

Abstract
Inflammation participates in tissue repair through multiple mechanisms including directly regulating the cell fate of resident progenitor cells critical for successful regeneration. Upon surveying target cell types of the TNF ligand TWEAK, we observed that TWEAK binds to all progenitor cells of the mesenchymal lineage and induces NF-κB activation and the expression of pro-survival, pro-proliferative and homing receptor genes in the mesenchymal stem cells, suggesting that this pro-inflammatory cytokine may play an important role in controlling progenitor cell biology. We explored this potential using both the established C2C12 cell line and primary mouse muscle myoblasts, and demonstrated that TWEAK promoted their proliferation and inhibited their terminal differentiation. By generating mice deficient in the TWEAK receptor Fn14, we further showed that Fn14-deficient primary myoblasts displayed significantly reduced proliferative capacity and altered myotube formation. Following cardiotoxin injection, a known trigger for satellite cell-driven skeletal muscle regeneration, Fn14-deficient mice exhibited reduced inflammatory response and delayed muscle fiber regeneration compared with wild-type mice. These results indicate that the TWEAK/Fn14 pathway is a novel regulator of skeletal muscle precursor cells and illustrate an important mechanism by which inflammatory cytokines influence tissue regeneration and repair. Coupled with our recent demonstration that TWEAK potentiates liver progenitor cell proliferation, the expression of Fn14 on all mesenchymal lineage progenitor cells supports a broad involvement of this pathway in other tissue injury and disease settings.
Keywords: Fn14, inflammation, muscle, regeneration, TWEAK
Introduction
Inflammation is the first-line defensive mechanism to noxious insults ranging from microbial infections to physical injury (Nathan, 2002). Although excessive inflammation can have devastating consequences, in most cases inflammatory responses lead to effective clearance of pathogens and/or healing. In fact, depletion of inflammatory cell types such as macrophages has been shown to result in delayed repair response in injury models of a number of tissues, including muscle (Lescaudron et al, 1999), mesothelium (Mutsaers et al, 2002), and both the central (Kotter et al, 2001) and peripheral nerve systems (Luk et al, 2003). It is now well established that inflammation contributes to tissue repair by clearing debris, engulfment and digestion of apoptotic/necrotic cell bodies, epithelial closure and promoting angiogenesis (Nathan, 2002).
Much underappreciated until recently, the various cytokines secreted by inflammatory cells also directly influence the properties of progenitor cells that reside in tissues undergoing repair and regeneration (Duffield, 2003). For example, the macrophage-derived cytokine TNF promotes the proliferation of oligodendrocyte progenitors after cuprizone-induced demyelination (Arnett et al, 2001). Similarly, soluble factor(s) secreted by macrophages can also drive the proliferation of mesothelial cells (Mutsaers et al, 2002) and muscle satellite cells (Merly et al, 1999) during mesothelial and muscle regeneration, respectively. However, it has also been demonstrated that inflammatory cytokines block neural progenitor differentiation (Ekdahl et al, 2003; Monje et al, 2003), suggesting that the biological consequence of these inflammatory mediators on tissue repair and regeneration is likely a complex one.
The largely macrophage-derived cytokine TWEAK and its receptor Fn14 are relatively new additions to the TNF superfamily, which includes well-known modulators of cell proliferation, differentiation and apoptosis (Locksley et al, 2001). Originally identified as a weak inducer of cell death in tumor cell lines (Chicheportiche et al, 1997), TWEAK was subsequently shown to exert pleiotropic effects on a variety of cell types in vitro, including pro-angiogenic activities on endothelial cells (Lynch et al, 1999; Jakubowski et al, 2002) and pro-inflammatory activities on epithelial cells (Chicheportiche et al, 1997), astrocytes (Saas et al, 2000), dermal fibroblasts and synoviocytes (Chicheportiche et al, 2002). Conspicuously absent from the TWEAK-responding cell types are lymphocytes, and Fn14 expression on these cells has not been demonstrated. Interestingly, the TWEAK receptor Fn14 is an FGF-inducible gene and is highly upregulated in liver injury and regeneration and arterial wounding (Feng et al, 2000; Wiley et al, 2001), suggesting a regulatory role in settings of tissue injury and repair. Recently, we demonstrated that the TWEAK/Fn14 pathway is a potent inducer of liver progenitor cell proliferation in response to chemical injury in vivo (Jakubowski et al, 2005). However, the physiological relevance of the TWEAK/Fn14 pathway in other contexts remains elusive.
In this study, we explored the potential role of TWEAK/Fn14 pathway in modulating mesenchymal progenitor cell biology in vitro and in vivo in a model of skeletal muscle injury and repair. We showed that all progenitor cells of mesenchymal lineage express the TWEAK receptor Fn14. We further demonstrated that TWEAK promotes proliferation of both an established myoblast cell line and primary muscle myoblasts. Finally, by generating mice deficient in Fn14, we established that the TWEAK/Fn14 pathway is required for optimal muscle regeneration in vivo.
Results
Mesenchymal progenitor cells are a novel target cell type of TWEAK
In our effort to identify in vivo target cell types for TWEAK, we found that TWEAK binds to progenitor cells of the mesenchymal lineage, including human primary mesenchymal stem cells, skeletal muscle myoblasts and preadipocytes as well as chondrocyte and osteoblast precursors cultured in vitro (Figure 1A). We also demonstrated that the TWEAK receptor Fn14 was expressed on these progenitor cells by staining with the anti-human Fn14 monoclonal antibody ITEM-4 (Figure 1A). To see if these cells would respond to TWEAK, we examined NF-κB activation in mesenchymal stem cells and osteoblast precursors upon TWEAK treatment as it has previously been shown that Fn14 signal transduction is mediated through the TRAF-binding site in its cytoplasmic tail (Brown et al, 2003). As shown in Figure 1B, TWEAK induced robust NF-κB activation in both cell types as measured by the amount of activated p65 present in the cell lysate following TWEAK stimulation using the TranAM assay system, indicating that progenitor cells of the mesenchymal lineage are indeed TWEAK-responsive cells. The ability of TWEAK to induce NF-κB activation in these progenitor cells is further confirmed by transcription profiling study of mesenchymal stem cells treated with TWEAK. Even in the high serum culture condition (10% FBS), TWEAK induced robust transcriptional upregulation of well-known NF-κB-regulated genes, including TRAF1 and 3, NF-κB2 and RelB, which are regulators of NF-κB pathway themselves, and pro-survival genes such as A20 and c-IAP2, as well as cell adhesion genes such as ICAM-1 and VCAM-1n (Figure 1C). Importantly, under low (0.2%) or medium (2%) serum conditions, TWEAK also induced the expression of many cell cycle-related genes including cdc2, cyclin A2, survivin, MAD2, among others (Figure 1D and data not shown). These results therefore indicate that TWEAK may regulate cell fate decisions of progenitor cells.
Figure 1.
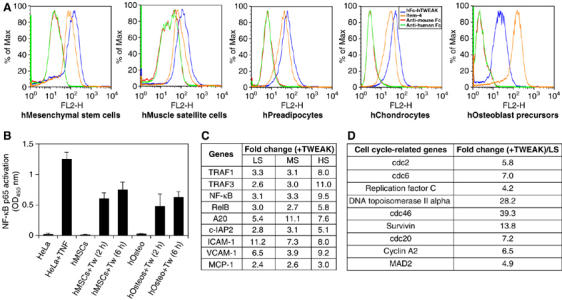
Human mesenchymal progenitor cells are a novel target cell type for TWEAK. (A) Human primary mesenchymal stem cells, skeletal muscle myoblasts, preadipocytes, chondrocytes and osteoblast precursors (Cambrex) were cultured according to the manufacturer's protocols. First-passage cells showed staining for TWEAK binding using Fc-TWEAK and for expression of Fn14 using the anti-hFn14 mAb ITEM-4. Anti-mouse and anti-human Fcs were used as negative controls, (B) NF-κB was activated in human mesenchymal stem cells (hMSCs) and osteoblast precursors (hOsteos) following 2 or 6 h of treatment with 100 ng/m TWEAK (Tw). Activation was measured using the TransAM NF-κB p65 activation assay system with cell lysates from normal and TNF-treated HeLa cells serving as negative and positive controls. The assays were carried out in triplicate and the data shown are representative of three independent experiments. (C) List of representative genes induced by TWEAK (100 ng/ml versus heat-inactivated TWEAK 100 ng/ml) in mesenchymal stem cells in low serum (LS: 0.2% FBS), moderate serum (MS: 2% FBS) and high serum (HS: 10% FBS). (D) List of some cell cycle-related genes induced by TWEAK (versus inactivated TWEAK) in mesenchymal stem cells cultured under low-serum conditions (0.2% FBS). Triplicate samples were analyzed for each condition and the fold changes were calculated using averages from triplicates. All fold changes reached statistical significance (P<0.01).
TWEAK promotes proliferation and inhibits terminal myogenesis of C2C12 cells
To further investigate how TWEAK might influence cell fate of progenitor cells, we took advantage of the well-established in vitro cell differentiation model of the mesenchymal lineage, C2C12 terminal myogenesis. We first confirmed that, similar to human skeletal muscle myoblasts, the murine C2C12 myoblasts also expressed Fn14 on their surface (Figure 2A and B). As expected, upon switching from regular growth medium (GM) (containing 15% FBS) to (DM), (containing 2% horse serum), mononuclear proliferating C2C12 cells enter into a well-characterized differentiation program that includes cell cycle arrest, fusion of mononuclear cells and formation of multinucleate myotubes (Figure 2C). In the presence of TWEAK, however, C2C12 cells largely remained as a monolayer of mononuclear cells even when placed in DM (Figure 2D). Both the anti-TWEAK blocking antibody ABG.11 and the hFn14-hFc-soluble TWEAK receptor effectively reversed the inhibition of myotube formation, confirming the specificity of our observation (Figure 2E and not shown). Importantly, addition of the anti-Fn14 blocking antibody P2D3 also was able to reverse the inhibitory effect of TWEAK (Figure 2F), demonstrating that the activity of TWEAK on C2C12 cells was mediated through the Fn14 receptor. As it is well documented that TNF is a potent inhibitor of myogenesis in vitro (Szalay et al, 1997; Miller et al, 1988), we decided to investigate if there was any potential crosstalk between the TNF and TWEAK pathways in the C2C12 myogenesis model. Although the soluble TNF receptor mTNFRI-Fc totally reversed the inhibition of C2C12 differentiation by TNF (Figure 2G and H), it had no effect on the inhibitory activity of TWEAK (Figure 2I). A similar result was also seen using anti-TNF or anti-TNFRI blocking antibodies (data not shown). Conversely, the anti-TWEAK blocking antibody ABG.11 and anti-Fn14 blocking antibody P2D3 both failed to block TNF's ability to inhibit C2C12 myogenesis (Figure 2J and not shown). Based on these observations, we concluded that although both TWEAK and TNF exhibit similar potent inhibitory effects on C2C12 differentiation, these two act independently of each other through different receptors.
Figure 2.
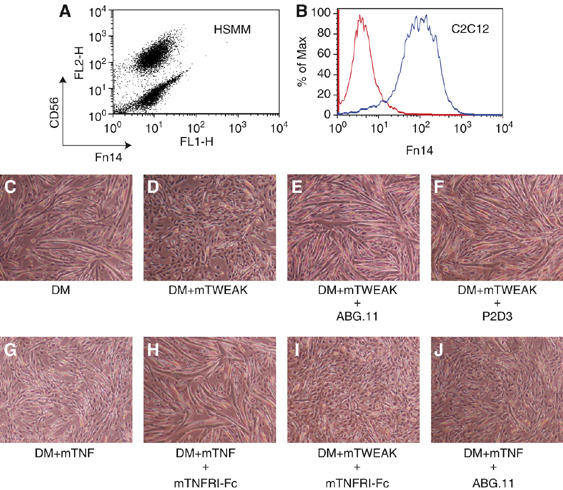
The TWEAK receptor Fn14 is expressed by myoblasts, and TWEAK inhibits differentiation of C2C12 cells. (A) FACS showed that most human skeletal muscle myoblasts (HSMM, from Cambrex) expressed Fn14, as well as the lineage marker CD56, on the cell surface. (B) Myoblasts of the mouse C2C12 line also expressed Fn14 on the surface. (C–J) Myotube formation by C2C12 cells in low-serum differentiation medium (DM) was assessed after addition of the following reagents: (C) no addition, myotube formation was extensive, (D) 100 ng/ml murine recombinant TWEAK, myotube formation inhibited; (E) 100 ng/ml mTWEAK+10 μg/ml anti-TWEAK mAb ABG.11, normal myotubes; (F) 100ng/ml mTWEAK+10 μg/ml anti-Fn14 mAb P2D3, normal myotubes; (G) 5 ng/ml mTNF; decreased myotubes; (H) 5 ng/ml mTNF+10 μg/ml mTNFRI-Fc, normal myotubes; (I) 5 ng/ml mTWEAK+10 μg/ml mTNFRI-Fc to block TNFR1, decreased myotubes; and (J) 5 ng/ml mTNF+10 μg/ml anti-TWEAK ABG.11, decreased myotubes. Phase contrast at 5 days after switching to DM with indicated additions; images are representative of at least 10 independent experiments.
Terminal myogenic differentiation of C2C12 cells requires exit from the cell cycle and expression of myogenic transcription factors such as myogenin, which in turn activate the transcription of an array of muscle-specific genes. To understand the molecular events underlying TWEAK's inhibitory effect on C2C12 myogenesis, we performed profiling studies comparing global gene expression patterns of C2C12 cells cultured in low-serum DM with or without TWEAK, and a number of representative genes with significant alteration in their expression levels are listed in Figure 3A. Upon analysis, we noticed that the expression levels of a large number of genes involved in cell cycle progression (i.e., cyclin D1 and c-myc), DNA/RNA synthesis (i.e., DNA polymerase A and TFIIIA) as well as chromosome remodeling (i.e., chromatin assembly factor) were dramatically increased with TWEAK stimulation, suggesting that these cells were mitotically active (Figure 3A). Using BrdU labeling, we confirmed that most C2C12 cells in DM in the absence of TWEAK are nonmitotic as expected. However, TWEAK-treated C2C12 cells were almost all BrdU positive, indicating that TWEAK stimulation potently promoted proliferation of these C2C12 cells even under low-serum condition (Figure 3B). Given that withdrawal from cell cycle is a well-established prerequisite for C2C12 terminal differentiation (Walsh and Perlman, 1997), it is likely that the perturbation of cell cycle arrest in these cells by TWEAK is responsible for the blockade in C2C12 myogenesis. The failure in myogenic progression in the presence of TWEAK was reflected at the molecular level by the decreased expression, based on both DNA array and Western blot analyses (Figure 3A and C), of the muscle-specific transcription factors myogenin and MyoD, which are critical for the terminal differentiation of C2C12 cells and govern the expression of many muscle-specific genes. Consequently, the overall expression levels of multiple muscle-related genes, such as the muscle structural proteins myosin heavy and light chains, as well as the muscle metabolic gene carbonic anhydrase, were significantly suppressed by TWEAK treatment (Figure 3A). This suppression of muscle-specific gene expression by TWEAK was accompanied by drastic phenotype differences between the TWEAK-treated and untreated cells. Whereas the majority of TWEAK-treated C2C12 cells maintained the undifferentiated mononuclear morphology with few actin filaments even in low-serum DM, the untreated C2C12 cells fully differentiated and became fused multinucleated myotubes, as revealed by DAPI and phalloidin staining (Figure 3D).
Figure 3.

TWEAK blocks the myogenic program and prevents cell cycle arrest in C2C12 cells. (A) TWEAK induced expression level changes in many genes important for cell cycle control and myogenic differentiation,for example, the muscle-specific transcription factors myogenin and MyoD. Fold changes and s.d.s were calculated using data from duplicate samples that passed statistical tests. (B) C2C12 cells continued to proliferate in low-serum DM when treated with TWEAK as shown by the much larger percentage of cells that incorporated BrdU in TWEAK-treated than in untreated cultures. Cells were analyzed after culture in DM for 3 days with or without 100 ng/ml of Fc-TWEAK and after 12 h BrdU labeling. (C) Myogenin protein was much lower after TWEAK treatment as shown by immunoblots of C2C12 cells in DM in the absence (none) or presence of Fc-hTWEAK (hTweak, 100 ng/ml) or hTNFa (10 ng/ml). (D) Staining of actin filaments by fluorescent phalloidin (red) was decreased after TWEAK treatment of C2C12 cells cultured in DM for 5 days with or without 100 ng/ml of Fc-TWEAK as indicated. Nuclei were identified by DAPI stain (blue).
Generation of Fn14-deficient mice
The profound effect of TWEAK on the established myoblast line C2C12 suggested that the TWEAK/Fn14 pathway may indeed be an important pathway regulating progenitor cell biology. To better understand the physiological relevance of this pathway, we decided to generate mice deficient in the TWEAK receptor Fn14. Using the standard gene-targeting strategy, an ∼10-kb Kpn1 genomic DNA fragment containing the entire mouse Fn14 gene was isolated and a targeting vector was designed to delete the first two exons (encoding amino acids 1–66), which contain the entire extracellular ligand-binding domain of Fn14 (Supplementary Figure S1a). Embryonic stem cell clones undergoing successful homologous recombination (8 out of 136) were identified by both genomic Southern blot and PCR analysis (Supplementary Figure S1a and data not shown) and injected into the C57BL/6 blastocysts to generate chimeras. Homozygous Fn14 null mice were obtained through standard breeding schemes (see Materials and methods). Homozygous Fn14−/− mice appear normal and do not have any obvious developmental defect in muscle or other tissues (Supplementary Figure S1e and unpublished data). To confirm the presence of a null mutation, we derived wild-type, heterozygous and homozygous mouse embryonic fibroblast (MEF) clones and assayed for both Fn14 mRNA and protein surface expression. As shown in Supplementary Figure S1b and c, both Northern blot and RT–PCR analysis clearly indicated the lack of Fn14 mRNA in Fn14−/− MEF clones. Importantly, FACS staining also demonstrated that TWEAK no longer bound to individually derived Fn14−/− clones (Supplementary Figure S1d), confirming the lack of Fn14 on the surface of these cells and that Fn14 is the only receptor for TWEAK. The residual low-frequency binding detected by Fc-TWEAK in Fn14−/− (also seen in Fn14+/+) MEF clones appears to be nonspecific, as we could not compete it off using the soluble receptor Fn14-Fc (not shown).
TWEAK/Fn14 pathway regulates the proliferative potential of primary mouse muscle myoblasts
Our in vitro C2C12 results strongly suggested that the TWEAK/Fn14 pathway might also regulate the properties of primary myogenic precursor cells such as muscle satellite cells and their committed myoblast progeny. Following confirmation that more than 95% of cells in our primary myoblasts preparation do indeed express Fn14 on the surface (not shown), we examined the direct effect of TWEAK on the growth and differentiation characteristics of mouse primary myoblasts isolated from the limb muscles of 6 to 8-week-old mice. In GM, addition of TWEAK resulted in an enhanced proliferation of the primary myoblasts as reflected by the presence after 3 days of growth of ∼2 × more mononuclear myoblasts in TWEAK-treated than in untreated cultures (Figure 4A and B). The proliferative effect of TWEAK was specific because the proliferation rate returned to basal level upon treatment with the TWEAK-neutralizing reagents anti-TWEAK antibody or Fn14-Fc (Figure 4C and not shown). Inclusion of TWEAK in the DM of primary myoblasts, on the other hand, resulted in significantly reduced myotube formation and the appearance of dying cells (Figure 4D and E). This cell death response of primary myoblasts was quite different from the response of C2C12 myoblasts, which remained alive and capable of proliferation in the TWEAK-containing DM for at least a week. Although we do not yet understand the precise reason(s) for the different responses of C2C12 and primary muscle cells, it is known that immortalized myogenic cell lines have mutations in cell cycle regulatory genes (Nowak et al, 2004); so it is not surprising that primary cells would behave differently than an established cell line in both proliferative potential and susceptibility to apoptosis.
Figure 4.

The TWEAK/Fn14 pathway regulates proliferation and differentiation of primary muscle myoblasts. TWEAK promoted the growth (A–C) and inhibited the differentiation (D, E) of primary myoblasts from wild-type mice. (A–C) After 3 days of proliferation in growth medium, cultures of primary myoblasts treated with 100 ng/ml Fc-TWEAK (B) contained ∼2 × more cells than untreated cultures (A) or cultures treated with both Fc-TWEAK and Fn14-Fc to block receptor function (C). (D, E) After 4 days in low-serum differentiation medium, untreated cultures of primary myoblasts formed multinucleate myotubes (D), whereas myotubes were rarely found in TWEAK-treated cultures (E). (F) Primary myoblasts isolated from Fn14-deficient mice produced fewer progeny than wild-type myoblasts in culture. Results from two independent experiments (exp. no. 1 and exp. no. 2) are shown. In each experiment, myoblasts were obtained from two individual wild-type (WT) and two individual Fn14−/− (KO) mice. Cells were seeded at the same initial cell densities. (G, H) Phase-contrast images of myotubes formed by myoblasts isolated from WT and Fn14-deficient mice.
Primary myoblasts isolated from Fn14-deficient mice produced fewer viable progeny in culture than did myoblasts from wild-type mice (Figure 4F). To rule out the possibility that the proliferation of wild-type myoblasts was ‘artificially' enhanced by exogenous TWEAK from the chicken extracts used in the culture media or TWEAK produced by myoblasts in an autocrine manner, we included the soluble Fn14-Fc decoy receptor in the wild-type myoblasts culture and observed no reduction in the number of progeny (data not shown). Based on these results, we conclude that the TWEAK/Fn14 pathway is intrinsically required for the full proliferative potential of mouse myoblasts. Interestingly, while Fn14-deficient myoblasts retained the ability to differentiate into myotubes when cultured in low-serum DM, we have consistently noticed morphological differences in myotubes fused from wild-type versus Fn14−/− myoblasts in that the Fn14−/−myoblasts seemed to form thicker myotubes upon fusion (Figure 4G and H). The underlying molecular basis for this difference is not yet known.
TWEAK/Fn14 pathway participates in muscle regeneration in vivo
Muscle satellite cells are adult muscle precursor cells residing quiescently under the basal lamina of the muscle fiber. Although not required for early muscle development, these cells are believed to be the main, if not the only, contributor to muscle regeneration in response to damage or increase in workload. Upon activation, satellite cells are capable of proliferating to produce committed myoblasts as progeny, which then repair injured muscle either by fusing with injured pre-existing myofibers or by forming entirely new myofibers (Hawke and Garry, 2001). Although histologically well characterized, the stepwise program of muscle regeneration is incompletely understood at the molecular level and a limited number of regulating factors have been identified to date (Morgan and Partridge, 2003). Based on our culture results, we hypothesized that the TWEAK/Fn14 pathway might be a novel regulator of muscle regeneration in vivo, and we tested this idea by comparing regeneration of wild-type versus Fn14−/− muscle in vivo following injury induced by the snake venom cardiotoxin (Couteaux et al, 1988).
We first examined the expression patterns of TWEAK and Fn14 mRNAs following cardiotoxin injection in the tibialis anterior (TA) muscle of wild-type and Fn14−/− mice using in situ hybrization analysis. The levels of both TWEAK and Fn14 were minimal in the undamaged wild-type muscle tissue (Figure 5A and G, quantified in Figure 5M), and TWEAK mRNA expression was also low in undamaged Fn14−/− muscle (Figure 5B). As expected, Fn14 mRNA was essentially undetectable in FN14−/− muscles (Figure 5H, J, L and M).
Figure 5.

TWEAK and Fn14 mRNAs increased upon injury, but increased TWEAK persists longer in Fn14−/− than in wild-type muscles and is produced by macrophages. (A–L) In situ hybridzation of TWEAK (A–F) and Fn14 (G–L) mRNAs in control (no inj) and cardiotoxin-treated wild-type and Fn14−/− muscles at 3 or 5 days post injection as indicated. Scale bar in (A) represents 100 μm. (M) At day 3 after cardiotoxin treatment, quantification of grain density showed that TWEAK mRNA was significantly (P<0.01, n=4) increased several-fold in both wild-type (gray bars) and Fn14−/− (blue bars) muscles. At day 5, in constrast, TWEAK persisted at a high level in Fn14−/− muscles, but returned to near the initial level in wild-type muscles. Fn14 mRNA was also increased by injury at days 3 and 5 in wild-type muscles (green bars, right panel). Error bars=s.d. No Fn14 signal above background was found in Fn14−/− muscles (not shown). (N) Macrophages are the major source of TWEAK expression in regenerating muscles. At 3 days after cardiotoxin, TA muscles (n=3) were dissociated into single-cell suspensions and three cell preparations were made: (i) cells positive for the macrophage marker Mac-1+ were purified using Mac-1 magnetic beads (macrophages, blue bars); (ii) myogenic cells were purified on a Percoll gradient (myoblasts, red bars); and (iii) Mac-1-negative cells, which were cells that did not bind to the Mac-1 beads (flow-through, green bars). RNA was isolated from each cell fraction and quantitative PCR was used to measure the amount of TWEAK mRNA in each cell type using an equal amount of input RNA and normalized versus the amount of GAPDH mRNA. TWEAK mRNA expression was significantly higher (*P<0.05, n=3) in macrophages than in myoblasts or flow-through cells. Error bars=s.d. (O) mRNA levels of TNF/TNFRs and EDA/XEDAR with and without cardiotoxin in wild-type and Fn14−/− mice. At 3 days after cardiotoxin (CTX), total RNA was isolated from TA muscles (n=3) and quantitative PCR was used to measure the mRNA levels for TNF, TNFR1, TNFR2, EDA and XEDAR. Statistical significance was not achieved between wild-type and Fn14-deficient mice.
Three days after cardiotoxin injury, expression levels of both TWEAK and Fn14 were significantly increased in the damaged areas of wild-type muscles (Figure 5C, I and M) and TWEAK mRNA to approximatly the same level in Fn14−/− as in wild-type muscles (Figure 5D and M). The similar increase in TWEAK mRNA in wild-type and Fn14−/− muscles at 3 days after cardiotoxin treatment was confirmed by quantitative PCR (not shown). By day 5, TWEAK expression had subsided considerably in wild-type muscles, whereas the level of Fn14 remained as high at day 5 as at day 3 (Figure 5E, K and M). The finding that increased TWEAK expression persisted for a longer period of time in injured muscles of Fn14 −/− mice than in wild-type mice (Figure 5E versus F) suggests that inactivation of the TWEAK receptor altered the injury response to cardiotoxin in the Fn14-deficient mice.
Two lines of evidence suggested that macrophages were the major source of the increased TWEAK expression in injured wild-type and Fn14−/− muscles. First, bright-field analysis revealed a clear association of TWEAK expression with infiltrating inflammatory cells, whereas Fn14 expression seemed to adopt a much more diffuse pattern (data not shown). Second, using a quantitative PCR assay, we found that macrophages (i.e., Mac-1-positive cells) isolated from both wild-type and Fn14−/− TA muscles at 3 days after cardiotoxin injection expressed relatively large amounts of TWEAK mRNA (Figure 5N). In contrast, very little TWEAK mRNA was found in either the non-macrophage cell fraction (i.e., Mac-1-negative cells) or in myogenic cells purified by Percoll gradient fractionation (Figure 5N).
To begin to determine if signaling by additional members of the TNF superfamily might also be altered in FN14-deficient compared with wild-type mice, we carried out quantitative PCR measurements to measure in regenerating TA muscles (n=3) the relative levels of the mRNAs encoding two TNF family ligands, TNFα and EDA (ectodysplasin), and their respective receptors (TNFR1, TNFR2, and XEDAR), as these two TNF ligand/receptor pathways have previously been suggested to play a role in muscle regeneration (Guttridge et al, 2000; Newton et al, 2004). As shown in Figure 5O, we indeed found that the levels of these mRNAs were significantly induced following cardiotoxin-induced injury, but did not differ significantly between the wild-type and Fn14-deficient muscles at 3 days, although there seems to be a trend of increased expression in the Fn14 knockout mice, suggestive of potential compensatory mechanism (see Discussion).
Defective inflammatory response and muscle regeneration in Fn14−/− mice following cardiotoxin-induced muscle injury
We next assessed muscle regeneration in response to cardiotoxin injury in both wild-type and Fn14 knockout mice using histological analysis. Similar to previous reports, cardiotoxin injection in the TA muscles caused considerable damage to muscle fibers, triggering a robust regenerative process that resulted in almost complete regeneration of wild-type muscles by 2 weeks after injury (Figure 6A–D). When mice deficient in Fn14 were injected with cardiotoxin, however, regeneration appeared to be delayed compared with that seen in wild-type muscles (Figure 6E–H). For example, large numbers of newly formed muscle fibers with their ‘signature' centrally located nuclei appeared in TA muscles of wild-type mice at 5 and 7 days following cardiotoxin injection, whereas such regenerated myofibers were rare in Fn14−/− mice at these times (Figure 6B, C, F and G). By day 14, the regenerative process appeared to be nearly complete in wild-type mice as shown by the clearance of inflammation and the replacement of damaged myofibers by the newly formed central nucleate fibers. In Fn14−/− mice, however, signs of residual muscle damage remained along with regenerated fibers, and some inflammatory cells also persisted (Figure 6D and H).
Figure 6.

Fn14−/− mice have delayed muscle regeneration. Cardiotoxin-treated muscles at indicated days after injury of wild-type (A–D) and Fn14−/− (E–H) mice are shown. Detailed views of B and F are shown in (I) and (J) respectively to show regenerating, central nucleate myotubes, some of which are indicated by arrows. Images are representative of three independent experiments with seven mice per genotype in total. Scale bar: 200 μm (A–C, E–G), 100 μm (D, H). (K) The number of central nucleated myotubes in Fn14−/− mice was significantly lower than in wild-type muscles at 5 and 7 days, but not 14 days, after cardiotoxin. Error bars=s.d.; **=P<0.01 by Student's t-test; n as indicated. (L) Myofibers that express embryonic myosin heavy chain (eMyHC), an additional marker of regenerating myotubes (dark brown stain), were more abundant in wild-type than in Fn14−/− muscles at 4 days after injury.
To further analyze the delayed regeneration in Fn14−/− compared with wild-type muscles, we quantified the number of central nucleate myofibers and examined expression of the embryonic isoform of myosin heavy chain (eMyHC) at different stages of regeneration. Myofibers in adult animals that have centrally located nuclei or that express eMyHC are known to be in the process of regeneration (e.g., DiMario et al, 1991; Pavlath et al, 2003). Confirming that regeneration was delayed in the Fn14−/− muscles, central nucleate myofibers were significantly less abundant in regenerating Fn14−/− than in regenerating wild-type muscles at both 5 and 7 days after injury (Figure 6I–K). At 14 days after injury, in contrast, the Fn14−/− and wild-type muscles had similar numbers of central nucleate fibers. Furthermore, fibers that expressd eMyHC were less abundant in Fn14−/− muscles than in wild-type muscles at 4 days after injury (Figure 6L and M) and similar results were seen at 5 and 7 days after injury (not shown).
With the interplay between inflammation and tissue regeneration in mind, we explored the inflammatory response following cardiotoxin in wild-type and Fn14−/− mice. Massive infiltration of inflammatory cells, mainly macrophages and neutrophils, is an early hallmark event of cardiotoxin-induced muscle regeneration which peaks around 3 days after injection (Figure 7A). In Fn14−/− mice injected with cardiotoxin, H&E staining revealed not only a significantly reduced presence of such infiltrates at day 3, but also a 2-day delay of peak infiltration in Fn14−/− muscles to day 5 (Figure 6E and F). This observation was consistent with our in situ analysis, which showed that TWEAK mRNA (likely produced by infiltrating inflammatory cells) persisted at 5 days post injury in Fn14−/− but not in wild-type, mice (Figure 5E, F and M).
Figure 7.
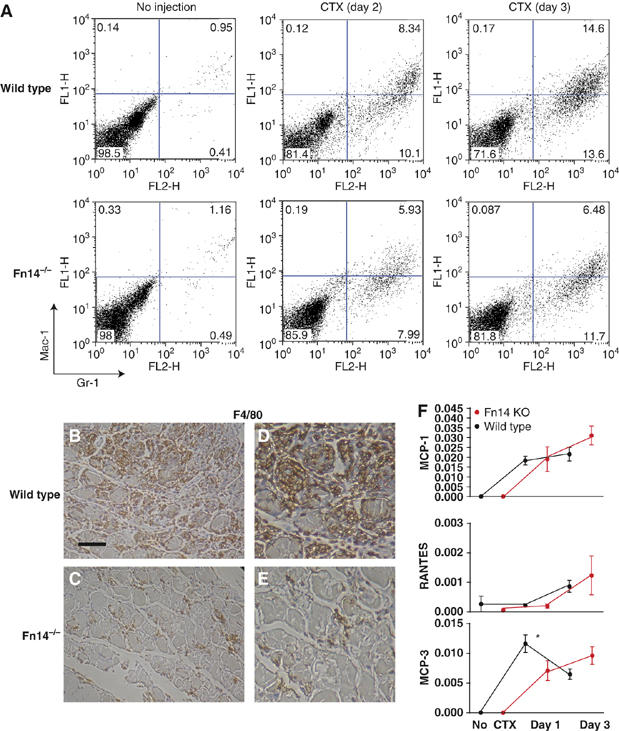
Altered inflammatory response in Fn14−/− muscles in response to cardiotoxin (CTX) injury. (A) FACS based on myeloid markers Mac-1 and Gr-1 showed that inflammatory cells were more abundant in wild-type than in Fn14−/− muscles at 2 and 3 days post injury. Slightly more cells were consistently recovered from wild-type than Fn14−/− mice at all time points; the same number of cells (10 000) are shown in each dot plot. For each genotype, cells from two mice were pooled for each time point and the results shown are representative of three independent experiments. (B–E) F4/80 expression (brown staining) of muscle tissue cross-sections shows higher numbers of inflammatory cells in wild-type (B) than in Fn14−/− (C) muscles on day 3 following cardiotoxin. Detailed views of (B) and (C) are shown in (D) and (E), respectively. Scale bar: 100 μm (B, C), 50 μm (D, E). (F) mRNA levels of MCP-1, MCP-3 and RANTES with and without cardiotoxin injection in wild-type and Fn14-deficient mice. At 1 and 3 days after cardiotoxin, total RNA was isolated from TA muscles (n=3) and quantitative PCR was used to quantify mRNAs. Asterisk indicates that the difference for MCP-3 expression at day 1 reached statistical significance (P<0.05, by Student's t-test).
To additionally assess, both qualitatively and quantitatively, the altered inflammatory response seen in Fn14−/− mice, we dissociated TA muscles using pronase digestion at different time points following cardiotoxin injection, and we carried out FACS analyses to identify inflammatory cells in the resulting single-cell suspensions. FACS analyses showed that very few inflammatory cells were present in uninjured TA muscles in either wild-type or Fn14−/− mice, as assessed by double staining with Mac-1 and Gr-1 (Figure 7A), which are markers for myeloid lineage inflammatory cells including monocytes and neutrophils (Lagasse and Weissman, 1996). Following cardiotoxin injection, wild-type muscles showed a very large increase of Mac-1+/Gr-1+ infiltrating cells with the percentage increasing from roughly 1% of the cells in uninjured muscles to 28% of the cells at day 3 after injury. Infiltrating cells also increased in injured Fn14−/− TA muscles, but the increase was smaller than in wild-type muscles, as Mac-1+/Gr-1+ cells accounted for less than 20% of the total isolated cells on day 3 from injured Fn14−/− TA muscles. The difference between inflammation in wild-type and Fn14−/− muscles was even more pronounced in the Mac-1hi/Gr-1+ subpopulation (marking monocytes/macrophages in a more activated state; Lagasse and Weissman, 1996), where the percentage in wild-type TA was more than double that in the Fn14−/− TA at 3 days after injury (14.6 versus 6.5%). We further confirmed our observation histologically by staining with the more specific monocyte/macrophage marker F4/80. On day 3, extensive F4/80+ infiltrates were located around the damaged muscle fibers in the wild-type muscles (Figure 7B and D). In Fn14−/− mice, however, the number of F4/80+ infiltrating cells was much less than in wild-type mice at day 3 (Figure 7C and E).
Because chemokines play a critical role in regulating the recruitment of inflammatory cells and TWEAK can potently induce chemokine production from C2C12 cells based on our profiling study (Figure 3), we conducted quantitative RT–PCR experiments to determine the temporal expression of several key chemokines following cardiotoxin injection in both wild-type and Fn14 knockout mice. As shown in Figure 7F, RANTES does not seem to play a key role in the immediate early phase following cardiotoxin injection, as no increase in its expression level was seen after day 1. However, the levels of MCP-1 were increased comparably in both wild-type and Fn14−/− mice, whereas the expression of MCP-3 was significantly lower in the absence of Fn14 at day 1 post cardiotoxin-induced injury, suggesting that MCP-3 might be a key mediator for the recruitment of inflammatory infiltrates in this model. Not surprisingly, the levels of MCP-3, MCP-1 and RANTES all trended higher in Fn14−/− mice at day 3 compared with wild-type mice, likely contributing to the delayed inflammation in these mice.
Discussion
In this study, we identified mesenchymal precursor cells as a novel target cell type for the inflammatory cytokine TWEAK, showing Fn14 expression on mesenchymal stem cells and all progenitor cells of this lineage. Using muscle myoblasts as an experimental system, we demonstrated that TWEAK stimulated proliferation of primary myoblasts and that Fn14-deficient myoblasts exhibited reduced proliferative capacity in vitro. In addition, TWEAK altered the differentiating capacity of both C2C12 and primary skeletal myoblast cells. Furthermore, our in vivo studies demonstrated upregulation of TWEAK and Fn14 expression within injured muscle tissue and delayed muscle regeneration in Fn14-deficient animals. Thus, the TWEAK/Fn14 pathway is an important modulator of the proliferative and differentiating potentials of skeletal muscle progenitor cells. In addition, we also defined TWEAK/Fn14 as a new biological pathway that regulates the properties of mesenchymal stem cells, herein showing TWEAK-induced NF-κB activation, as well as expression of pro-survival, pro-proliferative and cell adhesion genes.
The past few years have seen tremendous progress with respect to understanding the potential of mesenchymal progenitors in tissue repair and regeneration. However, our relatively poor understanding of factors governing their survival, homing, proliferation and differentiation presents a major challenge for a successful transition into clinical applications. In addition to TWEAK's proliferative effect on muscle myoblasts, we have also observed that TWEAK could potently inhibit the terminal differentiation of human osteoblast precursors, as well as adipogenesis and chondrogenesis of human mesenchymal stem cells in culture (B Browning and TS Zheng, unpublished results). Moreover, our profiling study indicated that TWEAK could induce the expression of antiapoptotic as well as cell adhesion molecules in mesenchymal stem cells, thus potentially affecting their homing and survival capacity in vivo. The ability of the TWEAK/Fn14 pathway to broadly regulate these critical properties of mesenchymal precursor cells might therefore present an opportunity of ‘TWEAKing' existing approaches for cell-based regenerative therapies, particularly in skeletal, cardiac and joint injury settings (Barry, 2003). This notion is consistent with our recent finding that TWEAK also potentiates the proliferation of liver progenitor cells (Fausto, 2005; Jakubowski et al, 2005), suggesting a fundamental involvement of this pathway in modulating progenitor cell behavior.
We found that activation of the TWEAK/Fn14 pathway inhibited differentiation and increased proliferation of both the immortalized C2C12 muscle cell line and primary myogenic cells. Our finding is consistent with previous demonstration that activation of the NF-κB pathway could drive cell proliferation and inhibit C2C12 differentiation through regulation of cyclin D1 (Guttridge et al, 1999). While our paper was under initial review, Dogra et al (2006) published their study, which also demonstrated that C2C12 differentiation is inhibited by TWEAK, and that TWEAK activates the NF-κB pathway and decreases the amount of MyoD. The results of Dogra et al are consistent with ours, and we additionally demonstrated the effect of TWEAK on different types of mesenchymal progenitor cells, identified differences between the responses of C2C12 and primary muscle cells to TWEAK, prepared Fn14−/− mice to inactivate the TWEAK pathway and identified a significant role for the TWEAK/Fn14 pathway in regulating skeletal muscle regeneration after injury in vivo.
Identification of the TWEAK/Fn14 pathway as a regulator of muscle myoblast cell proliferation and differentiation following muscle injury in vivo is significant. Previously, a number of soluble factors, such as members of the FGF family, PDGF, TGFβ, myostatin, IGF, HGF and TNF, have been implicated in the proliferation and differentiation of muscle precursor cells based on in vitro studies. Studies using knockout mice, however, have not always provided unequivocal support of the in vitro observations, possibly owing to the presence of redundant mechanisms in vivo. For example, as a well-documented cachetic factor on muscle fibers, TNF has been reported to inhibit differentiation, promote proliferation, and induce apoptosis of myoblast cells in vitro (Stewart et al, 2004); however, muscle regeneration occurs normally in TNF−/− mice (Collins and Grounds, 2001), a result that is in marked contrast to the delayed regeneration in mice lacking the TWEAK receptor Fn14 (this study). A further complication comes from the fact that most of these factors also exert potent effects on many other cell types including mature muscle fibers, making interpretation of their direct effect on muscle precursor cells from in vivo studies less straightforward. Based on our in situ data, Fn14 expression in quiescent muscle tissue seems to be very low, but drastically increases following cardiotoxin-induced muscle damage. Coupled with the observation that no apparent developmental defect in the muscle tissues was seen in either the Fn14 knockout mice or TWEAK overexpressing transgenic mice (LC Burkly, unpublished results), it appears that TWEAK/Fn14 is a unique pathway that becomes specifically engaged in muscle myoblast proliferation only during injury settings. Supporting this hypothesis, dramatic upregulation of Fn14 mRNA was also observed in the denervated muscle tissues of the mutant SOD transgenic model of ALS (J Lincecum and S Perrin, unpublished results).
Although delayed by several days, regeneration in Fn14−/− mice following cardiotoxin injury eventually became as complete as in wild-type muscles, showing that the TWEAK/Fn14 pathway is one of the multiple pathways that likely contribute to muscle regeneration. In many biological systems, wholly or partially redundant mechanisms are commonly found. One example is the transcriptional regulation of myogenesis by MyoD and Myf-5 during development (Rudnicki et al, 1993). Although we clearly demonstrated that TWEAK and TNF could inhibit C2C12 myogenesis independent of each other in vitro, it is still tempting to speculate that TWEAK and TNF could synergize with and potentially compensate for each other in vivo, given that they both are potent activators of the NF-κB pathway. In fact, it has been previously shown that TWEAK and TNF could synergistically induce the production of chemokines and cytokines in dermal fibroblasts (Chicheportiche et al, 2002). The intriguing possibility of such synergism between TNF and TWEAK in regulating muscle regeneration and other biological systems in vivo remains to be investigated. In addition, although not statistically significant, there seems to be some level of compensatory activation of the TNF/TNFR and EDA/XEDAR pathways in Fn14 knockout mice following cardiotoxin injury according to our quantitative RT–PCR analysis, a finding that reinforces the complex and often redundant nature of many biological responses. However, our study clearly shows that TWEAK and TNF are two independent ligand/receptor pathways important for the regulation of biological properties of mesenchymal and muscle cells; our study is therefore consistent with the results from a complete survey of binding between TNF family receptors and ligands, which showed that TWEAK bound only to Fn14 and TNF bound only to TNFR1 and TNFR2 (Bossen et al, 2006).
The mechanism by which Fn14 deficiency resulted in delayed muscle regeneration following cardiotoxin injection is likely two-fold. First, based on our in vitro observations with the C2C12 line and isolated primary muscle myoblasts, it is likely that, similar to the reduced proliferative capacity seen with isolated Fn14−/− myoblasts in culture, the expansion of myoblasts triggered by cardiotoxin was less than optimal in Fn14−/− mice, resulting in delayed muscle regeneration. Although we did observe delayed BrdU incorporation in Fn14−/− mice compared with wild-type mice (data not shown) using a pulsing protocol that is reported to label only myoblasts (Yan et al, 2003), we were unable to demonstrate experimentally that those BrdU-labeled cells were all myoblasts. As a result, we cannot conclude with absolute certainty that myoblast proliferation was directly affected in cardiotoxin-injured Fn14−/− mice. The second possible mechanism for delayed muscle regeneration could be due to the reduced inflammatory response in Fn14−/− mice, as inflammation could certainly contribute positively to efficient muscle regeneration by a number of means, including removal of debris from damaged fibers and secretion of soluble factors that promote muscle regeneration.
Although seemingly independent events, the proliferation/activation of myoblasts and inflammation during muscle regeneration might actually be tightly coupled, as suggested by the compelling evidence for active crosstalks between muscle precursor cells and inflammatory infiltrates in a recent study (Chazaud et al, 2003). We believe that the TWEAK/Fn14 pathway could very well be a mediator of such cellular interactions and one can envision a scenario of how the TWEAK/Fn14 pathway may function as a positive regulator of muscle precursor cell properties during injury-triggered muscle regeneration (Figure 8). Following cardiotoxin injection, necrotic muscle fibers can release a number of soluble factors, including chemokines that will recruit neutrophils and macrophages (Tsivitse et al, 2005), as well as FGFs (D'Amore, 1990), which would induce Fn14 expression on muscle precursor cells. As a result, TWEAK produced by the early inflammatory infiltrates would engage Fn14-expressing muscle precursor cells, resulting in not only their proliferation, but also enhanced production of chemokines important for recruitment of additional inflammatory cells that are beneficial to the repair response, thus creating a positive feedback loop promoting muscle regeneration. This notion is consistent with our C2C12 profiling result demonstrating that TWEAK induced drastic upregulation of several chemokines critical for macrophage recruitment including MCP-1, MCP-3, RANTES and MIP-1 (Figure 3A), whose expressions are significantly induced following cardiotoxin-induced muscle injury (Hirata et al, 2003). In fact, our real-time PCR analysis confirmed that the initial MCP-3 expression following cardiotoxin injury was much reduced in the absence of Fn14, and suggests that MCP-3-mediated chemotaxis is an important downstream trigger for the TWEAK/Fn14-induced inflammatory response amplification loop.
Figure 8.

A working model for regulation of muscle regeneration by the TWEAK/Fn14 pathway. Cardiotoxin-induced injury triggers release of soluble factors that recruit early inflammatory cells and induce Fn14 expression on muscle precursor cells. Secreted by infiltrating inflammatory cells, TWEAK engages Fn14 and promotes the proliferation and activation of myoblasts, which can in turn contribute to a robust inflammatory response by producing additional chemotactant. The TWEAK/Fn14 pathway therefore positively contributes to muscle regeneration by promoting cellular crosstalk between inflammatory cells and muscle precursor cells.
It has traditionally been thought that inflammatory responses following tissue injury are critical for the clearance of damaged tissue debris and removal of necrotic/apoptotic cells. A number of more recent studies, however, provided growing evidence, suggesting that inflammation can also influence tissue repair by directly acting on the regenerating cell pool (Mutsaers et al, 2002; Arnett et al, 2003; Monje et al, 2003). In our current study, we demonstrated TWEAK's ability to drive the expansion of muscle myoblasts in vitro and apparently in vivo in response to injury, presenting a concrete example of how a cytokine derived from inflammatory cells can participate in tissue repair and regeneration. Given the broad expression of Fn14 on progenitor cells of the mesenchymal lineage and its rapid upregulation in many injury settings, we propose that the physiological function of the TWEAK/Fn14 pathway is to participate in acute phase tissue repair in multiple biological systems by promoting progenitor cell proliferation. On the other hand, we have also observed that TWEAK potently inhibited the terminal differentiation of muscle myoblasts, as well as other mesenchymal progenitor cells. One could therefore envision that under pathological conditions where inflammatory responses persist, prolonged presence of TWEAK could potentially block the differentiation of progenitor cells and impede efficient tissue repair. The complex physiological and pathological consequences of TWEAK activity suggest that manipulation of the TWEAK system may present therapeutic opportunities for cell-based regenerative therapy and in various inflammatory diseases such as arthritis and atherosclerosis, where extensive tissue remodeling and regeneration occur.
Materials and methods
Proteins and antibodies
The construct for Fc-TWEAK was generated by replacing the Baff portion of a previously described Fc-Baff construct with a cDNA fragment encoding soluble TWEAK. A stable 293T cell line expressing the construct was then generated for protein production. The hamster anti-TWEAK antibody ABG.11 and anti-human Fn14 antibody ITEM-4 were generated as previously described (Jakubowski et al, 2002; Nakayama et al, 2003). The blocking anti-Fn14 antibody P2D3 was generated by immunizing Fn14 knockout mice with Fn14-transfected cells. The following antibodies were purchased from the indicated sources: anti-mouse TNF G281-2626, PE-anti-human CD56, FITC-anti-mouse Mac-1 and PE-anti-mouse Gr-1 (BD Biosciences), anti-myogenin F5D (Santa Cruz Biotechnology).
Cell culture
C2C12 cells (ATCC) were seeded at 2 × 105 cells/well in six-well plates in growth medium (GM; DMEM supplemented with 15% FBS). The following day, cells were washed with PBS once and switched to low-serum differentiation medium (DM: DMEM supplemented with 2% horse serum). Primary muscle satellite cells were isolated from limb muscles of 6- to 8-week-old Fn14−/− mice and wild-type control mice (n=4, for each group) as described (Dominov et al, 1998, 2001; Girgenrath et al, 2005). See Supplementary data for additional details. Primary myoblasts were induced to differentiate by replacing GM with (DM that consisted of DMEM without chicken embryo extract and with only 2% serum. Human primary cells were all purchased from Cambrex (Walkersville, MD) and cultured according to the supplier's protocol.
Cardiotoxin injection
Cardiotoxin injection was performed as before (Yan et al, 2003). Briefly, 50 μl of 10 mM cardiotoxin (Sigma) was injected into the TA muscles of the right legs of wild-type or Fn14-deficient mice of 129/sv background, whereas the left legs were used as uninjected control. At the indicated time points, TA muscles were harvested and fixed in 4% paraformaldehyde. For BrdU incorporation, mice were injected intraperitoneally with 1 mg of BrdU 6 hours before harvesting the muscles.
Profiling analysis
For transcription profiling of TWEAK's effect on C2C12 cells, 1.2 × 106 C2C12 cells were seeded in 100-mm cell culture dishes in GM overnight before being switched to differentiation medium. Total RNA from C2C12 cells cultured in GM and DM at different time points was extracted using TriZol reagent (Invitrogen) and further purified by RNAeasy (Qiagen). Duplicate samples for each sample point were subjected to profiling using the Affymetrix mouse chip MG-U74A according to the protocol recommended by the manufacturer. For profiling of TWEAK's effect on MSCs, 2 × 106 cells were cultured in 100-mm cell culture dishes in regular DMEM culture medium containing different serum concentrations (10, 2 or 0.2%). After overnight culture, TWEAK (100 ng/ml) or heat-inactivated TWEAK (100ng/ml) were added in triplicate plates for each treatment condition and cells were harvested 18 h later for profiling using the Affymetrix human chip U133.
FACS
Fc-TWEAK binding to and Fn14 surface expression on primary cells were analyzed by FACS staining according to the standard protocol. Briefly, cells were harvested by incubating in 5 mM EDTA/PBS solution at 37°C until the cells detached. Cells were suspended in FACS buffer (PBS with 1% FBS) and stained with either 100 ng/ml of Fc-TWEAK or 1 mg/ml of ITEM-4 for 45 min, followed by PE-conjugated goat anti-human or mouse Fc secondary antibodies. Samples were read on a FACS Calibur and analyzed using FlowJo. For FACS of muscle dissociates, TA muscles were dissected, fat and connective tissues were removed, and minced muscles were incubated in 0.2 mg/ml Pronase solution (Roche Diagnostics) at 37°C for 1 h. After terminating digestion by washing with DMEM, the dissociates were recovered by sequentially filtering through 100 μm and 40 μm cell strainers (BD Labware) and used for staining with anti-Mac-1 and anti-Gr-1 antibodies.
NF-κB activation assay
NF-κB activation was measured (n=3) using the TransAM NF-κB p65 assay system (Active Motif) according to the manufacturer's protocol.
For details on quantitative PCR, generation of Fn14−/− mice, in situ hybridization and histological analyses, see Supplementary data.
Supplementary Material
Supplementary Information
Acknowledgments
We sincerely thank L Evangelisti and S Shulga-Morskaya for embryonic stem cell culture and injection; M Wang and J Lincecum for in situ analysis; T Crowell and H Gardner for histological analysis; D Gong for protein purification; D McCrann, J Shearston, S Perrin and S Szak for gene profiling analysis. This work was supported in part by NIH Grant HL39727 (JAW) and Grants AR49496, ES11384 and HL64641 (JBM).
References
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP (2001) TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 4: 1116–1122 [DOI] [PubMed] [Google Scholar]
- Arnett HA, Wang Y, Matsushima GK, Suzuki K, Ting JP (2003) Functional genomic analysis of remyelination reveals importance of inflammation in oligodendrocyte regeneration. J Neurosci 23: 9824–9832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry FP (2003) Biology and clinical applications of mesenchymal stem cells. Birth Defects Res Part C Embryo Today 69: 250–256 [DOI] [PubMed] [Google Scholar]
- Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, Hertig S, Ambrose C, Tschopp J, Schneider P (2006) Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem 281: 13964–13971 [DOI] [PubMed] [Google Scholar]
- Brown SA, Richards CM, Hanscom HN, Feng SL, Winkles JA (2003) The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappaB activation. Biochem J 371: 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazaud B, Sonnet C, Lafuste P, Bassez G, Rimaniol AC, Poron F, Authier FJ, Dreyfus PA, Gherardi RK (2003) Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J Cell Biol 163: 1133–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, Garcia I, Browning JL (1997) TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 272: 32401–32410 [DOI] [PubMed] [Google Scholar]
- Chicheportiche Y, Chicheportiche R, Sizing I, Thompson J, Benjamin CB, Ambrose C, Dayer JM (2002) Proinflammatory activity of TWEAK on human dermal fibroblasts and synoviocytes: blocking and enhancing effects of anti-TWEAK monoclonal antibodies. Arthritis Res 4: 126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RA, Grounds MD (2001) The role of tumor necrosis factor-alpha (TNF-alpha) in skeletal muscle regeneration. Studies in TNF-alpha(−/−) and TNF-alpha(−/−)/LT-alpha(−/−) mice. J Histochem Cytochem 49: 989–1001 [DOI] [PubMed] [Google Scholar]
- Couteaux R, Mira JC, d'Albis A (1988) Regeneration of muscles after cardiotoxin injury. I. cytological aspects. Biol Cell 62: 171–182 [PubMed] [Google Scholar]
- D'Amore PA (1990) Modes of FGF release in vivo and in vitro. Cancer Metast Rev 9: 227–238 [DOI] [PubMed] [Google Scholar]
- DiMario JX, Uzman A, Strohman RC (1991) Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev Biol 148: 314–321 [DOI] [PubMed] [Google Scholar]
- Dogra C, Changotra H, Mohan S, Kumar A (2006) Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J Biol Chem 281: 10327–10336 [DOI] [PubMed] [Google Scholar]
- Dominov JA, Dunn JJ, Miller JB (1998) Bcl-2 expression identifies an early stage of myogenesis and promotes clonal expansion of muscle cells. J Cell Biol 142: 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominov JA, Houlihan-Kawamoto CA, Swap CJ, Miller JB (2001) Pro- and anti-apoptotic members of the Bcl-2 family in skeletal muscle: a distinct role for Bcl-2 in later stages of myogenesis. Dev Dyn 220: 18–26 [DOI] [PubMed] [Google Scholar]
- Duffield JS (2003) The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci (London) 104: 27–38 [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci USA 100: 13632–13637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausto N (2005) Tweaking liver progenitor cells. Nat Med 11: 1053–1054 [DOI] [PubMed] [Google Scholar]
- Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, Peifley KA, Winkles JA (2000) The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol 156: 1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath M, Kostek CA, Miller JB (2005) Diseased muscles that lack dystrophin or laminin-alpha2 have altered compositions and proliferation of mononuclear cell populations. BMC Neurol 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr (1999) NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19: 5785–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr (2000) NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289: 2363–2366 [DOI] [PubMed] [Google Scholar]
- Hawke TJ, Garry DJ (2001) Myogenic satellite cells: physiology to molecular biology. J Appl Physiol 91: 534–551 [DOI] [PubMed] [Google Scholar]
- Hirata A, Masuda S, Tamura T, Kai K, Ojima K, Fukase A, Motoyoshi K, Kamakura K, Miyagoe-Suzuki Y, Takeda S (2003) Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am J Pathol 163: 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, Browning B, Michaelson JS, Baestcher M, Wang B, Bissell DM, Burkly LC (2005) TWEAK induces liver progenitor cell proliferation. J Clin Invest 115: 2330–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski A, Browning B, Lukashev M, Sizing I, Thompson JS, Benjamin CD, Hsu YM, Ambrose C, Zheng TS, Burkly LC (2002) Dual role for TWEAK in angiogenic regulation. J Cell Sci 115: 267–274 [DOI] [PubMed] [Google Scholar]
- Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ (2001) Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 35: 204–212 [DOI] [PubMed] [Google Scholar]
- Lagasse E, Weissman IL (1996) Flow cytometric identification of murine neutrophils and monocytes. J Immunol Methods 197: 139–150 [DOI] [PubMed] [Google Scholar]
- Lescaudron L, Peltekian E, Fontaine-Perus J, Paulin D, Zampieri M, Garcia L, Parrish E (1999) Blood borne macrophages are essential for the triggering of muscle regeneration following muscle transplant. Neuromuscul Disord 9: 72–80 [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104: 487–501 [DOI] [PubMed] [Google Scholar]
- Luk HW, Noble LJ, Werb Z (2003) Macrophages contribute to the maintenance of stable regenerating neurites following peripheral nerve injury. J Neurosci Res 73: 644–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR (1999) TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem 274: 8455–8459 [DOI] [PubMed] [Google Scholar]
- Merly F, Lescaudron L, Rouaud T, Crossin F, Gardahaut MF (1999) Macrophages enhance muscle satellite cell proliferation and delay their differentiation. Muscle Nerve 22: 724–732 [DOI] [PubMed] [Google Scholar]
- Miller SC, Ito H, Blau HM, Torti FM (1988) Tumor necrosis factor inhibits human myogenesis in vitro. Mol Cell Biol 8: 2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302: 1760–1765 [DOI] [PubMed] [Google Scholar]
- Morgan JE, Partridge TA (2003) Muscle satellite cells. Int J Biochem Cell Biol 35: 1151–1156 [DOI] [PubMed] [Google Scholar]
- Mutsaers SE, Whitaker D, Papadimitriou JM (2002) Stimulation of mesothelial cell proliferation by exudate macrophages enhances serosal wound healing in a murine model. Am J Pathol 160: 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Ishidoh K, Kojima Y, Harada N, Kominami E, Okumura K, Yagita H (2003) Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK-induced cell death. J Immunol 170: 341–348 [DOI] [PubMed] [Google Scholar]
- Nathan C (2002) Points of control in inflammation. Nature 420: 846–852 [DOI] [PubMed] [Google Scholar]
- Newton K, French DM, Yan M, Frantz GD, Dixit VM (2004) Myodegeneration in EDA-A2 transgenic mice is prevented by XEDAR deficiency. Mol Cell Biol 24: 1608–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak JA, Malowitz J, Girgenrath M, Kostek CA, Kravetz AJ, Dominov JA, Miller JB (2004) Immortalization of myogenic cells does not require loss of p16INK4a, p19ARF, or p53 and is accelerated by inactivation of Bax. BMC Cell Biol 5: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlath GK, Dominov JA, Kegley KM, Miller JB (2003) Regeneration of skeletal muscles with altered timing of expression of the myogenic bHLH factor MRF4. Am J Pathol 162: 1685–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R (1993) MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75: 1351–1359 [DOI] [PubMed] [Google Scholar]
- Saas P, Boucraut J, Walker PR, Quiquerez AL, Billot M, Desplat-Jego S, Chicheportiche Y, Dietrich PY (2000) TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia 32: 102–107 [PubMed] [Google Scholar]
- Stewart CE, Newcomb PV, Holly JM (2004) Multifaceted roles of TNF-alpha in myoblast destruction: a multitude of signal transduction pathways. J Cell Physiol 198: 237–247 [DOI] [PubMed] [Google Scholar]
- Szalay K, Razga Z, Duda E (1997) TNF inhibits myogenesis and downregulates the expression of myogenic regulatory factors myoD and myogenin. Eur J Cell Biol 74: 391–398 [PubMed] [Google Scholar]
- Tsivitse SK, Mylona E, Peterson JM, Gunning WT, Pizza FX (2005) Mechanical loading and injury induce human myotubes to release neutrophil chemoattractants. Am J Physiol Cell Physiol 288: C721–729 [DOI] [PubMed] [Google Scholar]
- Walsh K, Perlman H (1997) Cell cycle exit upon myogenic differentiation. Curr Opin Genet Dev 7: 597–602 [DOI] [PubMed] [Google Scholar]
- Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA, Fanslow WC (2001) A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 15: 837–846 [DOI] [PubMed] [Google Scholar]
- Yan Z, Choi S, Liu X, Zhang M, Schageman JJ, Lee SY, Hart R, Lin L, Thurmond FA, Williams RS (2003) Highly coordinated gene regulation in mouse skeletal muscle regeneration. J Biol Chem 278: 8826–8836 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information