

Abstract
Initiation of DNA synthesis involves the loading of the MCM2–7 helicase onto chromatin by Cdt1 (origin licensing). Geminin is thought to prevent relicensing by binding and inhibiting Cdt1. Here we show, using Xenopus egg extracts, that geminin binding to Cdt1 is not sufficient to block its activity and that a Cdt1–geminin complex licenses chromatin, but prevents rereplication, working as a molecular switch at replication origins. We demonstrate that geminin is recruited to chromatin already during licensing, while bulk geminin is recruited at the onset of S phase. A recombinant Cdt1–geminin complex binds chromatin, interacts with the MCM2–7 complex and licenses chromatin once per cell cycle. Accordingly, while recombinant Cdt1 induces rereplication in G1 or G2 and activates an ATM/ATR-dependent checkpoint, the Cdt1–geminin complex does not. We further demonstrate that the stoichiometry of the Cdt1–geminin complex regulates its activity. Our results suggest a model in which the MCM2–7 helicase is loaded onto chromatin by a Cdt1–geminin complex, which is inactivated upon origin firing by binding additional geminin. This origin inactivation reaction does not occur if only free Cdt1 is present on chromatin.
Keywords: Cdt1, DNA replication licensing, geminin, prereplication complex, rereplication
Introduction
Within each cell cycle, chromatin is faithfully replicated exactly once. To ensure that no part of the genome is rereplicated, chromatin must acquire replication competence before S phase in the so-called licensing reaction (Blow and Laskey, 1988). Entry into S phase then blocks further licensing for the remaining cell cycle (reviewed in Machida et al, 2005; DePamphilis et al, 2006). Chromatin licensing takes place at the end of mitosis, beginning with the binding of the ORC complex to chromatin at multiple potential origins of replication, and is followed by the assembly of two other factors, Cdc6 and Cdt1 (reviewed in Bell and Dutta, 2002). The latter interacts with the MCM2–7 DNA helicase complex (Tanaka and Diffley, 2002; Yanagi et al, 2002; Cook et al, 2004; Ferenbach et al, 2005) and loads it onto chromatin to form prereplication complexes (pre-RCs). The resulting licensed replication origins are activated at the start of S phase in a reaction mediated by two main kinase/cyclin systems, CDC7/Dbf4 and cyclinE/CDK2.
Cdt1 is ubiquitinylated at the onset of S phase, and its consequent destruction is one mechanism which may prevent the relicensing of replication origins (Liu et al, 2004; Nishitani et al, 2004, 2006; Sugimoto et al, 2004; Arias and Walter, 2005; Li and Blow, 2005; Hu and Xiong, 2006; Senga et al, 2006). In metazoans, relicensing is further inhibited by an additional mechanism involving geminin, a protein that binds to Cdt1 and blocks the loading of the MCM2–7 helicase onto chromatin (McGarry and Kirschner, 1998; Wohlschlegel et al, 2000; Nishitani et al, 2001; Quinn et al, 2001; Tada et al, 2001). Geminin is degraded by the anaphase-promoting complex (APC), suggesting that its destruction at the end of mitosis liberates Cdt1 and enables a new round of origin licensing. However, the mechanism of licensing is probably more complex, as recent reports have shown that polyubiquitination of geminin in anaphase does not necessarily destroy the protein. Instead, it becomes incapable of interacting with Cdt1, whereas subsequent nuclear import of the protein in G1 restores its ability to bind to and block Cdt1 (Hodgson et al, 2002; Li and Blow, 2005).
The molecular event that reactivates geminin upon nuclear import remains unknown. It is also unclear why a part of the geminin pool in egg extracts is already complexed with Cdt1 in the absence of chromatin and what the function of this complex might be during the licensing reaction (Hodgson et al, 2002; Maiorano et al, 2004; Yoshida et al, 2005). In addition to its negative role in the control of DNA replication, geminin was recently shown to be required for cell cycle progression, as it complexes Cdt1 during mitosis and protects it from degradation, thereby maintaining sufficient amounts of Cdt1 for the licensing of replication origins in the following cell cycle (Ballabeni et al, 2004). This result is consistent with the unexpected finding that geminin is present at high levels in fast-dividing cells and also in cancer cells, contrary to what one might expect from its established role in blocking licensing and DNA replication (Wohlschlegel et al, 2002; Gonzalez et al, 2004; reviewed in Pitulescu et al, 2005).
In Xenopus, depletion of geminin results in relicensing and limited rereplication (Yoshida et al, 2005). This result was explained by assuming that geminin must be present before the degradation of Cdt1 in order to prevent rereplication. Adding recombinant Cdt1 during G2 is also sufficient to give rise to relicensing and massive rereplication (Arias and Walter, 2005; Li and Blow, 2005; Maiorano et al, 2005; Yoshida et al, 2005).
In this study, we show that geminin does not necessarily block the activity of Cdt1, but rather controls it through formation of a Cdt1–geminin complex, which acts as a molecular switch, turning DNA replication origins on or off. During licensing, geminin is recruited to chromatin together with Cdt1 and does not block licensing activity. Indeed, a complex of recombinant Cdt1 and geminin is capable of loading the MCM2–7 helicase onto chromatin to license DNA for replication. Furthermore, whereas the addition of free Cdt1 either in G1 or G2 leads to rereplication and an ATM/ATR-dependent checkpoint response, adding the active Cdt1–geminin complex, even in large excess, induces neither rereplication nor a checkpoint response. The stoichiometry of the Cdt1–geminin complex is critical for its licensing activity, as the addition of more geminin to the complex blocks its ability to load the MCM2–7 helicase onto chromatin. Taken altogether, these data suggest that a Cdt1–geminin complex, and not free Cdt1, is the natural MCM2–7 loader whose activity can be precisely regulated throughout the cell cycle by changing its stoichiometry, explaining geminin's essential function in cell cycle progression.
Results
A fraction of geminin binds to chromatin already during the licensing reaction in a Cdt1-dependent manner
Western blot analysis has shown that the bulk of geminin in Xenopus egg extracts binds to chromatin at the onset of S phase (Maiorano et al, 2004; Arias and Walter, 2005; Li and Blow, 2005; Yoshida et al, 2005). Accordingly with these findings, using immunofluorescence, we also detected significant loading of geminin onto chromatin at S-phase onset (here at 40 min), followed by a decrease at the end of S phase/G2 (Figure 1A). However, we reproducibly detected earlier binding of geminin to chromatin, within 5 min after adding sperm chromatin to egg extract (Figure 1A). The early chromatin binding of geminin was not detected in a control reaction using a nonspecific antibody (Figure 1B, left panel, pre-immune), nor when geminin was depleted from the extract (Figure 1B, right panel and Supplementary Figure S1), demonstrating that the observed signal was geminin-specific. We next confirmed geminin binding during the first 5 min after sperm chromatin addition by Western blotting. Figure 1C shows that geminin is recruited to chromatin with the same kinetics as Cdt1 and before the loading of the MCM2–7 complex. Interestingly, further recruitment of bulk geminin occurs at onset of S phase, which appears to be synchronized with the previously reported polyubiquitination of Cdt1 (Figure 1C, Cdt1 long exposure, Arias and Walter, 2005). Finally, we tried to estimate the amount of Cdt1 and geminin bound to chromatin by a quantification experiment shown in Figure 1D. Chromatin was purified after different incubation times and the signal intensities of Cdt1 and geminin were compared to dilutions of the corresponding recombinant proteins (see Figure 2A) loaded onto the same gel. Whereas the Cdt1 signal remained constant during the time course of the experiment, geminin levels increased considerably at a time corresponding to the onset of DNA synthesis. From these and other independent experiments, the Cdt1–geminin ratio was estimated to be about 1:2.4 during licensing and to exceed 1:4 after initiation. We have previously shown that the mean inter-origin spacing is around 20 kbp in such replication experiments (Lemaitre et al, 2005). We estimate that five Cdt1 molecules and 12 geminin molecules are bound per origin during licensing. After initiation, about 20 geminin molecules are bound per origin while the Cdt1 level remains unchanged. We conclude that Cdt1 and geminin are recruited synchronously to chromatin during the licensing reaction, and that the stoichiometric ratio of geminin to Cdt1 is increased after initiation of DNA replication.
Figure 1.
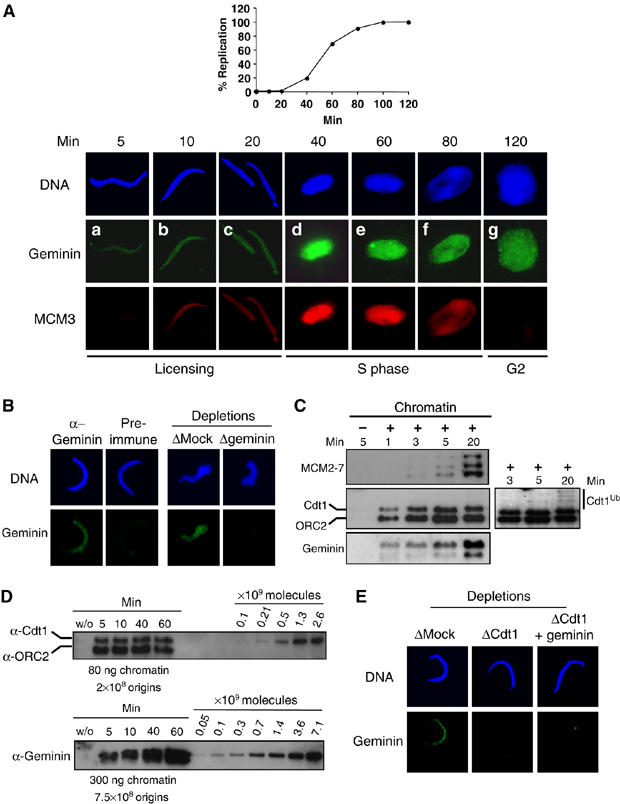
A fraction of geminin assembles in a Cdt1-dependent manner during licensing onto chromatin. (A) Upper panel: kinetics of the replication reaction performed as described in Materials and methods and expressed as the percentage of replicated sperm DNA compared to the total input DNA. Lower panel: sperm chromatin incubated in egg extract for indicated times and further detergent extracted and processed for immunofluorescence. Geminin was visualized by FITC (green) and MCM3 by Texas red (red). DNA was stained with Hoechst and is shown in blue. (B) Left panel: immunofluorescence of chromatin purified as in (A) after 5 min incubation in egg extract and stained either with geminin antibody or with preimmune serum. Right panel: immunofluorescence of chromatin after 5 min incubation in egg extract that was either Mock- or geminin-depleted. (C) Western blot analysis of Cdt1, MCM2–7, geminin and ORC2 (as a loading control) on chromatin purified after the indicated time. Also shown is a Mock purification (first lane), without added sperm chromatin, to determine background staining by non-chromatin bound proteins. A longer exposure of the Cdt1 blot is also presented to show polyubiquitination of Cdt1 at the onset of S phase. (D) Western blot analysis of purified chromatin following the indicated times of incubation in egg extract (S-phase entry at 40 min). A dilution series of recombinant Cdt1 and geminin was loaded on the same gel. The amount of loaded chromatin is indicated, as well as the corresponding number of origins (Lemaitre et al, 2005). (E) Geminin signal on chromatin incubated 5 min in egg extract, either Mock-depleted, Cdt1-depleted, or Cdt1-depleted and complemented with geminin before addition of sperm chromatin.
Figure 2.
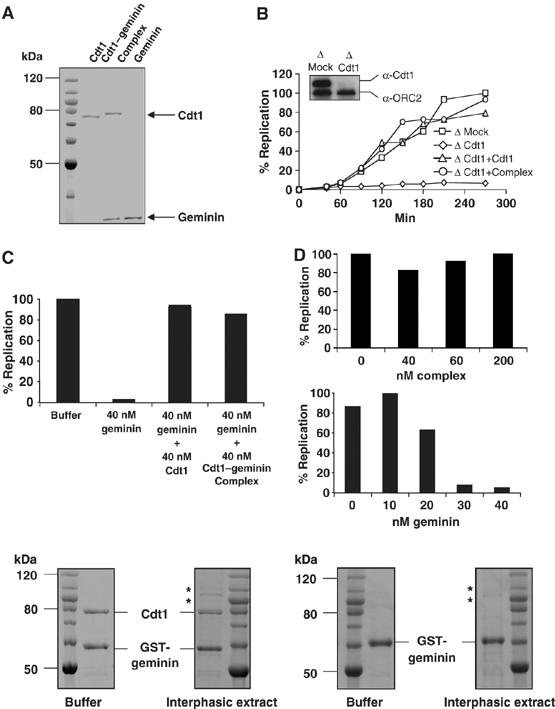
The recombinant Cdt1–geminin complex is active in licensing DNA for replication. (A) Coomassie-stained SDS–PAGE of purified Cdt1, Cdt1–geminin complex and geminin. Also shown is a molecular weight marker. Protein concentrations were determined by comparison with BSA dilution series. (B) Kinetics of DNA replication in either Mock-depleted extract (squares) or in Cdt1-depleted extract supplemented by buffer (diamonds), 40 nM Cdt1 (triangles) or 40 nM Cdt1–geminin complex (circles). The insert shows a Western blot for Cdt1 and ORC2 (as loading control) of Mock- and Cdt1-depleted extract. (C) Percentage of replicated sperm chromatin after 100 min in egg extract supplemented by buffer, 40 nM geminin, 40 nM geminin plus 40 nM Cdt1, or 40 nM geminin plus 40 nM Cdt1–geminin complex. (D) Percentage of replicated sperm chromatin after 100 min in egg extract supplemented by buffer or by increasing amounts of Cdt1–geminin complex (upper panel) or by geminin (lower panel). (E) Incubation of Cdt1–geminin complex immobilized by GST-geminin to GSH-Sepharose (left panels) or GST-geminin alone immobilized in the same way (right panels) either in buffer or in interphasic egg extract for 40 min at room temperature (Coomassie stain). Additional bands visible after incubation in egg extract are proteins that bound nonspecifically to the GSH-Sepharose (marked by asterisks).
As geminin is present in egg extracts both in a free form as well as in a complex with Cdt1 (Hodgson et al, 2002; Maiorano et al, 2004; Yoshida et al, 2005), depleting Cdt1 only removes the Cdt1-bound geminin pool. If the endogenous Cdt1–geminin complex is recruited to chromatin, then depletion of Cdt1 should leave only free geminin in the extract, unable to bind chromatin. Figure 1E shows that this is indeed the case: Cdt1 depletion abolished geminin binding to chromatin during licensing, strongly suggesting that geminin can only be recruited to chromatin when it is complexed with Cdt1. In agreement with this result, addition of an excess of free geminin to the Cdt1-depleted extract failed to restore geminin binding to chromatin (Figure 1E, right panel and Supplementary Figure S2). Further evidence for an endogenous Cdt1–geminin complex bound to chromatin during licensing was obtained from co-immunoprecipitation experiments. Sperm chromatin was assembled in egg extract for 10 min, purified and salt-washed to remove Cdt1 as described (Maiorano et al, 2000a, 2000b). Co-immunoprecipitations from the soluble fraction, performed with Cdt1 and geminin antibodies, showed that both proteins co-precipitate in both cases (immunoprecipitation of geminin co-precipitates Cdt1 and immunoprecipitation of Cdt1 co-precipitates geminin, Supplementary Figure S3).
From these experiments, we conclude that geminin is recruited in a complex with Cdt1 to chromatin during the licensing reaction.
A Cdt1–geminin complex is active in origin licensing
To investigate more in detail the recruitment of the Cdt1–geminin complex to chromatin, we expressed and purified recombinant free Cdt1, free geminin, as well as the Cdt1–geminin complex. To purify the complex, we coexpressed both proteins and used a double-affinity tag strategy (His6-tagged Cdt1 and GST-TEV-tagged geminin) to ensure that no free Cdt1- or geminin subunits were present in the final purified fraction (Figure 2A, see also Materials and methods. Although the complex can also be assembled from individual purified proteins in vitro (Supplementary Figure S4), we found coexpression more efficient to obtain a stable, homogeneous complex). Figure 2A shows the purified recombinant proteins used in this study. The Cdt1 protein in the Cdt1–geminin complex lane migrates slightly slower on SDS–PAGE than free Cdt1, as expected due to its His6-tag. Further characterization of the purified complex by dynamic light scattering demonstrated that the complex is a homogeneous and monodispersed particle with a diameter of about 13.5 nm (Supplementary Figure S5 and Discussion).
We next addressed whether the Cdt1–geminin complex would be active in licensing. To test this, egg extracts were depleted of Cdt1 and then complemented by either free recombinant Cdt1 or by the Cdt1–geminin complex. As shown in Figure 2B, a Cdt1-depleted extract did not replicate sperm chromatin, but DNA replication could be restored by addition of 40 nM free Cdt1, as reported before (Maiorano et al, 2000b). Importantly, addition of 40 nM of Cdt1–geminin complex to the Cdt1-depleted extracts restored DNA replication with the same efficiency as addition of free Cdt1. Moreover, like free Cdt1, the Cdt1–geminin complex also restored block of DNA replication in egg extracts treated with an excess of free geminin (Figure 2C). Finally, Figure 2D shows that the Cdt1–geminin complex did not block DNA replication even when added at high concentrations to egg extracts (up to 200 nM), whereas a 30 nM addition of free geminin led to complete inhibition of DNA replication. We conclude that the Cdt1–geminin complex is active in licensing chromatin for DNA replication.
These observations also confirm that the Cdt1–geminin complex is stable, as its disassembly would produce, apart from free Cdt1, free geminin, which could block licensing. To further address the stability of the Cdt1–geminin complex, it was bound to GSH-Sepharose via the GST-tag on geminin and washed with more than 1000 volumes of buffer. No loss of Cdt1 was detected (see below and data not shown), and no dissociation was observed upon addition of up to 2 M MgCl2, confirming previous results showing that the complex is stable up to 4 M urea (Tada et al, 2001; Saxena et al, 2004 and data not shown).
The tight binding of Cdt1 to geminin was also not affected upon incubation of the immobilized complex in egg extract (Figure 2E). The possibility that, during this incubation, recombinant Cdt1 was exchanged for endogenous Cdt1 is unlikely for two reasons. First, we did not detect the presence of the faster-migrating endogenous Cdt1 after recovery of the Sepharose-bound complex. Second, recombinant geminin that was similarly immobilized to GSH-Sepharose did not bind detectable amounts of endogenous Cdt1 under the same conditions (Figure 2E, right panel). These results confirm that Cdt1 and geminin form a highly stable complex, which is as active as free Cdt1 in replication licensing.
The Cdt1–geminin complex is recruited to chromatin and interacts with the MCM2–7 complex
We further examined the licensing activity of the Cdt1–geminin complex by analyzing the binding of geminin and the MCM2–7 helicase to chromatin in the presence of identical amounts of Cdt1, geminin, or the Cdt1–geminin complex. Adding free Cdt1 did not change the level of geminin bound to chromatin, whereas adding free geminin slightly increased it (Figure 3, compare E, F and G). In contrast, adding the same amount of the Cdt1–geminin complex caused a dramatic increase of geminin on chromatin (Figure 3H). Importantly, while a small amount of additional free geminin on chromatin blocked MCM2–7 loading, a much higher level recruited to chromatin as part of the Cdt1–geminin complex did not interfere with MCM2–7 loading (Figure 3, compare K and L). These data confirm that geminin is efficiently recruited to chromatin during licensing only when it is added in a complex with Cdt1. They also confirm the high stability of the Cdt1–geminin complex in egg extract, as free geminin is poorly recruited to chromatin (Figure 3, compare G and H). Similar results were obtained using a high-speed extract that is devoid of membranes and unable to support DNA replication, in agreement with the conclusions that the observed reactions occurred during licensing, before initiation of DNA synthesis (data not shown).
Figure 3.
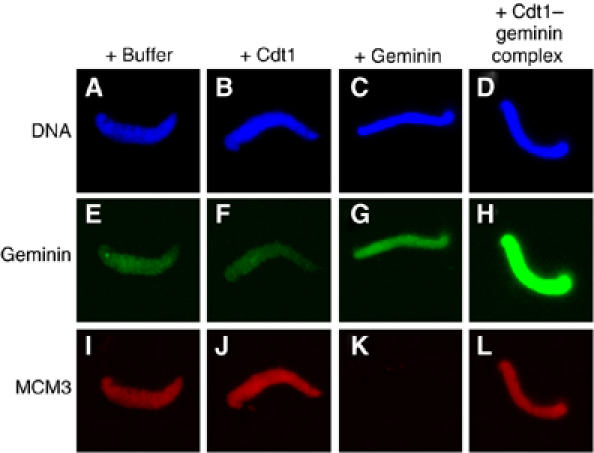
Geminin is massively recruited to chromatin without blocking MCM2–7 loading only if added in a complex with Cdt1. Immunofluorescence of geminin (FITC, green) and MCM3 (Texas red, red) on sperm chromatin (Hoechst, blue) after 5 min incubation in egg extract. As indicated, equal amounts of Cdt1, geminin or the Cdt1–geminin complex were added to the egg extract 10 min before the addition of sperm chromatin.
Together, these data show that the Cdt1–geminin complex can be recruited to chromatin during licensing to load the MCM2–7 complex, and that the amount of geminin that is bound to chromatin before S phase depends on the Cdt1–geminin complex present in the extract.
Whereas in yeast a preformed, non-chromatin bound complex of Cdt1 and MCM2–7 has been reported (Tanaka and Diffley, 2002), there has not been any evidence for such a complex in vertebrates. However, in vitro reconstitution experiments using recombinant and purified metazoan proteins were successfully employed to detect such a direct interaction, and it was further suggested that geminin binding would sterically prevent Cdt1 from interacting with the MCM2–7 complex (Yanagi et al, 2002; Cook et al, 2004; Lee et al, 2004; Ferenbach et al, 2005). We therefore tested the ability of the Cdt1–geminin complex to interact with the MCM2–7 complex under in vitro conditions. The MCM2–7 complex was purified from egg extracts as previously described (Maiorano et al, 2000a) and incubated with GSH-Sepharose bound Cdt1, geminin or Cdt1–geminin complex. As shown in Supplementary Figure S6, the MCM2–7 complex could interact equally well with Cdt1 and with the Cdt1–geminin complex, but not with geminin alone. These data confirm that, as for Cdt1, the Cdt1–geminin complex can bind directly to the MCM2–7 proteins in vitro.
Cdt1 recruitment to chromatin without geminin induces a checkpoint response
It has been reported that the addition of recombinant Cdt1 to egg extract in G1 induces limited rereplication, while its addition in G2 induces massive rereplication and checkpoint activation (Arias and Walter, 2005; Li and Blow, 2005; Maiorano et al, 2005; Yoshida et al, 2005). We reproducibly observed that adding free Cdt1 to egg extract before the addition of sperm chromatin significantly slowed down DNA replication. Interestingly, such an effect was not observed when the Cdt1–geminin complex was added (Figure 4A, left panel). The addition of caffeine counteracted this Cdt1-specific slow down of replication, suggesting that a surplus of free Cdt1, but not of the Cdt1–geminin complex, may induce a checkpoint response during the first round of DNA replication (Figure 4A, right panel).
Figure 4.
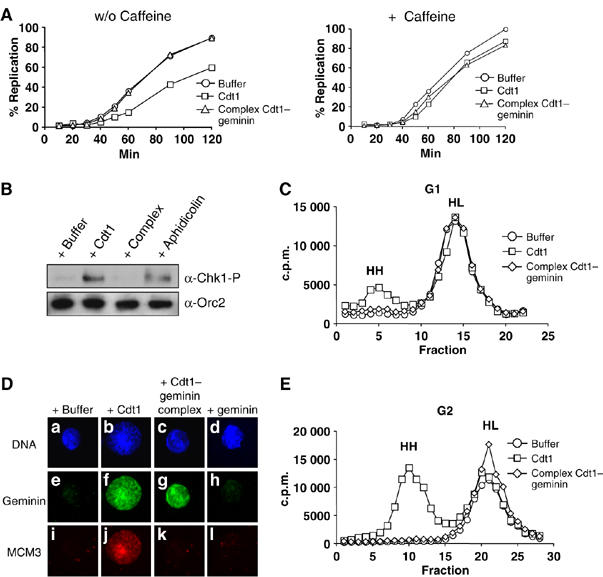
Free Cdt1, but not the Cdt1–geminin complex, leads to rereplication and checkpoint activation. (A) Left panel: kinetics of sperm chromatin replication supplemented with buffer (circles), 40 nM free Cdt1 (rectangles) or 40 nM Cdt1–geminin complex (triangles). Right panel: the same reactions were performed in the presence of 5 mM caffeine. (B) Western blot analysis of purified nuclei with a phospho-Chk1 antibody. Nuclei were purified after 100 min incubation of sperm chromatin in egg extract supplemented with either buffer, free Cdt1, Cdt1–geminin complex (all 40 nM), or Aphidicolin (50 μg/ml). A Western blot with ORC2 antibody as loading control is also shown. (C) Analysis of replicated DNA half-substituted with BrdUTP (‘heavy–light', HL) or double-substituted after rereplication (‘heavy–heavy', HH) on a CsCl gradient. Before sperm chromatin addition, reactions were supplemented either with buffer (circles), free Cdt1 (rectangles) or Cdt1–geminin complex (diamonds). (D) Immunofluorescence of G2 chromatin (120 min after sperm chromatin addition) and 20 min after addition of either buffer, free Cdt1, Cdt1–geminin complex, or free geminin. DNA is shown in blue stained with Hoechst, geminin in green (FITC) and MCM3 in red (Texas red). (E) Analysis of replicated DNA half-substituted with BrdUTP after one round of replication (‘heavy–light', HL) or double-substituted after rereplication (‘heavy–heavy', HH) on a CsCl gradient in reactions supplemented in G2 (120 min after sperm DNA was added) either with buffer (circles), free Cdt1 (rectangles) or Cdt1–geminin complex (diamonds).
To confirm our observations, we supplemented egg extracts with equivalent amounts of free Cdt1 or Cdt1–geminin complex, added sperm chromatin, isolated nuclei at the end of S phase and tested for checkpoint activation by detection of phosphorylated Chk1 (Guo et al, 2000; Liu et al, 2000; Capasso et al, 2002). As controls, parallel reactions were supplemented with either buffer or 50 μg/ml aphidicolin. Only those reactions supplemented with aphidicolin or free Cdt1 showed phosphorylation of Chk1, indicating checkpoint activation (Figure 4B). We next tested for rereplication by using a CsCl gradient to detect double BrdUTP-substituted DNA. As shown in Figure 4C, the addition of free Cdt1 before the initiation of DNA synthesis gave rise to partial rereplication. In contrast, no rereplication (and no checkpoint activation, see above) was detected when the Cdt1–geminin complex was added. These data strongly suggest that disturbing the Cdt1:geminin ratio by adding free Cdt1 before the onset of S phase results in rereplication, activates an ATM/ATR-dependent checkpoint response during S phase, and thus delays DNA replication. Therefore, the recruitment of the Cdt1–geminin complex during licensing to chromatin appears essential to prevent rereplication during S phase.
The activity of the Cdt1–geminin complex is faithfully regulated by the cell cycle
We and others recently reported that Cdt1 added during G2 can cross the nuclear membrane and induce massive rereplication (Arias and Walter, 2005; Li and Blow, 2005; Maiorano et al, 2005; Yoshida et al, 2005). Given that the Cdt1–geminin complex is as efficient as free Cdt1 in licensing, we tested whether its addition in G2 would similarly induce rereplication. We purified G2 chromatin 20 min after adding geminin, Cdt1 or the Cdt1–geminin complex and found that addition of free Cdt1 or the Cdt1–geminin complex, but not free geminin, induced the rerecruitment of geminin to chromatin (Figure 4D, compare e–h). Rerecruitment of Cdt1 to chromatin in G2 also led to the rerecruitment of the MCM2–7complex and to rereplication (Figure 4D and E). In contrast, the addition of the Cdt1–geminin complex in G2 induced neither the rerecruitment of the MCM2–7 complex to chromatin (Figure 4D, compare j and k) nor rereplication, as seen by the absence of double BrdUTP-substituted DNA (Figure 4E). Interestingly, the rerecruitment of geminin to chromatin induced by the addition of free Cdt1 during G2 could neither prevent relicensing nor rereplication (Figure 4D, compare f, j and g, k).
We conclude that free Cdt1 on chromatin causes uncontrolled licensing in both G1 and G2, resulting in rereplication. In contrast, the Cdt1–geminin complex is faithfully controlled in both G1 and G2. In G1, it licenses chromatin only once for replication, and in G2 it does not allow (re-) licensing of chromatin, thereby preventing a second round of replication within the same cell cycle.
The stoichiometry of the Cdt1–geminin complex determines its activity
The above findings prompted us to ask how the Cdt1–geminin complex could be active in licensing without promoting relicensing, and how geminin can block the loading of the MCM2–7 complex onto chromatin when appropriate despite the Cdt1–geminin complex being active. One possibility could be that the nuclear environment inactivates the Cdt1–geminin complex. We have previously shown that MCM2–7 loading is blocked following accumulation of geminin on chromatin (Maiorano et al, 2004). Two different oligomerization states of geminin have been reported. The first, a dimer of either a free geminin domain or a geminin domain complexed with a Cdt1 geminin-binding domain, was identified by crystallization studies (Lee et al, 2004; Saxena et al, 2004; Thépaut et al, 2004). Whereas the second (using small angle scattering and electron microscopy to visualize full-length geminin) was shown to be a tetramer (Okorokov et al, 2004; Thépaut et al, 2004). We explored the possibility that such changes of the Cdt1–geminin complex stoichiometry could account for its inactivation.
We have shown that the Cdt1–geminin complex is stable in solution. However, we also showed that further recruitment of geminin to chromatin occurs after initiation of DNA synthesis. Thus, we investigated whether the complex might change its stoichiometry when incubated with an excess of free geminin. Figure 5A shows that the active Cdt1–geminin complex can incorporate additional geminin, which was detectable through the use of a higher molecular weight myc-tagged form of free geminin in the binding assay outlined in Figure 5A. Binding of the additional geminin produced an ‘I-complex' (for Inactive complex, see below). In parallel, immobilized Cdt1–geminin complex was incubated with buffer only and processed similarly, and named the ‘A-complex' (for Active complex, see below). An immobilized, unrelated control GST-protein (the yeast Seh1 nucleoporin), as well as GST-geminin itself were unable to bind additional free geminin (Supplementary Figure S7), showing that the association of free geminin with the complex was specific. Quantification of Cdt1 and geminin (untagged and myc-tagged) on the Coomassie-stained SDS–PAGE shown in Figure 5A indicated that the Cdt1–geminin A-complex has a Cdt1:geminin stoichiometry of 1:3, and that this stoichiometry was changed upon incubation with myc-tagged geminin by the addition of one geminin molecule per complex (I-complex, 1:4 stoichiometry). It is essential to point out that the experimental design used in these experiments precludes the possible loss and exchange of geminin from the complex, as the complex is immobilized on the Sepharose via the GST-tag on geminin itself. Interestingly, the expression and purification of the Cdt1–geminin complex from coexpressed recombinant proteins revealed the A-complex, suggesting that this stoichiometry is preferred in vitro.
Figure 5.
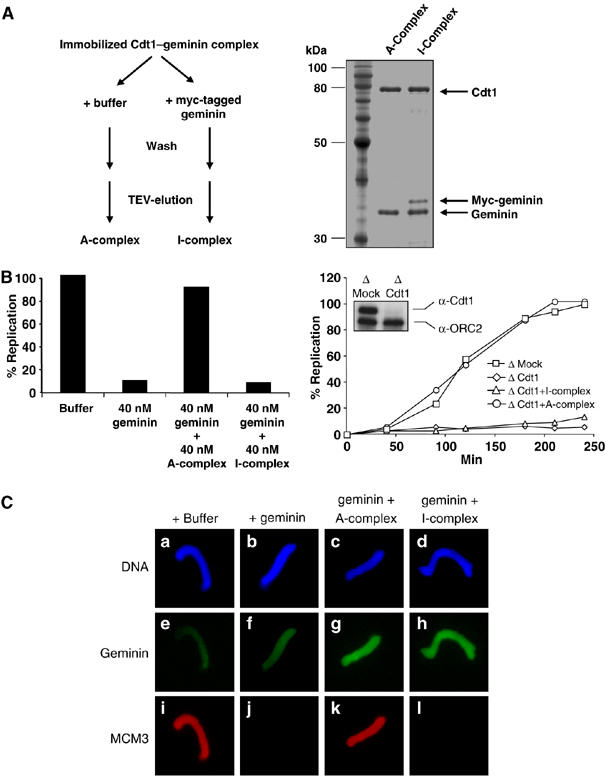
The stoichiometry of the Cdt1–geminin complex determines its active and inactive state. (A) Left panel: diagram explaining the formation of the ‘A'- and ‘I'–Cdt1–geminin complex. Right panel: SDS–PAGE of the active A-complex and the inactive I-complex, which contains additional bound myc-tagged geminin. Also shown is a molecular weight marker. (B) Left panel: percentage of replicated sperm chromatin after 100 min in egg extract supplemented by buffer, 40 nM geminin, 40 nM geminin plus 40 nM A-complex, or 40 nM geminin plus 40 nM I-complex. Right panel: kinetics of DNA replication in either Mock-depleted extract (squares) or in Cdt1-depleted extract supplemented by buffer (diamonds), 40 nM A-complex (circles) or 40 nM I-complex (triangles). The insert shows a Western blot for Cdt1 and ORC2 (as loading control) of Mock- and Cdt1-depleted extract. (C) Immunofluorescence of sperm chromatin incubated for 20 min in egg extract complemented with buffer or equal amounts of either geminin alone, the A–Cdt1–geminin complex or I–Cdt1–geminin complex in addition to geminin. DNA is shown in blue (Hoechst), geminin in green (FITC) and MCM3 in red (Texas red).
We then tested whether the change in the stoichiometry of the Cdt1–geminin complex could account for its inactivation at the onset of S phase. As shown in Figure 5B, the I-complex was unable to counteract a geminin-induced block of DNA replication, in contrast to the A-complex. Furthermore, in contrast to the A-complex, the I-complex could not restore DNA replication in a Cdt1-depleted extract (Figure 5B right panel). Unlike the A-complex, the I-complex also failed to recruit the MCM2–7 helicase onto chromatin (Figure 5C, compare k and l). Together, these data suggest that an active Cdt1–geminin complex is recruited to chromatin during licensing and is converted to its inactive form upon origin firing through the introduction of additional geminin to the complex, resulting in the inactive I-complex. These data are consistent with the significant recruitment of geminin to chromatin just at the onset of S phase after the initial recruitment during pre-RC formation (see Discussion).
Discussion
In this study, we provide evidence that geminin is recruited to chromatin together with Cdt1 during the formation of prereplication complexes and demonstrate that a complex of Cdt1 and geminin is competent in licensing chromatin for DNA replication. We also show that in G1 or G2, free Cdt1, but not the Cdt1–geminin complex, can cause rereplication and checkpoint activation. Finally, our results suggest that a switch in the ratio of Cdt1 versus geminin in the complex controls whether Cdt1 is active or inactive in recruiting the MCM2–7 helicase to chromatin and licensing DNA for replication. Thus, the presence of geminin in a complex with Cdt1 does not necessarily block the licensing activity of Cdt1, but rather allows its activity to be controlled before, during and after S phase. We propose a model in which an active Cdt1–geminin complex is recruited to chromatin during origin licensing, which is then inactivated following origin firing through a reaction, which includes both the recruitment of additional geminin to the complex and further Cdt1 degradation (Figure 6). This regulation of the licensing reaction by a dynamic stoichiometry of the Cdt1–geminin complex may explain recent confusing data concerning the control of DNA replication during cell cycle progression and cell differentiation by geminin.
Figure 6.
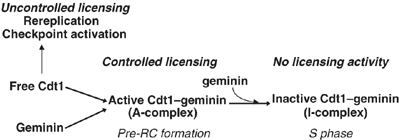
Functional model of the Cdt1/geminin licensing switch on chromatin. Free Cdt1 can license DNA replication origins, but its activity is not controlled and leads to rereplication and S-phase checkpoint activation. In contrast, the Cdt1–geminin A-complex licenses origins in a precisely controlled manner, without rereplication or checkpoint activation. After nuclear import renders free geminin competent to interact with Cdt1 (Hodgson et al, 2002), the active A-complex incorporates additional geminin forming the I-complex blocked in further licensing activity. Degradation of Cdt1 and geminin during S phase and mitosis resets the system for another cycle.
Geminin controls the activity of Cdt1 during the entire cell cycle
We and others have reported that endogenous Cdt1–geminin complexes are present in egg extracts in the absence of chromatin (Wohlschlegel et al, 2000; Hodgson et al, 2002; Maiorano et al, 2004; Yoshida et al, 2005). Hodgson et al (2002) estimated that 24% of endogenous geminin in an egg extract kept at room temperature is complexed with Cdt1, and Yoshida et al (2005) reported that 70% of the endogenous Cdt1 in an interphasic egg extract could be codepleted with geminin (for a review of the ratio of geminin and Cdt1, see Pitulescu et al, 2005). However, the biological significance of this complex remained unexplained.
Here, we showed that a Cdt1–geminin complex is not only recruited to chromatin during licensing, but that this complex is also competent to recruit the MCM2–7 complex to chromatin and to license the genome for replication. These data show that the previously proposed simple model, that geminin binding always blocks Cdt1, has to be revised. This may explain why, in proliferating and transformed cells, geminin levels are high and are compatible with high proliferation activity (Wohlschlegel et al, 2002; Pitulescu et al, 2005), as during late embryonic development, when high levels of geminin are maintained during cell proliferation (Del Bene et al, 2004; Luo et al, 2004). It has been reported that geminin binds and antagonizes Brg1, a member of the SWI/SNF chromatin remodeling complex, keeping genes responsible for neuronal differentiation in a repressive state and thus maintaining cell proliferation (Seo et al, 2005). Importantly, this interaction with Brg1 does not prevent geminin from binding to Cdt1. Therefore, geminin can modulate both cell proliferation and differentiation by interacting with different partners. In the above situation, high levels of geminin are necessary to maintain cell proliferation and are at the same time competent to interact with Cdt1 (Seo et al, 2005).
Significantly, the activity of the Cdt1–geminin complex, but not of Cdt1 alone, can accurately be controlled throughout the cell cycle. An excess of free Cdt1 over geminin (either by adding a surplus of Cdt1 or by decreasing/depleting geminin) leads to genomic instability. Consistent with our findings, in several cell lines, the silencing of geminin leads to rereplication, DNA damage and resulting Chk1-dependent checkpoint activation, and cell cycle arrest (McGarry, 2002; Melixetian et al, 2004; Zhu et al, 2004). Although bulk geminin is degraded in cells during mitosis (McGarry and Kirschner, 1998; Nishitani et al, 2001), recent results suggest that specifically geminin and Cdt1, which form a complex during mitosis, are not degraded (Ballabeni et al, 2004). If geminin is silenced, mitotic Cdt1 degradation is complete so that cells cannot even enter the following cell cycle (Ballabeni et al, 2004). These data suggest that, in somatic cells, an excess of free Cdt1 imposes a high risk of genetic stability, and that the cell actively senses and regulates the Cdt1/geminin ratio. Thus, accurate control of the balance between Cdt1 and geminin is essential for cell survival and genetic stability. Using gene expression profiling in cancer diagnostics, Cdt1 and geminin have become important targets (for a review, see Montanari et al, 2006). We suggest that the evaluation of their ratio might be as important to predict clinical outcome as the simple evaluation of their absolute levels.
Free Cdt1 is not bound by geminin before replication starts
We observed that adding free Cdt1 to egg extract does not increase the amount of geminin on chromatin before S-phase entry (Figure 3). This is consistent with results suggesting that free geminin cannot interact with Cdt1 unless it is first modified by nuclear import (Hodgson et al, 2002). Consistently, when Cdt1 was added at the beginning of a replication reaction, a strong increase of chromatin-bound geminin (compared with a reaction not supplemented with Cdt1) was detected only after nuclear membrane formation and after initiation of DNA replication had begun (Supplementary Figure S8). This suggests that, at least during early development, the amount of the Cdt1–geminin complex does not change before nuclear formation and onset of S phase, but only after replication starts. This explains the presence of preassembled Cdt1–geminin complexes in egg extracts and could also explain why, in somatic cells, only geminin-complexed Cdt1 might not be degraded during mitosis.
Our biochemical data show that the Cdt1–geminin complex is highly stable in solution as well as in an egg extract. Dissociation of the complex would produce free Cdt1 upon addition of the complex that would cause a checkpoint activation (Figure 4B), which is not observed. Dissociation of the complex would also produce three times more of free geminin, which would block Cdt1. The stability of the complex is further illustrated by the observation that addition of free Cdt1 does not increase geminin on chromatin during licensing, and that addition of free geminin leads only to a small increase of geminin on chromatin (Figure 3). In this case, geminin is only recruited through chromatin-bound Cdt1 or Cdt1–geminin complexes, as geminin cannot bind to chromatin independently of Cdt1 (Figure 1E and Maiorano et al, 2004). Thus, the strong recruitment of geminin to chromatin upon addition of the Cdt1–geminin complex can only be explained by the recruitment to chromatin of the complex as an entity.
The massive geminin recruitment to chromatin after onset of S phase may explain the inactivation of all chromatin-bound Cdt1, either free or complexed with geminin, at origins that have fired or on those that were not selected to fire. The Cdt1:geminin ratio on chromatin exceeds a 1:4 value during S phase, possibly through additional chromatin-dependent recruitment or oligomerization reactions of geminin on chromatin (see below).
A structural model for the molecular licensing switch
Based on a crystal structure of a complex of the interacting domains of Cdt1 and geminin, Lee et al (2004) proposed a model in which geminin's C-terminus would shield the Cdt1 domain that, in its free form, interacts with the MCM2–7 complex. Here, we show that full-length geminin molecules can bind to full-length Cdt1 without disturbing the ability of Cdt1 to interact with the MCM2–7 complex. Thus, binding of geminin to Cdt1 per se is not sufficient to inhibit the recruitment of the MCM2–7 helicase to chromatin. Instead, additional geminin molecules must be assembled during a second step in order to inhibit MCM2–7 loading. The Cdt1–geminin complex may function as a molecular switch to achieve, firstly in its ‘on' state, the licensing and subsequent activation of replication origins. Secondly, once origins have fired, the complex would turn ‘off' the licensing activity, through recruitment of additional geminin. Interestingly, such a two-step loading mechanism of DNA replication proteins has recently also been suggested for MCM10, which appears to initially bind to chromatin in an early step essential for recruiting Cdc45. To initiate DNA synthesis, polα-primase bound to MCM10 is recruited at a later step, probably through the formation of MCM10 oligomers on chromatin (Ricke and Bielinsky, 2004).
The Cdt1–geminin complex appears to have an elongated structure, as its Stokes-radius, as obtained from gel-filtration experiments, corresponds to higher molecular masses determined using other techniques such as glycerol gradients or equilibrium sedimentation (Hodgson et al, 2002; Lee et al, 2004; Maiorano et al, 2004; Saxena et al, 2004; Ferenbach et al, 2005). We determined the diameter of the recombinant Cdt1–geminin complex by dynamic light scattering to be 13.5 nm, which is well in the range of an elongated complex composed of three geminin molecules and one molecule of Cdt1. Based on sedimentation equilibrium experiments, geminin alone was reported to be a dimer (Benjamin et al, 2004), although small-angle scattering experiments using a crystallized fragment of geminin suggested a dimer of dimers (Thépaut et al, 2004). The electron microscopic structure of full-length geminin shows a tetramer made up from two pairs of dimers forming a key-like structure, in which the N-terminal portions of the four molecules form a closed hole that is large enough to encircle double-stranded DNA (Okorokov et al, 2004). Thus, geminin can exist in different association states, which is consistent with our findings of the different stoichiometries of geminin bound to Cdt1 presented here. The intriguing molecular structure of the geminin tetramer, which inactivates licensing, suggests a model in which once a fully assembled tetramer is formed, a closed ring around the DNA mechanically blocks both the loading of the MCM2–7 helicase and the melting of the DNA double strand.
Materials and methods
Xenopus egg extracts and replication reactions
Egg extracts were prepared as described previously (Menut et al, 1999). Upon thawing, egg extracts were supplemented with cycloheximide (250 μg/ml) and an energy regeneration system (10 μg/ml creatine kinase, 10 mM creatine phosphate, 1 mM ATP, 1 mM MgCl2). If recombinant proteins were added, the supplemented egg extracts were kept for 10 min at room temperature before sperm chromatin addition. To follow DNA replication by incorporation of α-[32P]dNTP into newly replicated DNA, 1 μl of α-[32P]dNTP (3000 Ci/mmol) was added to a standard reaction of 50 μl. Percentages were calculated relative to the amount of template sperm nuclei in control reactions. For density substitution experiments, standard replication reactions were supplemented with 0.1 mM BrdUTP. Egg extracts were depleted of Cdt1 as previously described (Maiorano et al, 2000b). To analyze semisubstituted (heavy–light, replicated) and fully substituted (heavy–heavy, re-replicated) DNA, cesium chloride density centrifugation was performed as described (Maiorano et al, 2005).
Cloning, expression and purification of recombinant proteins
Xenopus Cdt1 protein was expressed in Escherichia coli as a GST-TEV-tagged recombinant protein. A previously described construct (Maiorano et al, 2005) coding for a His-tagged Xenopus Cdt1 was cut with BamHI/SacI, and a GST-TEV fragment was inserted in frame. Xenopus gemininDEL (McGarry and Kirschner, 1998) was cloned into a pET24-GST-TEV vector (Lutzmann et al, 2002) as an NheI/BamHI fragment. The Cdt1–gemininDEL complex was coexpressed by cotransformation of the vectors coding for the His-tagged Cdt1 (Maiorano et al, 2005) and GST-TEV tagged geminin (this study). Myc-tagged gemininDEL was expressed from the pPROEXHT-GST-TEV vector, the myc-tag was introduced by PCR 5′ to the geminin ORF.
All constructs were expressed in E. coli BL21-codon plus RIL cells (Stratagene) and grown under standard conditions (Lutzmann et al, 2002).
Frozen E. coli cells containing the expressed proteins were thawed and lysed by sonification in Dicis-buffer (300 mM NaCl, 150 mM KOAc, 20 mM Tris, pH 7.5, 2 mM MgCl2, 10% Glycerol, 0.01% NP40). During cell lysis, the Dicis-buffer was complemented with protease inhibitors (Complete, Roche) and 0.1% NP40. After centrifugation (15 min, 15 000 r.p.m. in an SS34 rotor), the supernatant was incubated either with Glutathione-Sepharose 4B beads (Amersham Bioscience) or Ni-NTA Sepharose (Qiagen) for 45 min at 4°C. After binding, the GSH beads were first washed with 20 volumes of Dicis-Buffer and then with five volumes of Dicis-buffer containing 1 mM of DTT. TEV cleavage was performed in one volume of Dicis-buffer+1 mM DTT at RT for 1 h. Ni-NTA beads were washed with Dicis-buffer+20 mM imidazol and eluted stepwise with Dicis-buffer supplemented with 50, 100 and finally 150 mM imidazol. To purify the coexpressed Cdt1–geminin complex, after cell lysis, the supernatant was first subjected to a Ni-NTA affinity-purification as described above, and the imidazol eluates were then pooled and directly subjected to GSH-Sepharose affinity purification. Finally, the complex was eluted by TEV cleavage of the GST-TEV-tagged geminin protein complexed by Cdt1. All eluted proteins or protein complexes were aliquoted, frozen in liquid nitrogen and stored at −80°C.
Nuclei and chromatin isolation
Nuclei were isolated diluting the replication reaction five-fold with buffer XB (10 mM HEPES–KOH pH 7.7, 100 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 5% sucrose). The diluted reaction was kept at 4°C for 2 min before nuclei were isolated by centrifugation (6000 g, 5 min, 4°C) through a 0.7 M sucrose cushion in XB. Chromatin was obtained by diluting replication reactions with five volumes of XB+0.3% Triton X-100 keeping them for 5 min at 4°C, and purifying chromatin through a sucrose cushion as described above.
Immunofluorescence
Immunofluorescence was performed as described earlier (Maiorano et al, 2005). Coverslips were mounted on slides with MOVIOL (Aldrich), dried for 2 h at 37°C and analyzed with a Zeiss inverted microscope.
Stability test of the Cdt1–geminin complex in egg extract and buffer
Cdt1–geminin complex immobilized to GSH-Sepharose was incubated either in XB buffer or in interphasic egg extract for 40 min at room temperature. The beads were then extensively washed to remove unbound material and the proteins eluted using SDS sample buffer and separated by SDS–PAGE.
Antibodies
The geminin antibody was described in Maiorano et al (2004) and the Cdt1 antibody in Maiorano et al (2000a). Phospho-Chk1 antibody was purchased from Cell Signalling.
Supplementary Material
Supplementary Figures
Supplementary Figures
Acknowledgments
We thank D. Fisher for sharing unpublished results and critical reading of the manuscript. We are also grateful to A Padilla (CBS Montpellier) for performing dynamic light scattering analysis of the Cdt1–geminin complex. Malik Lutzmann is supported by a Liebig-Stipendium des Fonds der chemischen Industrie Deutschland. This work was supported by the CNRS, the Association pour la Recherche contre le Cancer (ARC) and the Ligue contre le Cancer.
References
- Arias EE, Walter JC (2005) Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev 19: 114–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabeni A, Melixetian M, Zamponi R, Masiero L, Marinoni F, Helin K (2004) Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO J 23: 3122–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Benjamin JM, Torke SJ, Demeler B, McGarry TJ (2004) Geminin has dimerization, Cdt1-binding, and destruction domains that are required for biological activity. J Biol Chem 279: 45957–45984 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Laskey RA (1988) A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature 332: 546–548 [DOI] [PubMed] [Google Scholar]
- Capasso H, Palermo C, Wan S, Rao H, John UP, O'Connell MJ, Walworth NC (2002) Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J Cell Sci 115: 4555–4564 [DOI] [PubMed] [Google Scholar]
- Cook JG, Chasse DA, Nevins JR (2004) The regulated association of Cdt1 with minichromosome maintenance proteins and Cdc6 in mammalian cells. J Biol Chem 279: 9625–9633 [DOI] [PubMed] [Google Scholar]
- Del Bene F, Tessmar-Raible K, Wittbrodt J (2004) Direct interaction of geminin and Six3 in eye development. Nature 427: 745–749 [DOI] [PubMed] [Google Scholar]
- DePamphilis ML, Blow JJ, Gosh S, Saha T, Noguchi K, Vassilev A (2006) Regulating the licensing of DNA replication origins in metazoa. Curr Opin Cell Biol 18: 231–239 [DOI] [PubMed] [Google Scholar]
- Ferenbach A, Li A, Brito-Martins M, Blow JJ (2005) Functional domains of the Xenopus replication licensing factor Cdt1. Nucleic Acids Res 33: 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MA, Tachibana KE, Chin SF, Callagy G, Madine MA, Vowler SL, Pinder SE, Laskey RA, Coleman N (2004) Geminin predicts adverse clinical outcome in breast cancer by reflecting cell-cycle progression. J Pathol 204: 121–130 [DOI] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG (2000) Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 14: 2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson B, Li A, Tada S, Blow JJ (2002) Geminin becomes activated as an inhibitor of Cdt1/RLF-B following nuclear import. Curr Biol 12: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Xiong Y (2006) An evolutionarily conserved function of proliferating cell nuclear antigen for Cdt1 degradation by the Cul4-Ddb1 ubiquitin ligase in response to DNA damage. J Biol Chem 281: 3753–3756 [DOI] [PubMed] [Google Scholar]
- Lee C, Hong B, Choi JM, Kim Y, Watanabe S, Ishimi Y, Enomoto T, Tada S, Kim Y, Cho Y (2004) Structural basis for inhibition of the replication licensing factor Cdt1 by geminin. Nature 430: 913–917 [DOI] [PubMed] [Google Scholar]
- Lemaitre JM, Danis E, Pasero P, Vassetzky Y, Mechali M (2005) Mitotic remodeling of the replicon and chromosome structure. Cell 123: 787–801 [DOI] [PubMed] [Google Scholar]
- Li A, Blow JJ (2005) Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA rereplication in Xenopus. EMBO J 24: 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Li X, Yan F, Zhao Q, Wu X (2004) Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J Biol Chem 279: 17283–17288 [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Luo L, Yang X, Takihara Y, Knoetgen H, Kessel M (2004) The cell cycle regulator geminin inhibits Hox function through direct and polycomb-mediated interactions. Nature 427: 749–753 [DOI] [PubMed] [Google Scholar]
- Lutzmann M, Kunze R, Buerer A, Aebi U, Hurt E (2002) Modular self-assembly of a Y-shaped multiprotein complex from seven nucleoporins. EMBO J 21: 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Hamlin JL, Dutta A (2005) Right place, right time, and only once: replication initiation in metazoans. Cell 123: 13–24 [DOI] [PubMed] [Google Scholar]
- Maiorano D, Krasinska L, Lutzmann M, Méchali M (2005) Recombinant Cdt1 induces rereplication of G2 nuclei in Xenopus egg extracts. Curr Biol 15: 146–153 [DOI] [PubMed] [Google Scholar]
- Maiorano D, Lemaitre JM, Méchali M (2000a) Stepwise regulated chromatin assembly of MCM2–7 proteins. J Biol Chem 275: 8426–8431 [DOI] [PubMed] [Google Scholar]
- Maiorano D, Moreau J, Méchali M (2000b) XCdt1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature 404: 622–625 [DOI] [PubMed] [Google Scholar]
- Maiorano D, Rul W, Méchali M (2004) Cell cycle regulation of the licensing activity of Cdt1 in Xenopus laevis. Exp Cell Res 295: 138–149 [DOI] [PubMed] [Google Scholar]
- McGarry TJ (2002) Geminin deficiency causes a Chk1-dependent G2 arrest in Xenopus. Mol Biol Cell 13: 3662–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry TJ, Kirschner MW (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93: 1043–1053 [DOI] [PubMed] [Google Scholar]
- Melixetian M, Ballabeni A, Masiero L, Gasparini P, Zamponi R, Bartek J, Lukas J, Helin K (2004) Loss of geminin induces rereplication in the presence of functional p53. J Cell Biol 165: 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menut S, Lemaitre JM, Hair A, Méchali M (1999) DNA replication and chromatin assembly using Xenopus egg extracts. In: Advances in Molecular Biology: A Comparative Methods Approach to the Study of Oocytes and Embryos, Richter JD (ed), pp. 198–226. Oxford: Oxford University Press [Google Scholar]
- Montanari M, Macaluso M, Cittadini A, Giordano A (2006) Role of geminin: from normal control of DNA replication to cancer formation and progression? Cell Death Differ 13: 1052–1056 [DOI] [PubMed] [Google Scholar]
- Nishitani H, Lygerou Z, Nishimoto T (2004) Proteolysis of DNA replication licensing factor Cdt1 in S-phase is performed independently of geminin through its N-terminal region. J Biol Chem 279: 30807–30816 [DOI] [PubMed] [Google Scholar]
- Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, Lygerou Z, Nishimoto T (2006) Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J 25: 1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Taraviras S, Lygerou Z, Nishimoto T (2001) The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J Biol Chem 276: 44905–44911 [DOI] [PubMed] [Google Scholar]
- Okorokov AL, Orlova EV, Kingsbury SR, Bagneris C, Gohlke U, Williams GH, Stoeber K (2004) Molecular structure of human geminin. Nat Struct Mol Biol 11: 1021–1022 [DOI] [PubMed] [Google Scholar]
- Pitulescu M, Kessel M, Luo L (2005) The regulation of embryonic patterning and DNA replication by geminin. Cell Mol Life Sci 62: 1425–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn LM, Herr A, McGarry TJ, Richardson H (2001) The Drosophila geminin homolog: roles for geminin in limiting DNA replication, in anaphase and in neurogenesis. Genes Dev 15: 2741–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, Bielinsky AK (2004) Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol Cell 16: 173–185 [DOI] [PubMed] [Google Scholar]
- Saxena S, Yuan P, Dhar SK, Senga T, Takeda D, Robinson H, Kornbluth S, Swaminathan K, Dutta A (2004) A dimerized coiled-coil domain and an adjoining part of geminin interact with two sites on Cdt1 for replication inhibition. Mol Cell 15: 245–258 [DOI] [PubMed] [Google Scholar]
- Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A (2006) PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem 281: 6246–6252 [DOI] [PubMed] [Google Scholar]
- Seo S, Herr A, Lim JW, Richardson GA, Richardson H, Kroll KL (2005) Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev 19: 1723–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto N, Tatsumi Y, Tsurumi T, Matsukage A, Kiyono T, Nishitani H, Fujita M (2004) Cdt1 phosphorylation by cyclin A-dependent kinases negatively regulates its function without affecting geminin binding. J Biol Chem 279: 19691–19697 [DOI] [PubMed] [Google Scholar]
- Tada S, Li A, Maiorano D, Méchali M, Blow JJ (2001) Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat Cell Biol 3: 107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Diffley JF (2002) Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2–7 during G1 phase. Nat Cell Biol 4: 198–207 [DOI] [PubMed] [Google Scholar]
- Thépaut M, Maiorano D, Guichou JF, Auge MT, Dumas C, Méchali M, Padilla A (2004) Crystal structure of the coiled-coil dimerization motif of geminin: structural and functional insights on DNA replication regulation. J Mol Biol 342: 275–287 [DOI] [PubMed] [Google Scholar]
- Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A (2000) Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290: 2309–2312 [DOI] [PubMed] [Google Scholar]
- Wohlschlegel JA, Kutok JL, Weng AP, Dutta A (2002) Expression of geminin as a marker of cell proliferation in normal tissues and malignancies. Am J Pathol 16: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi K, Mizuno T, You Z, Hanaoka F (2002) Mouse geminin inhibits not only Cdt1–MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J Biol Chem 277: 40871–40880 [DOI] [PubMed] [Google Scholar]
- Yoshida K, Takisawa H, Kubota Y (2005) Intrinsic nuclear import activity of geminin is essential to prevent re-initiation of DNA replication in Xenopus eggs. Genes Cells 10: 63–73 [DOI] [PubMed] [Google Scholar]
- Zhu W, Chen Y, Dutta A (2004) Rereplication by depletion of geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol Cell Biol 24: 7140–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figures