteoclast differentiation requires TAK1 and MKK6 for NFATc1 induction and NF-kappaB transactivation by RANKL. Cell Death Differ 13: 1879–1891 [DOI] [PubMed] [Google Scholar]
- Ha H, Kwak HB, Lee SK, Na DS, Rudd CE, Lee ZH, Kim HH (2003) Membrane rafts play a crucial role in receptor activator of nuclear factor kappaB signaling and osteoclast function. J Biol Chem 278: 18573–18580 [DOI] [PubMed] [Google Scholar]
- Hotokezaka H, Sakai E, Kanaoka K, Saito K, Matsuo K, Kitaura H, Yoshida N, Nakayama K (2002) U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. J Biol Chem 277: 47366–47372 [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Wu CB, Sun CC, Liao CH, Lau YT, Yang CM (2006) Sphingosine-1-phosphate induces COX-2 expression via PI3K/Akt and p42/p44 MAPK pathways in rat vascular smooth muscle cells. J Cell Physiol 207: 757–766 [DOI] [PubMed] [Google Scholar]
- Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem 278: 46832–46839 [DOI] [PubMed] [Google Scholar]
- Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T (2002) Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem 277: 41147–41156 [DOI] [PubMed] [Google Scholar]
- Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S (2004) Transactivation of sphingosine-1-phosphate receptors by FcepsilonRI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med 199: 959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneider NC, Lindner J, Feistritzer C, Sturn DH, Mosheimer BA, Djanani AM, Wiedermann CJ (2004) The immune modulator FTY720 targets sphingosine-kinase-dependent migration of human monocytes in response to amyloid beta-protein and its precursor. FASEB J 18: 1309–1311 [DOI] [PubMed] [Google Scholar]
- Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y (2000) Prostaglandin E2 induces expression of receptor activator of nuclear factor-kappa B ligand/osteoprotegrin ligand on pre-B cells: implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res 15: 1321–1329 [DOI] [PubMed] [Google Scholar]
- Kitano M, Hla T, Sekiguchi M, Kawahito Y, Yoshimura R, Miyazawa K, Iwasaki T, Sano H (2006) Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum 54: 742–753 [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J (2001) Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 20: 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMontagne K, Littlewood-Evans A, Schnell C, O'Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau G, Billy E, Theuer A, Hla T, Wood J (2006) Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res 66: 221–231 [DOI] [PubMed] [Google Scholar]
- Lee SE, Woo KM, Kim SY, Kim HM, Kwack K, Lee ZH, Kim HH (2002) The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone 30: 71–77 [DOI] [PubMed] [Google Scholar]
- Lee ZH, Kim HH. (2003) Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun 305: 211–214 [DOI] [PubMed] [Google Scholar]
- Li X, Pilbeam CC, Pan L, Breyer RM, Raisz LG. (2002a) Effects of prostaglandin E2 on gene expression in primary osteoblastic cells from prostaglandin receptor knockout mice. Bone 30: 567–573 [DOI] [PubMed] [Google Scholar]
- Li X, Udagawa N, Itoh K, Suda K, Murase Y, Nishihara T, Suda T, Takahashi N (2002b) p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 143: 3105–3113 [DOI] [PubMed] [Google Scholar]
- Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem 275: 19513–19520 [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13: 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Alfred H, Merrill AH Jr, Milstien S, Spiegel S (2005) SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem 280: 37118–37129 [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M (2000) Involvement of p38 mitogen-activated protein kinase signalling pathway in osteoclastogenesis mediated by receptor activator of NFκ-B ligand (RANKL). J Biol Chem 275: 31155–31161 [DOI] [PubMed] [Google Scholar]
- Matsuo K, Galson DL, Zhao C, Peng L, Laplace C, Wang KZ, Bachler MA, Amano H, Aburatani H, Ishikawa H, Wagner EF (2004) Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem 279: 26475–26480 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S (2003) Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell 11: 113–126 [DOI] [PubMed] [Google Scholar]
- Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J (1999) Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4: 353–362 [DOI] [PubMed] [Google Scholar]
- Olivera A, Spiegel S (1998) Sphingosine kinase. assay and product analysis. Methods Mol Biol 105: 233–242 [DOI] [PubMed] [Google Scholar]
- Park SW, Sung MW, Heo DS, Inoue H, Shim SH, Kim KH (2005) Nitric oxide upregulates the cyclooxygenase-2 expression through the cAMP-response element in its promoter in several cancer cell lines. Oncogene 24: 6689–6698 [DOI] [PubMed] [Google Scholar]
- Ramsay RG, Ciznadija D, Vanevski M, Mantamadiotis T (2003) Transcriptional regulation of cyclo-oxygenase expression: three pillars of control. Int J Immunopathol Pharmacol 16: 59–67 [PubMed] [Google Scholar]
- Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P (2002) Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J 21: 6505–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JN, Kim I, Lee JS, Koh GY, Lee ZH, Kim HH (2002) A novel zinc finger protein that inhibits osteoclastogenesis and the function of tumor necrosis factor receptor-associated factor 6. J Biol Chem 277: 8346–8353 [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4: 397–407 [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 20: 345–357 [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, Koshihara Y, Oda H, Nakamura K, Tanaka S (2000) Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum 43: 259–269 [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3: 889–901 [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP (2003) Genetic regulation of osteoclast development and function. Nat Rev Genet 4: 638–649 [DOI] [PubMed] [Google Scholar]
- Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, Choi Y (1999) TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell 4: 1041–1049 [DOI] [PubMed] [Google Scholar]
- Wu G, Luo J, Rana JS, Laham R, Sellke FW, Li J (2006) Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc Res 69: 512–519 [DOI] [PubMed] [Google Scholar]
- Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, D'Andrea RJ, Gamble JR, Vadas MA (2002) Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem 277: 7996–8003 [DOI] [PubMed] [Google Scholar]
- Xu K, Robida AM, Murphy TJ (2000) Immediate-early MEK-1-dependent stabilization of rat smooth muscle cell cyclooxygenase-2 mRNA by Galpha(q)-coupled receptor signaling. J Biol Chem 275: 23012–23019 [DOI] [PubMed] [Google Scholar]
- Yopp AC, Randolph GJ, Bromberg JS (2003) Leukotrienes, sphingolipids, and leukocyte trafficking. J Immunol 171: 5–10 [DOI] [PubMed] [Google Scholar]
- Zaric J, Ruegg C (2005) Integrin-mediated adhesion and soluble ligand binding stabilize COX-2 protein levels in endothelial cells by inducing expression and preventing degradation. J Biol Chem 280: 1077–1085 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables