

Abstract
Most organisms use glutathione to regulate intracellular thiol redox balance and protect against oxidative stress; protozoa, however, utilize trypanothione for this purpose. Trypanothione biosynthesis requires ATP-dependent conjugation of glutathione (GSH) to the two terminal amino groups of spermidine by glutathionylspermidine synthetase (GspS) and trypanothione synthetase (TryS), which are considered as drug targets. GspS catalyzes the penultimate step of the biosynthesis—amide bond formation between spermidine and the glycine carboxylate of GSH. We report herein five crystal structures of Escherichia coli GspS in complex with substrate, product or inhibitor. The C-terminal of GspS belongs to the ATP-grasp superfamily with a similar fold to the human glutathione synthetase. GSH is likely phosphorylated at one of two GSH-binding sites to form an acylphosphate intermediate that then translocates to the other site for subsequent nucleophilic addition of spermidine. We also identify essential amino acids involved in the catalysis. Our results constitute the first structural information on the biochemical features of parasite homologs (including TryS) that underlie their broad specificity for polyamines.
Keywords: binding site, glutathionylspermidine, mechanism, structure, trypanothione
Introduction
Parasitic diseases, such as Chagas' disease, African sleeping sickness and several widespread illnesses known collectively as leishmaniasis, cause millions of human deaths each year worldwide. Lack of suitable drugs or vaccines is a major concern. In contrast to bacterial or viral infections, development of effective antiparasitic chemotherapy has been hindered by the close similarities between parasite and host metabolisms. Many of the existing drugs suffer from poor efficacy, host toxicity or/and drug resistance. To aid the development of new drugs, a special emphasis should be placed on a metabolic pathway in parasites that differs from or does not exist in the host (Fairlamb and Cerami, 1992; Krauth-Siegel et al, 2003; Müller et al, 2003).
Spermidine (N-(3-aminopropyl)-1,4-diaminobutane) and glutathione (GSH, γGlu-Cys-Gly) are present at high concentrations (0.1−10 mM) in most cells. Spermidine is a polycationic molecule that interacts with proteins, phospholipids and nucleic acids (Marton and Pegg, 1995), affecting primarily cell proliferation and differentiation (Tabor and Tabor, 1984; Pegg, 1986; Wang, 1995). GSH, a primary antioxidant, is important in maintaining the redox balance as well as in reductively scavenging reactive oxygen species (Meister and Anderson, 1983). Notably, the enzyme glutathione reductase keeps GSH in a reduced/activated form. In contrast, protozoal parasites of the genera Trypanosoma and Leishmania lack GSH reductase and GSH peroxidase activities (Boveris et al, 1980; Fairlamb and Cerami, 1985; Penketh et al, 1987). These pathogenic parasites instead employ trypanothione (bis(glutathionyl)spermidine) to defend against oxidative stress (Shames et al, 1986). The analogous enzymes trypanothione reductase and trypanothione peroxidase exist exclusively in the Kinetoplastida (Fairlamb and Cerami, 1992). Thus, trypanothione-related metabolism appears to be an attractive target for therapeutic intervention.
There are two biosynthetic steps to produce trypanothione from GSH and spermidine; the initial reaction requires glutathionylspermidine synthetase (GspS) to catalyze the coupling of GSH and spermidine to form glutathionylspermidine (Gsp) (Henderson et al, 1990; Smith et al, 1992). Gsp is then conjugated with another GSH to produce trypanothione by trypanothione synthetase (TryS) (Oza et al, 2002a, 2002b, 2003; Comini et al, 2003). Each step involves an amide bond formation that requires prior phosphorylation of the carboxy (C) terminus of GSH by ATP. Escherichia coli produces only the metabolic intermediate Gsp, but not trypanothione. The corresponding enzyme, GspS, was identified more than four decades ago (Dubin, 1959; Tabor and Tabor, 1975). Although the biological function of the E. coli GspS remains obscure, previous work indicates that the enzyme has a second activity to hydrolyze Gsp back to GSH and spermidine (Bollinger et al, 1995). The two activity domains are separate in this bifunctional protein: the amidase domain is located at the N terminus and the synthetase domain is at the C terminus (Kwon et al, 1997). Interdomain communication negatively regulates the amidase activity in the E. coli enzyme (Lin et al, 1997b).
Some parasites, such as Trypanosoma cruzi and Trypanosoma brucei, use one enzyme to synthesize Gsp and trypanothione, but others (e.g., Crithidia fasciculata) utilize two separate enzymes for this purpose (Oza et al, 2002a, 2002b, 2003; Comini et al, 2003). The majority of these proteins are bifunctional, having both amidase and synthetase activities, which suggests the importance of regulating the physiological concentrations of substrate and/or product. The scarcity of homologous sequences and the lack of any structural information have impeded our understanding of the Gsp- or trypanothione-related enzymes.
Herein we report the determination of five X-ray crystal structures of E. coli GspS, including the protein/substrate, protein/product and protein/inhibitor complexes. In particular, during crystallization, the nanomolar phosphinate inhibitor became phosphorylated to generate the phosphinophosphate intermediate at the active site despite its limited stability (t1/2=25 min) (Chen et al, 1997; Lin et al, 1997a). These results clarify the mechanistic details of the synthetase reaction and will contribute to our understanding of structural and functional differences within the TryS enzyme family.
Results
Overall structure
We obtained five crystal structures: apo_GspS and the GspS_AMPPNP, GspS_GSH_ADP, GspS_inhibitor and GspS_ADP complexes. All these structures contain a dimeric GspS in each asymmetric unit (Figure 1). Each of the final refined structures includes 571–603 total residues per GspS monomer, with some disordered regions at the N terminus (1–10) and in some surface loop regions (536–542, 547–563 in apo_GspS, and 30–40, 455–457 in the GspS complexes). The overall structure description is based on the information from the GspS_GSH_ADP structure that has the highest resolution (2.2 Å) and is more intact in the refined model.
Figure 1.
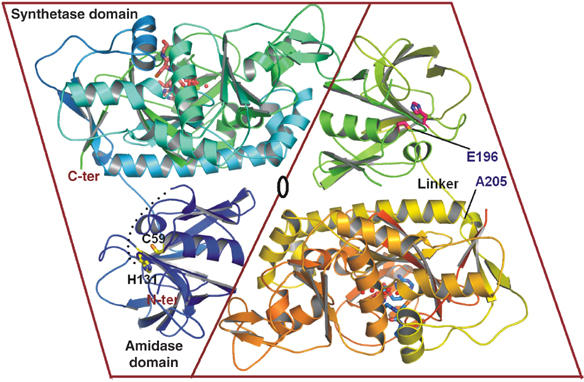
Overall structure of E. coli glutathionylspermidine synthetase/amidase. A ribbon diagram of the overall structure of E. coli GspS, showing two monomers in the asymmetric unit, and a pseudo-two-fold axis between the two monomers. The amidase domain (N-terminal 1–195), synthetase domain (C-terminal residues 206–619) and linker region (Glu196 to Ala205) are labeled. Active sites of the synthetase domain are revealed by the substrates represented as sticks (ADP and GSH) and spheres (Mg2+). Side chains of catalytic residues Cys59 and His131 in the amidase domain are designated in the same way. The dash represents a portion of the undefined region (residue 30–40) in the solved structure. The ribbon figures were drawn using PyMOL.
The globular structure reveals a mixed α/β fold with a size of 30 × 35 × 40 Å3 in the N-terminal amidase domain and an equilateral triangle shape in the C-terminal synthetase domain with the sides of the triangle of ∼60 Å and thickness of 30 Å (Figure 2A). Residues 196–205 between the two domains are defined as the linker region. The N-terminal amidase domain has an open-sandwich topology comprising two central α-helices (α2 and α3) surrounded by four (β1, β2, β3 and β4) and eight (β5, β6, β7, β8, β9, β10, β11 and β12) antiparallel twisted strands, as shown in Figure 2B. As we will demonstrate, the C-terminal synthetase domain belongs to the ATP-grasp superfamily (Murzin, 1996) and is structurally similar to that of human glutathione synthetase (PDB code: 2HGS) (Polekhina et al, 1999), despite no obvious sequence homology. The synthetase domain is composed of three main structural units, including (1) an antiparallel β-sheet (strands β15, β16, β29, β30 and β31; green in Figure 2B), together with α6 (green), α7, α8 (gray), α14 and α15 (yellow) packing on one side of the sheet, α4, α5 (gray), α9, α10 (blue), β13, β14 and β32 (gray) packing on the other side; (2) a parallel β-sheet (β17, β18, β21 and β22; red in Figure 2B) together with α11, α13 (red), α12, β19 and β20 (gray) and (3) a lid domain (orange in Figure 2B) composed of an antiparallel sheet of β23, β24, β25, β26, β27 and α16.
Figure 2.
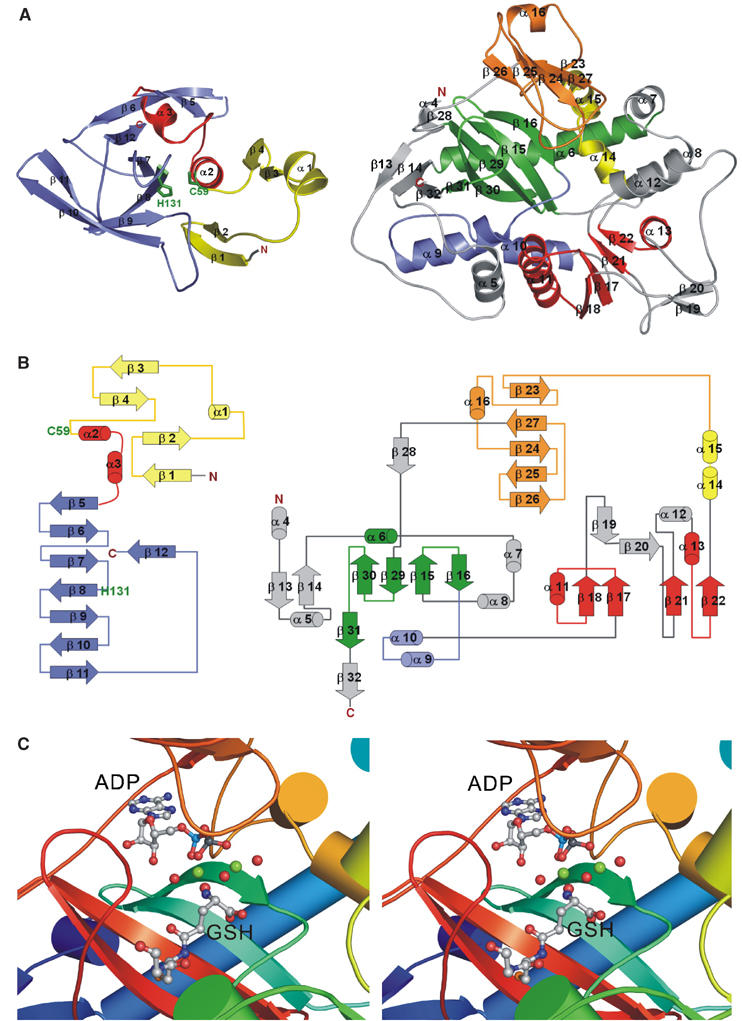
Structure analysis of the two activity domains in E. coli GspS. (A) Folding of the amidase domain (left, residues 1–195) and synthetase domain (right, residues 206–619). The amidase domain contains two central α-helices (red) that are surrounded by four and eight antiparallel twisted strands (yellow and blue, respectively). The synthetase domain mainly consists of antiparallel β-sheets (green), parallel β-sheets (red) and a lid domain (orange). Please see Results for the detailed description. (B) A topology diagram corresponding to each activity domain. The color codes for the secondary structural elements are identical to those in (A). (C) A stereo view of the catalytic region of the synthetase domain. The substrates ADP and GSH are shown as ball-and-stick structures and Mg-O as spheres (green–red).
The active site of the synthetase domain, clearly demarcated by the bound ligands in the complex structures, is located at the central antiparallel β-sheet and is surrounded by five loops (Figure 2C for stereo view); that is, P-loop (residues 535–543, designated in orange), loop1 (441–444, yellow), loop2 (332–338, cyan green), loop3 (601–609, red) and the D–E loop (387–392, green). As a part of the lid domain, P-loop (536AGRCGS542) is disordered in the apo_GspS structure, but forms a closed conformation when bound with substrate, product or inhibitor. Figure 3A and 3B show the surface charge potential of the synthetase active site of the GspS_GSH_ADP and GspS_inhibitor complexes, respectively. As shown in Figure 4A, P-loop, loop2 and loop3 have different conformations due to the binding of ATP and GSH.
Figure 3.
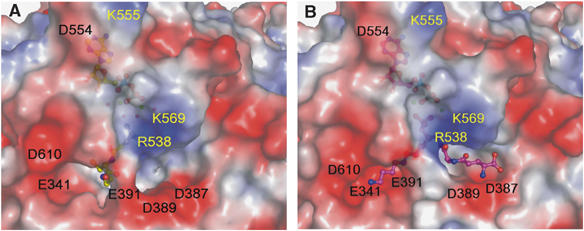
Substrate GSH (left) and inhibitor (right) binding in the Gsp synthetase domain and its electrostatic surface. Electrostatic surface representations of the GspS_GSH_ADP (A) and GspS_inhibitor (B) complexes. The colors red, white and blue indicate negative, neutral and positive charges, respectively.
Figure 4.
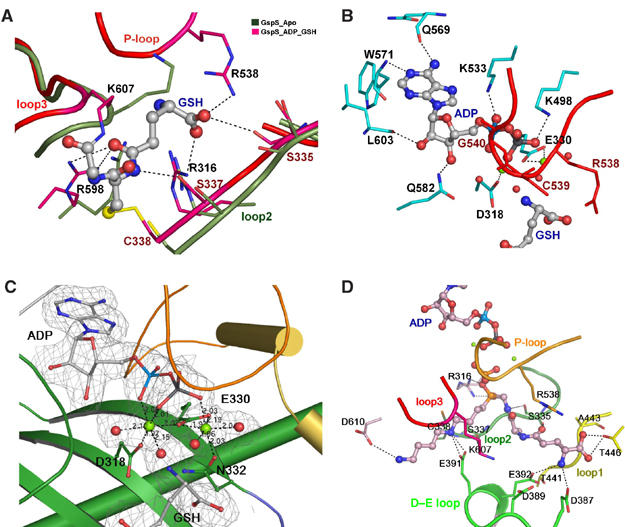
Details of the interactions of GspS with substrate/product or inhibitor in the synthetase domain. (A) Interaction of substrate with loops. Three loops are shown, including the P-loop (residues 536–542), loop2 (332–338) and loop3 (601–609). The loops are represented by ribbons, and side chains of the residues interacting with GSH are shown as thicker lines. GSH is shown as ball-and-stick structures. The figure shows the comparison of positions of the three loops and the corresponding side chains between the structures apo_GspS (olive-green) and GspS_GSH_ADP (magenta). Hydrogen bonds between GspS and GSH are depicted as black dotted lines. The disulfide bond between Cys338 and the Cys of GSH is shown in yellow. (B) ADP-binding site. ADP is represented as a ball-and-stick structure, and the interacting residues are shown by their side chains as thicker lines in cyan. The P-loop, including Arg538 and Gly540, is in red. Magnesium ions are shown as green spheres. (C) Magnesium-binding site. Mg2+ ions are shown as light green and water molecules as red. The side chains of the coordinating residues are presented as ball-and-stick structures. Coordination of Mg2+ ions is depicted by dashed lines, and the distances are listed. The 2Fo−Fc electron density map of Mg-ADP contoured at 1σ level is shown. (D) Details of the interactions between the phosphinate inhibitor and GspS. Five loops are presented here in different colors, including the P-loop in orange, loop1 in yellow, loop2 in forest green, loop3 in red and D–E loop in green. Residues making hydrogen bonds with the inhibitor are shown by thicker lines.
The amidase domain is a member of the cysteine, histidine-dependent amidohydrolases/peptidases (CHAP) superfamily (Bateman and Rawlings, 2003). It is a cysteine protease with Cys59 and His131 as the catalytic dyad, and these two amino acids are invariant among all GspS and TryS enzymes.
Dimerization
GspS exists as a dimer in solution, as supported by analytical ultracentrifugation (see supporting information). The sedimentation velocity of E. coli GspS estimates the molecular mass to be 138 kDa. Because the GspS polypeptide has a mass of 70 kDa, this result suggests that GspS should exist as a dimer in solution. Thus, the dimeric GspS structure in the asymmetric unit is considered as a functional dimer. The intersubunit contacts have a total buried surface area of 3400 Å2. The intersubunit interactions are between the amidase domain from one monomer and the synthetase domain from another monomer (Figure 1). Hydrophobic interactions between the two monomers are Leu15 with Ala424, Pro20 with Ala461, Ala114 with Ala460 and Leu303 with Val94. A salt-bridge interaction exists between Arg307 in one monomer and Asp49 in another monomer with a distance of 2.85 Å. Additionally, hydrogen bonds are observed in the dimeric interface, such as Tyr18 with Arg481, and Gln160 with Thr466.
ATP-binding site
ADP was located at the antiparallel β-sheet of GspS in a manner analogous to that observed in other ATP-grasp proteins (Fan et al, 1994, 1995; Polekhina et al, 1999; Thoden et al, 2000, Figure 2C for stereo view). The adenine ring is buried in a hydrophobic pocket that is shaped by Tyr329, Ala531, Leu570, Leu603, Val604 and Leu515. The exocyclic 6-amino group of the adenine base is hydrogen bonded with the main-chain oxygen of Gln569 and the N1 with the amide hydrogen of Trp571 (Figure 4B). The O2′ atom of the ribose forms hydrogen bonds with the main-chain oxygen of Leu603 and amide of Ile605, and the O3′ atom with Nɛ2 of Gln582. The negative charges on the α- and β-phosphates are compensated by two conserved residues, Lys498 and Lys533. Both ɛ-amino groups of Lys residues form salt bridges with Oδ− of ADP. The position of the γ-phosphate of ATP is deduced from the GspS_AMPPNP structure where Nη of Arg316 is close to the γ-phosphate within the hydrogen-bonding distance. The main-chain nitrogen of Gly540 and Cys539 in the P-loop interact with the β- and γ-phosphates, respectively.
Magnesium binding
Figure 4C and 4D shows the location of two magnesium ions in the complex structures. They are both bound in an octahedral geometry. Mg1 (left green ball in Figure 4C) is ligated by an α-phosphate oxygen and a β-phosphate oxygen atom of ADP, Oδ1 of Asp318, a carboxylate oxygen of Glu330 and two water molecules. The metal-ligand distances vary from 1.97 to 2.15 Å. The γ-carboxylate group of Glu330 also interacts with Mg2 (right green ball in Figure 4C) using both oxygen atoms (Mg–O distances are 2.19 and 1.97 Å). The other four ligands of Mg2 include a β-phosphate oxygen atom of ADP, Oδ of Asn332 and two water molecules, with the distances ranging from 1.96 to 2.04 Å. Glu330, a highly conserved residue in all ATP-grasp proteins, seems to play a vital role in enzyme catalysis, because it bridges between the two metal sites.
The transferred phosphate during phosphorylation of the inhibitor
In our previous report (Chen et al 1997; Lin et al, 1997a), the phosphinate analog of Gsp was found to exhibit an ATP-dependent, slow-binding inhibition against E. coli Gsp synthetase. The mixture of GspS, ATP and the phosphinate inhibitor was co-crystallized for structural analysis. In the final refined structure, ATP was found to be hydrolyzed to ADP. In addition, an extra phosphate was attached to the phosphinate oxygen, indicating that phosphorylation of the inhibitor was driven by ATP hydrolysis to give the tetrahedral phosphinophosphate that is bound at the active site. The intermediate mimics the tetrahedral adduct formed by the nucleophilic addition of spermidine to the acylphosphate (see Supplementary data).
The γ-phosphate in AMPPNP or transferred phosphate in phosphinophosphate interacts with both Mg2+ ions, the main-chain amide of Cys539 in the P-loop, and Nη of Arg316. Arg316 is an important residue that plays a role in the transfer of γ-phosphate from ATP and the stabilization of the anionic tetrahedral intermediate. Arg316 hydrogen bonds to the γ-phosphate of AMPPNP (Figure 5A) as well as the phosphinyl oxygens of the inhibitor (Figure 5B). The main-chain amide of Cys539 contacts the γ-phosphate in the GspS_AMPPNP structure and the transferred phosphate in the GspS_inhibitor structure. The interaction stabilizes the pentavalent phosphate intermediate in the phosphorylation step (Figure 5A and B). Bridging between the transferred phosphate and ADP, the two Mg2+ ions serve as Lewis acids to assist the phosphate transfer and compensate the resulting negative charges during catalysis.
Figure 5.
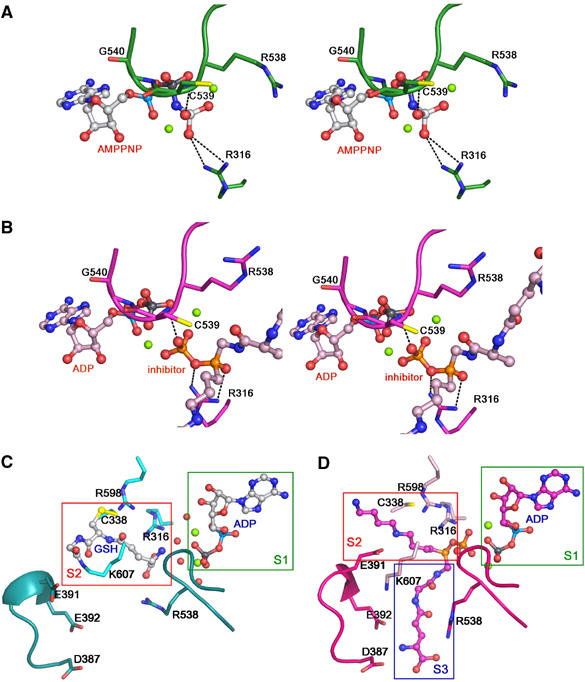
Two different binding sites of GSH indicated by comparing the complex structures. (A, B) A special emphasis is placed on the positions of the γ-phosphate and transferred phosphate. Ligands are drawn as ball-and-stick structures and Mg2+ as spheres. (A) The stereo view of the AMPPNP-binding site in the GspS_AMPPNP structure. The P-loop and the interacting residues are green. (B) The stereo view of the ADP and inhibitor-binding site in the GspS_inhibitor structure. The P-loop and the interacting residues are in magenta. (C, D) Comparison of the GspS_GSH_ADP and GspS_inhibitor structures with a special focus on the substrate-binding sites. Boxes show the substrate and the inhibitor-binding sites in the complex structures.
Furthermore, the Gsps_inhibitor structure was found similar to the complex structure of E. coli GSH synthetase with a phosphinyl peptide (Hiratake et al 1994; Hiratake, 2005). The peptide was phosphorylated, and ADP and the resulting phosphorylated phosphinate were located at the enzyme active site, as shown by the X-ray crystal structure analysis. Despite no obvious homology between this enzyme and E. coli GspS, both enzymes utilized the same residues to interact with the phosphinate and phosphates, including Arg316, D318 and E330 of GspS (corresponding to R210, D273 and E 281 of GSH synthetase, respectively).
Two GSH-binding sites
The first GSH-binding site (S2) is observed in the structure of GspS_GSH_ADP (Figure 5C). Surprisingly, the substrate is bound in the active site by forming a disulfide bond between its own Sγ atom and the Cys338 Sγ atom. The Gly portion of GSH also forms an isopeptide bond with Nζ of Lys607 (see Supplementary data).The orientation of the GSH binding in this structure is considered to be opposite to what it should adopt in the catalysis because the C-terminal carboxylate of Gly, serving as a nucleophile during the phosphate transfer, is located far from the ADP-binding site (S1). The formation of this isopeptide bond may be an accidental trap for nonproductive reactions in the absence of spermidine. GSH forms many hydrogen bonds with Arg316, Ser335, Ser337, Arg538, Arg598 and water molecules.
Formation of the mixed disulfide and the isopeptide, considered as a nonproductive mode, was observed only when the enzyme was incubated with ATP and GSH. In contrast, we never saw such formation in additional presence of spermidine (data not shown). The proteins were crystallized at pH 8.5 that is very different from the optimum pH (7.0). The basic condition gives 20% of the optimum activity at pH 7.0 and also favors the disulfide bonding formation.
The second GSH-binding site (S3) is revealed by the GspS_inhibitor structure (Figure 5D). The phosphinate inhibitor contains a tripeptide moiety of γ-Glu-Ala-Gly that is analogous to GSH (Figure 6). The tripeptide moiety interacts with several amino acids from the enzyme, including Ser335, Asp387, Glu392, Ala443 and Thr446 (Figure 4D).
Figure 6.
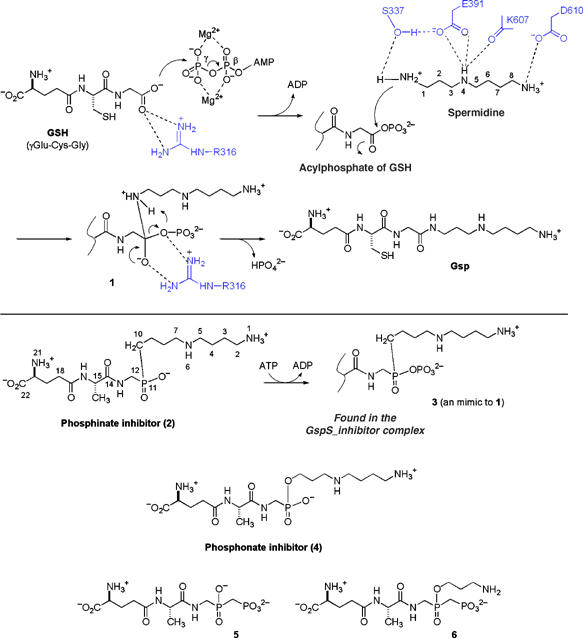
Proposed reaction mechanism of GspS in comparison with phosphorylation of the phosphinate inhibitor.
Spermidine binding
The possible spermidine-binding site is illustrated in the GspS_inhibitor structure, because the inhibitor is a Gsp analog containing GSH and spermidine. The interactions include hydrogen bonding between the terminal NH3+ group of inhibitor and Oδ1 of Asp610, as well as bidentate H-bonding of Glu391 Oɛ with the middle nitrogen of the spermidine moiety. The middle nitrogen is also within hydrogen-bonding distance with the main-chain oxygen of Lys607 (Figure 4D). The D–E loop was found to have different conformations between the GspS_GSH_ADP (Figure 5C) and GspS_inhibitor structures (Figure 5D). The four negatively charged residues Asp387, Asp389, Glu391 and Glu392 are closer to the S2 and S3 sites in the GspS_inhibitor structure in comparison with the GspS_GSH_ADP structure. (see Supplementary data). Notably, the side chain of Glu391 is positioned very differently in the two structures, revealing its key role in spermidine binding. The hydroxyl group of Ser337 is at a distance of 3.2 Å from the first carbon of the spermidine moiety of the inhibitor, indicating that the residue may facilitate the nucleophilic attack during catalysis by interacting with spermidine. Ser337 and Glu391 Oɛ2 form a hydrogen bond and are involved in the deprotonation of spermidine (Figure 6).
Discussion
Comparison of GspS with other ATP-grasp enzymes
Although GspS lacks any significant sequence identity or homology with other members of the ATP-grasp superfamily, our GspS crystal structures suggest that the enzyme indeed belongs to the superfamily. Based on published structures and structural comparisons among members, our consensus structure is related to human glutathione synthetase (hGS, Figure 7A and B), because the two structures can be superimposed to define the conserved structural units, including the parallel β-sheet, antiparallel β-sheet and lid domain (Figure 7C). Figure 7D shows structure-based alignment of E. coli GspS and hGS, revealing that a number of important residues and loops are conserved in the two proteins. The structure of E. coli GspS can be superimposed on hGS with an rms deviation of 320 Cα atoms of 3.8 Å. The most structurally similar units between the two structures are the parallel β-sheets (Figure 7C). hGS has two relatively long α-helices in the lid domain, in contrast to one short helix of E. coli GspS. The antiparallel β-sheet of hGS is more compact with extra and larger helices. As this domain facilitates substrate binding in both enzymes, the loose arrangement in GspS either accommodates larger substrates (or products) or undergoes significant conformational changes during catalysis.
Figure 7.
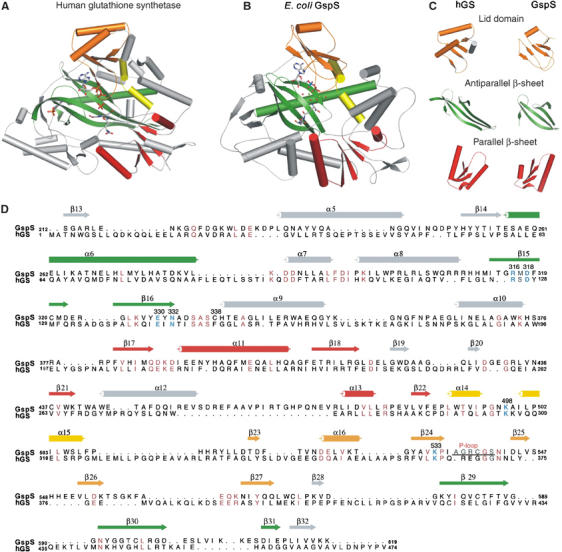
Comparison of hGspS and E. coli GspS domain. (A) Ribbon diagrams of hGS (PDB: 2HGS) and (B) E. coli GspS synthetase domain. (C) The comparisons of the lid domain, antiparallel β-sheet and parallel β-sheet units between the human glutathione synthetase and E. coli GspS synthetase domain. (D) Structure-based sequence alignment of E. coli GspS and hGS. The secondary structure and some residue numbering of E. coli GspS are shown above the alignment. All residues numbering of the two proteins are shown on both sides of each line. Catalytically related and conserved residues are shown in sky-blue; identical or similar residues are pink, and the P-loop defined in the E. coli GspS structure is indicated. The figure was produced using ALSCRIPT (Barton, 1993).
Two rare non-proline, cis-peptide bonds in the helical connection were found in our structure, including K56−W57 and F487−E488. K56−W57 is located in the N-terminal amidase domain. Interestingly, E488 corresponds to P295 in hGS, N114 in E. coli glutathione synthetase, G104 in biotin carboxylase and G85 in D-Ala-D-Ala ligase. Moreover, there is a great extent of variation in the residues forming the ATP-binding pocket; only a few residues are strictly conserved, such as Arg316, Lys498 and Lys533, which interact with the phosphates, E330 with Mg2+, and D318 and N332. The contributions of these comparative aspects to the GspS catalytic mechanism are discussed below.
The catalytic mechanism
The conjugation reaction of GSH with spermidine proceeds in two steps, which is analogous to other ATP-dependent ligases. It has been proposed that the C terminus of GSH is initially phosphorylated by γ-phosphate of ATP to form an acylphosphate, followed by the nucleophilic attack of N1-spermidine to the acylphosphate (Figure 6) (Chen et al, 1997; Chen and Coward, 1998). The resulting tetrahedral adduct then collapses to form an amide bond and breaks the C–O bond of the phosphate, leading to the formation of Gsp and release of inorganic phosphate, with ADP also being released after catalysis.
The five X-ray structures reported here are fully consistent with the proposed reaction mechanism. Among the structures, GspS_AMPPNP and GspS_GSH_ADP helped to delineate how GspS initially binds with the substrates ATP and GSH to catalyze the formation of acylphosphate, whereas the GspS_ADP structure provided the information on the enzyme/product complex. Each of these structures is necessary to obtain the full picture of the reaction mechanism. For example, without the information obtained from the GspS_inhibitor structure, it would be unclear how the reaction advances from the acylphosphate to form an amide bond. The phosphinate inhibitor (Figure 6, compound 2) was previously shown to be a remarkably potent inhibitor (Ki=3.2 μM and Ki*=7.8 nM, Lin et al 1997a), suggesting formation of phosphinophosphate (3), which dissociates from the enzyme GspS so slowly that it causes time-dependent inactivation (Chen et al 1997; Lin et al 1997a). Consistent with the inhibition study, the GspS_inhibitor structure reveals that the inhibitor reacts with ATP to produce ADP and the tight-binding phosphorylated intermediate. The resulting glimpse of the formation of the tetrahedral intermediate shows the nucleophilic attack of spermidine on the acylphosphate and subsequent amide-bond formation.
The comparison between these structures offers valuable information for determining the binding sites of the three substrates and demonstrates the progression of catalysis. ADP is stacked between two antiparallel β-sheets (S1 in Figure 5C and D). The cavity is covered by the disordered P-loop (not seen in apo_GspS). As discussed above, the main chains of C539 and G540 in the P-loop make contacts with the γ- and β-phosphates, respectively. Apparently, the loop serves to bind the nucleotide and prevents the intrusive hydrolysis of the acylphosphate intermediate by the surrounding solvent molecules. Examination of the GspS_GSH_ADP and GspS_inhibitor structures indicates that there are two possible binding sites for glutathione (S2 and S3 in Figure 5C and D), respectively. We propose that S2 is the initial GSH-binding site, and a subsequent conformational change triggered by ATP hydrolysis is likely involved in the translocation of the activated GSH to S3. The translocation has to occur to accommodate the other substrate, spermidine, at S2 for the next step of nucleophilic addition.
Our current data cannot exclude the possibility that spermidine and GSH may directly bind to S2 and S3, respectively, without the translocation. However, several pieces of supporting evidence favor the translocation mechanism. Firstly, Cys338 is located in S2 and is close to N6 (related to the spermidine moiety) in the GspS_inhibitor structure (see the inhibitor numbering in Figure 6). The Km values of C338A increase about 10-fold for GSH and four-fold for spermidine (Table I), suggesting that the residue is more involved in binding GSH. Secondly, consistent with the GspS_GSH_ADP structure, GSH was always found to reside at S2 in several attempts to co-crystallize GspS with GSH, even though the tripeptide was disordered and only a part of this molecule could be recognized in the electron density (data not shown). Thirdly, both E. coli GspS and parasite homologs (including TryS) show substrate inhibition with GSH, a phenomenon that can be explained by the translocation mechanism. High concentration (i.e., >2 mM) of GSH possibly leads to its simultaneous occupancy at both S2 and S3, which interferes with catalytic activity. Moreover, a significant movement of the D–E loop was found in connection with the translocation (Figure 5C and D and S2). The loop repositions to form multiple interactions with the inhibitor, including E391 with N6 (related to the spermidine moiety, see Figure 6), and E392 and D387 with N21 (related to the GSH moiety). In the GspS/substrate and GspS/product structures, the D–E loop adopts a different conformation from that in the GspS_inhibitor structure and is far from the S2- and S3-binding sites. As a result, after putative translocation of the acylphosphate from S2 to S3, the binding of spermidine to S2 likely induces the movement of the negatively charged loop to reposition the activated GSH and spermidine. The induced fit facilitates further catalytic steps (e.g., nucleophilic addition of spermidine to produce the tetrahedral intermediate).
Table 1.
Steady-state kinetic parameters for the activities of Gsps and its mutants
| GspS/mutant | Km, GSH (μM) | Km, spermidine (μM) | kcat/s |
|---|---|---|---|
| Wild type | 218±12 | 76±1 | 4.35±0.30 |
| C338A | 2098±152 | 406±38 | 4.28±0.35 |
| S335A | 794±71 | 126±15 | 3.31±0.17 |
| S337A | 224±3 | 76±3 | 2.49±0.21 |
| R538A | 1324±34 | 182±37 | 1.01±0.19 |
| T441A | 691±100 | 1610±137 | 0.25±0.05 |
| K607A | 251±29 | 153±14 | 4.55±0.19 |
| E392A | 7298±701 | 1008±92 | 0.75±0.05 |
| E391A | 407±42 | 4522±107 | 0.43±0.01 |
| R598A | 22.4±0.52 | 665±76 | 0.29±0.02 |
| R316E | —a | —a | —a |
| Values were determined by a previously reported spectrometric assay (see Materials and methods), which involved titration of a variable substrate in the presence of saturating concentrations of the other two substrates. | |||
| Not detected. |
A comparison of the GspS_inhibitor structure with the GspS_GSH_ADP structure reveals different conformations for the side chain of R538. In one conformation, R538 interacts with the γ-carboxylate of GSH (Figures 4A and 5C), whereas in the other conformation it faces the peptide part of the inhibitor (Figures 4D and 5B). The γ-carboxylate of GSH in the GspS_GSH_ADP structure occupies the same position as the phosphinate group of the inhibitor in the GspS_inhibitor structure. Because GSH reacts with ATP to form an acylphosphate intermediate at the initial step of the enzyme reaction, it is reasonable that the C-terminal carboxylate of GSH likely interacts with R538 and the resulting acyl phosphate interacts with the same residue during translocation.
In the complex structures, two Mg2+ cations bridge between ADP and inhibitor (or GSH). The phosphinate P–O bonds (P11–O) form a bidentate interaction with the guanidine group of R316. Because the inhibitor, 3, mimics the tetrahedral intermediate, we anticipate that 1 (Figure 6) would be oriented in an analogous manner during the catalytic cycle. Moreover, the proximity of the S337 hydroxyl group to the methylene group (C10, 3.2 Å) of 3 indicates a probable interaction with N1 of spermidine. The bifurcated hydrogen bond interactions of R316 with the carboxylic acid of GSH anchor and polarize a C-O bond to set up the other carboxylate oxygen atom to attack the γ-phosphate of ATP. The phosphate transfer step is assisted by the Mg2+ cations observed in the complex structures and yields the activated acylphosphate. Spermidine is oriented properly by multiple interactions, including D610 with N8, E391 and the main chain oxygen of K607 with N4, and S337 with N1 (see the spermidine numbering in Figure 6). It is not clear how the deprotonation of the N1 amino group of spermidine occurs, but the step is facilitated by the hydrogen bonding between S337 and E391. The deprotonation is essential to generate the nucleophilic NH2 form to attack the electrophilic carbonyl carbon of the acylphosphate. The reaction forms intermediate 1, in which the guanidine group of R316 stabilizes this high-energy species, similar to the observed interaction with 3. Intermediate 1 then decomposes to give the product Gsp with loss of phosphate.
A disulfide bond was found between GSH and C338 in the structure of GspS_GSH_ADP. The linkage likely contributes to the catalytic efficiency, but it is not necessarily required for the catalysis. The mutant C338A has a kcat of 4.28/s, very close to that of the wild type (4.35/s) (Table I). Likewise, GspS can accept the analogous substrates γGlu-Ala-Gly and γGlu-Ser-Gly with similar turnover numbers, but the Km values are 4- and 12-fold higher than that of GSH, respectively (data not shown). As a consequence, in the case where disulfide bond formation does not occur (i.e., GspS interacts with γGlu-Ala-Gly, or the mutant protein C338A interacts with GSH), the reaction still produces Gsp or its analog, but requires a higher substrate concentration to operate with similar efficiency.
Table I lists steady-state parameters for the activities of GspS and its mutants. The results support the claims made in the previous discussion. For example, the mutant R316E is completely inactive because the residue is essential in the formation and stabilization of the acylphosphate intermediate. Because E391 and E392 of the D–E loop are essential to participate in the binding interactions occurring at S2 and S3 sites, respectively, the Km values of their alanine mutants are much higher than the wild type, and both mutant proteins have low activity, as reflected by their turnover numbers. Therefore, these mutations result in tremendous loss in kcat/Km compared with the wild-type protein. T441 hydrogen bonds with the side chain of E392 (Figure 4D), and residues S335, C338 and R538 primarily affect substrate binding and/or translocation.
Comini et al (2005) studied the mutation of C. fasciculate TryS (Cf-TryS) at Arg553 (corresponding to R538 in E. coli GspS) and Arg613 (R598 in E. coli GspS), indicating that mutating each position resulted in marked reduction or abrogation of activity. The observation is consistent with our result. Arg553 of Cf-TryS, but not Arg613, was shown to be essential to the delayed proteolysis protected by the presence of substrates. The difference can be realized based on the structural information of E. coli GspS. R538 of E. coli GspS plays an indispensable role in the translocation as mentioned above. R598 of the same enzyme resides in the periphery of the S2 site and its role still remains unclear. Although the residue is located at the glycine-rich region that has been proposed previously to be a typical ATP-binding motif, R598 is not involved in the binding with ATP according to the resolved X-ray structures.
Examination of related parasite enzymes
In T. cruzi, only a single distinct enzyme (TryS) is involved in trypanothione biosynthesis (Oza et al, 2002b), that is, TryS catalyzes the consecutive conjugation of spermidine with two molecules of GSH to produce trypanothione. Unlike E. coli GspS, the T. cruzi enzyme has wide substrate specificity for polyamines, which has been attributed to selective pressures from the host under different environments (Oza et al, 2002b). To explain the wide substrate specificity of parasite TryS and the use of one enzyme instead of two (GspS and TryS), Oza et al (2003) hypothesized that an ancestral gsps gene, whose encoded protein exhibited narrow substrate specificity, may have undergone duplication and then diverged into two independent genes (gsps and trys). The subsequent loss of the gsps copy from the Trypanosoma genome thus led to the preservation of only trys (Oza et al, 2005). Owing to the significant sequence homology among E. coli GspS and parasite enzymes (e.g., there is 32% identity and 51.5% similarity between E. coli GspS and T. cruzi TryS by using Similarity Matrix PAM250), these intriguing evolutionary issues may be verified by the X-ray structures. Our results indicate that spermidine is oriented by the interaction of the three amino groups N1, N4 and N8 with the side chains of S337, E391 and D610 (Figure 6), respectively. In accordance with the homology alignment, the first two residues are strictly conserved; D610, however, is only conserved in prokaryotic GspS and not in the eukaryotic enzymes. In addition, TryS must have a larger binding site to accommodate Gsp, in comparison with GspS to hold spermidine. A homology model of T. cruzi TryS based on our GspS structures reveals an apparent difference. T. cruzi TryS may have an extra binding site that is located on the protein surface and near N1 of the phosphinate inhibitor (see the numbering in Figure 6). This site is thus considered as the extension of the S2 site and likely interacts with the GSH moiety of Gsp. Several residues of T. cruzi TryS are involved in the interactions, such as P613 (corresponding to D610 of E. coli GspS) and E611 (E608 of E. coli GspS). In contrast, this site is relatively shallow in E. coli GspS.
Furthermore, Comini and coworkers (2005) suggested three mechanistic possibilities to explain the dual activity of TryS, including a second reaction center in the enzyme, reorientation of initially formed Gsp to facilitate further conjugation with GSH at the same reaction center, and a flexible site to generate the activated GSH to ligate the tripeptide to either site of Gsp (or spermidine). Because our structural information supports only one ATP-binding site, the synthesis of trypanothione catalyzed by a dual functional TryS involves a conformational change of the lid domain to release the side product ADP, after formation of the intermediate Gsp. We thus favor the aforementioned second possibility, that is, to synthesize trypanothione directly from spermidine and GSH, the enzyme controls the open and closed conformation of the lid domain to load ATP twice and applies the same translocation to position GSH acylphosphate (see Supplementary data). To become further glutathionylated at the same reaction center that had glutathionylated spermidine, Gsp probably either flips within the active site or changes its position.
Design of simple but effective inhibitors
In accordance with the previous development of inhibitors against GspS, only phosphinate analogs of Gsp were found to react with ATP to produce a phosphinophosphate intermediate that is associated with a typical E·I⇆E·I* slow-binding isomerization. Compound 2 (Figure 6) gave a 410-fold gain in inhibitory potency from the initial Ki of 3.2 μM to the final Ki* of 7.8 nM. On the other hand, the phosphonate analog (4) did not undergo phosphorylation, and the affinity for GspS (Ki=6.0 μM) was ∼770-fold less than that of 2 (Chen et al, 1997). Furthermore, the phosphorylation step requires the presence of spermidine, as indicated by the poor inhibition of another simplified phosphinate (that contains a methyl group to replace the spermidine moiety), but it is not known if the entire spermidine group is involved in the binding. These observations support the idea that potent inhibition requires the consideration of the transferred γ-phosphate from ATP. It is thus necessary to incorporate an additional phosphate (or equivalent negative charge(s)) into designed molecules. As spermidine is at high concentration (0.1–10 mM) in most cells, compounds such as 5, which represent a simplified version of inhibitor 2, may be sufficient to inhibit the reaction in the presence of spermidine. Compound 6 will help to determine the importance of spermidine. Additionally, our structural information is also helpful to design potent inhibitors against parasite homologs (including GspS and TryS) because they and E. coli GspS share >50% sequence similarity.
Materials and methods
Protein preparation
The plasmid containing E. coli gene GspS was obtained from Professor Christopher T Walsh at Harvard Medical School. The recombinant plasmid was then transformed into E. coli BL21 (DE-3) (Novagen) for expression. The resulting protein was purified by following the reported procedure (Bollinger et al, 1995; Kwon et al, 1997). The protein was purified to give >95% homogeneity on the basis of SDS–PAGE and further confirmed by mass spectrometry. Se-Met GspS was prepared according to the method described previously (Guerrero et al, 2001) and purified as was the recombinant GspS.
Crystallization and data collection
GspS and the Se-Met GspS were dissolved (10 mg/ml) in 20 mM HEPES (pH 7.4) containing 1 mM EDTA and 5 mM DTT. The hanging-drop vapor diffusion method was applied for crystallization with a protein-to-reservoir ratio of 1:1. Using Hampton Research Crystallization kits, the initial screening generated the crystals of GspS in 0.1 M Tris–HCl (pH 8.5) containing 12% (v/v) PEG3350 and 0.5 M MgCl2, as well as the crystals of Se-Met GspS in 0.1 M trisodium citrate dihydrate (pH 5.6) containing 12% (v/v) PEG3350 and 0.6 M NaCl. The reservoir volume was 500 μl. High quality crystals were observed after 5 days at room temperature. Crystals were soaked in a cryoprotectant containing 20% (v/v) glycerol in the reservoir. The crystals of GspS in complex with substrate, product or inhibitor (2; the molecule was synthesize as previously described (Chen and Coward, 1998)) were obtained under conditions similar to that used for apo_GspS with slight modification.
The selenium multi-wavelength anomalous diffraction (MAD) data for GspS was collected at Taiwan beamline BL12B2 in SPring-8, Japan, with the wavelengths of peak (0.9797 Å), edge (0.9799 Å) and high remote (0.9537 Å). The ADSC Quantum 4R charge-coupled device and Oxford Cryostream cooler were used for data collection. Data for GspS_ADP and GspS_inhibitor were collected at SPXF (Synchrotron Protein Crystallography Facility)_BL13B in the National Synchrotron Radiation Research Center (at Hsinchu, Taiwan) using the ADSC Q315 CCD detector. GspS_GSH_ADP data were collected at Academia Sinica using a Rigaku MicroMax002 X-ray generator equipped with an R-Axis IV++ image plate detector. Details of the data collection statistics are shown in Table II.
Table 2.
Data collection and refinement statisticsa
| Apo_GspS | Se-Met-GspS | GspS_AMPPNP | GspS_ADP | GspS_ GSH _ADP | GspS_inhibitor | |||
|---|---|---|---|---|---|---|---|---|
| Space group | C2 | I23 | P1 | P1 | P1 | P1 | ||
| Unit cell parameters (Å) | a=149.27 | a=b=c=248.11 | a=60.01 | a=60.20 | a=59.91 | a=60.03 | ||
| b=92.96 | b=75.54 | b=75.61 | b=75.48 | b=75.29 | ||||
| c=108.30 | c=84.06 | c=84.44 | c=84.01 | c=84.66 | ||||
| β=109.37° | α=70.67° | α=70.17° | α=70.44° | α=70.09° | ||||
| β=74.36° | β=73.92° | β=74.17° | β=74.06° | |||||
| γ=78.22° | γ=77.77° | γ=78.06° | γ=77.55° | |||||
| Wavelength (Å) | 1.000 | 0.9797 (peak) | 0.9799 (edge) | 0.9537 (high remote) | 1.000 | 1.000 | 1.000 | 1.000 |
| Resolution (Å) | 50–2.2 (2.28–2.2) | 30–3.1 (3.21–3.1) | 50–3.06 (3.17–3.06) | 30–2.98 (3.09–2.98) | 30–2.7 (2.7–2.8) | 30–2.08 (2.08–2.15) | 30–2.2 (2.28–2.2) | 30–2.7 (2.7–2.8) |
| Total no. of reflections | 297 079 | 403 843 | 236 279 | 252 629 | 86 668 | 287 870 | 217 017 | 111 088 |
| No. of unique reflections | 70 953 | 45 503 | 47 722 | 51 551 | 34 912 | 75 954 | 64 297 | 35 215 |
| Completeness (%) | 99.7 (99.9) | 99.7 (98.8) | 100 (99.5) | 99.9 (99.5) | 96.4 (95.2) | 96 (92.6) | 95.7 (90.6) | 96.6 (96.8) |
| Redundancy | 4.1 (4.2) | 8.9 (8.8) | 5.0 (4.7) | 4.9 (4.6) | 2.5 (2.5) | 3.8 (3.3) | 3.3 (3.1) | 3.2 (3.2) |
| I/sigma | 18.8 (3) | 18.7 (6) | 22 (4.5) | 21.4 (3.9) | 11.1 (3) | 14.8 (3.4) | 26 (3) | 20.7 (6.4) |
| Rmergeb | 7.6 (48.6) | 11.4 (43.1) | 10.1 (47.7) | 10 (53.5) | 10.5 (44.3) | 9.5 (34.8) | 7.5 (35.7) | 8.4 (33.9) |
| Refinement statistics | ||||||||
| No. of reflections | 67 294 (5770)c | 29 772 (1929)e | 63 960 (4204)e | 61 422 (5085)c | 29 543 (2342)e | |||
| Resolution (Å)d | 50–2.2 (2.2–2.28) | 30–2.7 (2.7–2.8) | 30–2.1 (2.10–2.18) | 30–2.2 (2.2–2.28) | 30–2.8 (2.8–2.9) | |||
| Rworking | 17.55 (26.09) | 19.75 (32.72) | 17.35 (19.50) | 16.79 (25.98) | 17.30 (21.16) | |||
| Rfreec | 23.60 (32.65)f | 25.96 (39.56)f | 23.63 (25.54)g | 23.18 (34.81)f | 24.25 (30.85)g | |||
| No. of solvent molecules | 1224 | 462 | 911 | 1228 | 497 | |||
| R.m.s.d. bond length (Å) | 0.0197 | 0.0125 | 0.0117 | 0.0118 | 0.0081 | |||
| R.m.s.d. angles | 1.929 | 1.611 | 1.613 | 1.372 | 1.445 | |||
| Ramachandran plot (%) | ||||||||
| Most favored/allowed regions | 88.2/100 | 84.2/ 99.9 | 87.5/100 | 88.2/100 | 85.8/99.9 | |||
| Average B factor (Å2) | ||||||||
| Total | 39.63 | 18.11 | 31.41 | 32.58 | 29.70 | |||
| Substrate/product | 10.29 | 19.83 | 22.61 | 16.71 | ||||
| Inhibitor | 35.73 | |||||||
| All crystal forms have two molecules per asymmetric unit. | ||||||||
| Rmerge=SUM (ABS (I−〈I〉))/SUM (I). | ||||||||
| All positive reflections are used in the refinements. | ||||||||
| Values in parentheses are for the highest-resolution shell. | ||||||||
| Reflections, I>2σ. | ||||||||
| Rfree=R factor calculated using 5% of the reflection data chosen randomly and omitted from the start of refinement. | ||||||||
| Rfree=R factor calculated using 10% of the reflection data chosen randomly and omitted from the start of refinement. |
Model building and crystallographic refinement
Data were processed using the HKL software package (Otwinowski and Minor, 1997). Heavy atoms in the Se-substituted GspS crystals were located using the program SOLVE (Terwilliger and Berendzen, 1999), which was also used to calculate the phase angles. The MAD map at 3.3 Å was subjected to maximum-likelihood density modification followed by autotracing using RESOLVE (Terwilliger, 2000). An initial model was built using RESOLVE and XtalView (McRee, 1999). The model was improved by manual rebuilding using XtalView and O (Jones et al, 1991). The GspS protein model was finally refined to 2.2 Å using the native GspS data set. The structures of the enzyme/substrate, enzyme/product and enzyme/inhibitor complexes were determined by molecular replacement using the program CNS (Brünger et al, 1998) with the phase angle calculated from the refined model of apo_GspS. The model coordinates, topology and parameter files for ADP, AMPPNP and GSH (for CNS structure calculation) were taken from the HIC-UP www server (Kleywegt and Jones, 1998). The computational refinements, each started with simulated annealing protocol, were carried out using CNS. The refinement statistics are listed in Table II. All atoms are assumed to have full occupancies, and indeed the B factors of the ligands are all close to the values of the surrounding side chains. The criteria used in identifying water molecules are based on the well-defined 2Fo−Fc electron density map contoured at 1ó and the positions are generally near the protein atoms (within 6 Å).
Site-directed mutagenesis and activity assay
To produce GspS and corresponding mutant proteins more efficiently, the E. coli GspS gene was subcloned into the vector of pET28a (Novagen) that encodes an N-terminal His6 tag. The mutants were prepared using the QuickChange site-directed mutagenesis kit (Stratagene Co.). Each mutation was confirmed by DNA sequencing. The correct constructs were subsequently transformed to E. coli BL21 (DE3) for protein expression. All the His6-tagged proteins were purified by nickel affinity chromatography with the His Excellose Spin Kit (Yeastern Biotech). The procedures for cell culturing and lysis were similar to those used in previous reports (Bollinger et al, 1995; Kwon et al, 1997). Protein concentration was determined in accordance with the Bradford method (Bio-Rad Protein Assay kit) with bovine serum albumin as standard.
Activity measurements of Gsp synthetase and mutants were carried out using a continuous spectrometric assay. Formation of the product ADP (resulting from ATP hydrolysis) is coupled to oxidation of NADH through the activities of pyruvate kinase and lactate dehydrogenase (Oza et al, 2002a). The detailed procedures are based on a previously described method (Bollinger et al, 1995).
Supplementary Material
Supplementary information
Acknowledgments
We thank Dr Yu-Shan Huang and Mr Chien-Chang Tseng (NSRRC, National Syncrotron Radiation Research Center, Hsinchu, Taiwan) for technical support with data collections. We are grateful to the National Core Facility of Proteomics (Taipei, Taiwan) for crystallographic screening. This work was supported by grants from Academia Sinica (94C006-1 to CHL) and National Core Facility of High-Throughput Protein Crystallography Grant (NSC 95-3112-B-001-015 to AH-JW). The atomic coordinates and the structure factors have been deposited in the Protein Data Bank for structure (PDB ID for Apo_GspS, 2IOB; GspS_AMPPNP, 2IO7; GspS_ADP, 2IO8; GspS_GSH_ADP, 2IO9; GspS_inhibitor, 2IOA).
References
- Barton GJ (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng 6: 37–40 [DOI] [PubMed] [Google Scholar]
- Bateman A, Rawlings ND (2003) The CHAP domain: a large family of amidases including GSP amidase and peptidoglycan hydrolases. Trends Biochem Sci 28: 234–237 [DOI] [PubMed] [Google Scholar]
- Bollinger JM Jr, Kwon DS, Huisman GW, Kolter R, Walsh CT (1995) Glutathionylspermidine metabolism in Escherichia coli. Purification, cloning, overproduction, and characterization of a bifunctional glutathionylspermidine synthetase/amidase. J Biol Chem 270: 14031–14041 [DOI] [PubMed] [Google Scholar]
- Boveris A, Sies H, Martino EE, Docampo R, Turrens JF, Stoppani AO (1980) Deficient metabolic utilization of hydrogen peroxide in Trypanosoma cruzi. Biochem J 188: 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Chen S, Coward JK (1998) Investigations on new strategies for the facile synthesis of polyfunctionalized phosphinates: phosphinopeptide analogues of glutathionylspermidine. J Org Chem 63: 502–509 [DOI] [PubMed] [Google Scholar]
- Chen S, Lin CH, Walsh CT, Coward JK (1997) Novel inhibitors of trypanothione biosynthesis: synthesis and evaluation of a phosphinate analog of glutathionyl spermidine (GSP), a potent, slow-binding inhibitor of GSP synthetase. Bioorg Med Chem Lett 7: 505–510 [Google Scholar]
- Comini M, Menge U, Flohe L (2003) Biosynthesis of trypanothione in Trypanosoma brucei brucei. Biol Chem 384: 653–656 [DOI] [PubMed] [Google Scholar]
- Comini M, Menge U, Wissing J, Flohe L (2005) Trypanothione synthesis in crithidia revisited. J Biol Chem 280: 6850–6860 [DOI] [PubMed] [Google Scholar]
- Dubin T (1959) Evidence for conjugates between polyamines and glutathione in E. coli. Biochem Biophys Res Commun 1: 262–265 [Google Scholar]
- Fairlamb AH, Cerami A (1985) Identification of a novel, thiol-containing co-factor essential for glutathione reductase enzyme activity in trypanosomatids. Mol Biochem Parasitol 14: 187–198 [DOI] [PubMed] [Google Scholar]
- Fairlamb AH, Cerami A (1992) Metabolism and functions of trypanothione in the Kinetoplastida. Annu Rev Microbiol 46: 695–729 [DOI] [PubMed] [Google Scholar]
- Fan C, Moews PC, Shi Y, Walsh CT, Knox JR (1995) A common fold for peptide synthetases cleaving ATP to ADP: glutathione synthetase and D-alanine:D-alanine ligase of Escherichia coli. Proc Natl Acad Sci USA 92: 1172–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Moews PC, Walsh CT, Knox JR (1994) Vancomycin resistance: structure of D-alanine:D-alanine ligase at 2.3 Å resolution. Science 266: 439–443 [DOI] [PubMed] [Google Scholar]
- Guerrero SA, Hecht HJ, Hofmann B, Biebl H, Singh M (2001) Production of selenomethionine-labelled proteins using simplified culture conditions and generally applicable host/vector systems. Appl Microbiol Biotechnol 56: 718–723 [DOI] [PubMed] [Google Scholar]
- Henderson GB, Yamaguchi M, Novoa L, Fairlamb AH, Cerami A (1990) Biosynthesis of the trypanosomatid metabolite trypanothione: purification and characterization of trypanothione synthetase from Crithidia fasciculata. Biochemistry 29: 3924–3929 [DOI] [PubMed] [Google Scholar]
- Hiratake J (2005) Enzyme inhibitors as chemical tools to study enzyme catalysis: rational design, synthesis, and applications. Chem Record 5: 209–228 [DOI] [PubMed] [Google Scholar]
- Hiratake J, Kato H, Oda J (1994) Machanism-based inactivation of glutathione synthetase by phosphinic acid transition-state analogue. J Am Chem Soc 116: 12059–12060 [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1998) Databases in protein crystallography. Acta Crystallogr D 54: 1119–1131 [DOI] [PubMed] [Google Scholar]
- Krauth-Siegel RL, Meiering SK, Schmidt H (2003) The parasite-specific trypanothione metabolism of Trypanosoma and Leishmania. Biol Chem 384: 541–549 [DOI] [PubMed] [Google Scholar]
- Kwon DS, Lin CH, Chen S, Coward JK, Walsh CT, Bollinger JM Jr (1997) Dissection of glutathionylspermidine synthetase/amidase from Escherichia coli into autonomously folding and functional synthetase and amidase domains. J Biol Chem 272: 2429–2436 [DOI] [PubMed] [Google Scholar]
- Lin CH, Chen S, Kwon DS, Coward JK, Walsh CT (1997a) Aldehyde and phosphinate analogs of glutathione and glutathionylspermidine: potent, selective binding inhibitors of the E. coli bifunctional glutathionylspermidine synthetase/amidase. Chem Biol 4: 859–866 [DOI] [PubMed] [Google Scholar]
- Lin CH, Kwon DS, Bollinger JM Jr, Walsh CT (1997b) Evidence for a glutathionyl-enzyme intermediate in the amidase activity of the bifunctional glutathionylspermidine synthetase/amidase from Escherichia coli. Biochemistry 36: 14930–14938 [DOI] [PubMed] [Google Scholar]
- Marton LJ, Pegg AE (1995) Polyamines as targets for therapeutic intervention. Annu Rev Pharmacol Toxicol 35: 55–91 [DOI] [PubMed] [Google Scholar]
- McRee DE (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125: 156–165 [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME (1983) Glutathione. Annu Rev Biochem 52: 711–760 [DOI] [PubMed] [Google Scholar]
- Müller S, Liebau E, Walter RD, Krauth-Siegel RL (2003) Thiol-based redox metabolism of protozoan parasites. Trends Parasiol 19 (7): 320–328 [DOI] [PubMed] [Google Scholar]
- Murzin AG (1996) Structural classification of proteins: new superfamilies. Curr Opin Struct Biol 6: 386–394 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode: macromolecular crystallography Part A. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Oza SL, Ariyanayagam MR, Aitcheson N, Fairlamb AH (2003) Properties of trypanothione synthetase from Trypanosoma brucei. Mol Biochem Parasitol 131: 25–33 [DOI] [PubMed] [Google Scholar]
- Oza SL, Ariyanayagam MR, Fairlamb AH (2002a) Characterization of recombinant glutathionylspermidine synthetase/amidase from Crithidia fasciculata. Biochem J 364: 679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza SL, Shaw MP, Wyllie S, Fairlamb AH (2005) Trypanothione biosynthesis in Leishmania major. Mol Biochem Parasitol 139: 107–116 [DOI] [PubMed] [Google Scholar]
- Oza SL, Tetaud E, Ariyanayagam MR, Warnon SS, Fairlamb AH (2002b) A single enzyme catalyses formation of trypanothione from glutathione and spermidine in Trypanosoma cruzi. J Biol Chem 277: 35853–35861 [DOI] [PubMed] [Google Scholar]
- Pegg AE (1986) Recent advances in the biochemistry of polyamines in eukaryotes. Biochem J 234: 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penketh PG, Kennedy WP, Patton CL, Sartorelli AC (1987) Trypanosomatid hydrogen peroxide [corrected] metabolism. FEBS Lett 221: 421–437 [DOI] [PubMed] [Google Scholar]
- Polekhina G, Board PG, Gali RR, Rossjohn J, Parker MW (1999) Molecular basis of glutathione synthetase deficiency and a rare gene permutation event. EMBO J 18: 3204–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames SL, Fairlamb AH, Cerami A, Walsh CT (1986) Purification and characterization of trypanothione reductase from Crithidia fasciculata, a newly discovered member of the family of disulfide-containing flavoprotein reductases. Biochemistry 25: 3519–3526 [DOI] [PubMed] [Google Scholar]
- Smith K, Nadeau K, Bradley M, Walsh C, Fairlamb AH (1992) Purification of glutathionylspermidine and trypanothione synthetases from Crithidia fasciculata. Protein Sci 1: 874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor CW, Tabor H (1984) Polyamines. Annu Rev Biochem 53: 749–790 [DOI] [PubMed] [Google Scholar]
- Tabor H, Tabor CW (1975) Isolation, characterization, and turnover of glutathionylspermidine from Escherichia coli. J Biol Chem 250: 2648–2654 [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D 56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden JB, Blanchard CZ, Holden HM, Waldrop GL (2000) Movement of the biotin carboxylase B-domain as a result of ATP binding. J Biol Chem 275: 16183–16190 [DOI] [PubMed] [Google Scholar]
- Wang CC (1995) Molecular mechanisms and therapeutic approaches to the treatment of African trypanosomiasis. Annu Rev Pharmacol Toxicol 35: 93–127 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information