

Abstract
The kinesin-binding site on the microtubule has not been identified because of the technical difficulties involved in the mutant analyses of tubulin. Exploiting the budding yeast expression system, we succeeded in replacing the negatively charged residues in the α-helix 12 of β-tubulin with alanine and analyzed their effect on kinesin–microtubule interaction in vitro. The microtubule gliding assay showed that the affinity of the microtubules for kinesin was significantly reduced in E410A, D417A, and E421A, but not in E412A mutant. The unbinding force measurement revealed that in the former three mutants, the kinesin–microtubule interaction in the adenosine 5′-[β,γ-imido]triphosphate state (AMP-PNP state) became less stable when a load was imposed towards the microtubule minus end. In parallel with this decreased stability, the stall force of kinesin was reduced. Our results implicate residues E410, D417, and E421 as crucial for the kinesin–microtubule interaction in the strong binding state, thereby governing the size of kinesin stall force.
Keywords: kinesin, microtubule, mutant analysis, stall force, yeast
Introduction
Kinesin is a molecular motor involved in many cellular force-generating processes such as organelle transport and chromosome segregation (Vernos and Karsenti, 1996; Goldstein and Yang, 2000). Conventional kinesin contains two identical heavy chains and can move processively along a microtubule more than 1 μm in length without dissociation (Howard et al, 1989; Block et al, 1990). Processive movement of kinesin is explained by the hand-over-hand model, in which the motor maintains continuous contact with the microtubule as a result of alternating head catalysis of ATP (Hackney, 1994; Ma and Taylor, 1997). At each ATP hydrolysis cycle, kinesin makes an 8-nm step (equal to the size of tubulin dimer) towards the microtubule plus end (Hua et al, 1997; Schnitzer and Block, 1997), and this stepping motion is triggered by a conformational change in the ATP-bound head (Rice et al, 1999).
Docking of the crystal structure of kinesin into cryoelectron-microscopy maps of kinesin–microtubule complex indicated L8, L11, and α4/L12/α5 in the motor domain of kinesin as the structural key elements for microtubule binding in the presence of AMP-PNP, a non-hydrolyzable ATP analogue mimicking the ATP-bound state (Hirose et al, 1999; Hoenger et al, 2000; Kikkawa et al, 2000; Skiniotis et al, 2004). In this model, L8 may bind to α-helix 12 (H12) in β-tubulin, and L11 extends towards the H11–H12 loop in α-tubulin. α4/L12/α5 is also closely associated with H12 in β-tubulin and possibly with the C-terminus of β-tubulin, which is not defined in the crystal structure. The model indicates that kinesin–microtubule interaction might be mediated by electrostatic interactions. Some of the positively charged amino acids in these contact areas of kinesin potentially interact with the negatively charged surface of the microtubule, which is mainly composed of H12 in β-tubulin. Consistent with this assumption, using alanine-scanning mutagenesis of kinesin, several positively charged residues in L7/8, L11, and α4/L12/α5 have been identified as microtubule-interacting kinesin residues (Woehlke et al, 1997). However, the critical residues on tubulin have not been identified.
Proteolytic digestion of the C-terminal region of tubulin led to the reduced processivity of both single- and double-headed kinesin (Okada and Hirokawa, 2000; Thorn et al, 2000; Wang and Sheetz, 2000; Lakämper and Meyhöfer, 2005). The biochemical measurement of Kd of kinesin to both intact and protease-digested microtubule revealed that the negatively charged C-terminus of tubulin may be a binding partner for kinesin in the weak binding state (ADP state), but not in the strong binding state (AMP-PNP state) (Okada and Hirokawa, 2000; Skiniotis et al, 2004; Lakämper and Meyhöfer, 2005). A structural element other than the tubulin C-terminal region may serve as an interface that is specific for the strong binding state; the structural analyses have implicated H12 in β-tubulin as a potential candidate for this (Hirose et al, 1999; Hoenger et al, 2000; Kikkawa et al, 2000; Skiniotis et al, 2004). For elucidating the mechanism of kinesin motility, it is essential to identify the structural elements involved in each chemical state because the physicochemical properties inherent to each chemical state might be critically dependent on the interface configuration and the surface force type that works between the interfaces (Israelachvili, 1992; Okada and Hirokawa, 2000; Skiniotis et al, 2004; Lakämper and Meyhöfer, 2005).
Our lack of knowledge about kinesin interface on tubulin is attributed to the technical difficulties involved in mutant analysis of tubulin. Tubulin is a heterodimer composed of α- and β-polypeptides, each requiring a distinct set of chaperons for proper folding. Apart from this complexity, multiple α- and β-tubulin genes are found in most eukaryotic cells, with each subunit undergoing different post-translational modifications (Luduena, 1998). Consequently, it has been difficult to express and purify isotypically pure tubulin in biochemically useful amounts.
Here, we have used the budding yeast Saccharomyces cerevisiae to conduct mutant analyses on microtubules. S. cerevisiae contains only two α-tubulin genes (TUB1 and TUB3) and one β-tubulin gene (TUB2); since TUB3 is nonessential, it provides a potential source of isotypically pure tubulin (Bode et al, 2003). Using TUB3-null cells, we succeeded in generating a set of charged-to-alanine point mutations in the sequence coding for H12 in β-tubulin. Each of these mutated tubulins was purified from the cell lysate, polymerized into microtubules, and the effect of mutation was examined in microtubule gliding assay on conventional two-headed kinesin. This showed that the affinity of microtubule for kinesin was reduced in E410A, D417A, and E421A mutants. The measurement of the unbinding force (Kawaguchi and Ishiwata, 2001; Uemura et al, 2002) revealed that in the former three mutants, the kinesin–microtubule interaction in AMP-PNP state became less stable for minus end loading, whereas their interaction in ADP state was unaffected by the mutations. These results indicate that the negatively charged amino-acid residues E410, D417, and E421 in β-tubulin are crucial for the strong binding of kinesin to the microtubule.
Hereafter, we have abbreviated the α-helix and β-sheet in the tubulin structures as ‘H' and ‘S,' corresponding to α and β, respectively, in the kinesin structure, according to the original paper on the structure of microtubule (Nogales et al, 1998).
Results
Construction, expression, and purification of H12 mutated microtubules
To prepare isotypically pure tubulin, we used TUB3-null yeast cells, having only a single α- and β-tubulin gene, TUB1 and TUB2, respectively (see Supplementary Methods and Table SII). As the aim of this study was to examine the motility of kinesin along the mutated microtubules, it was necessary to obtain yeast microtubules that were stable at low concentrations required for in vitro motility assay (∼10 μg/ml). Hence, Taxol-binding ability was introduced into a β-tubulin gene by site-directed mutagenesis at five amino acids (Gupta et al, 2003), and the gene tub2-A19K-T23V-G26D-N227H-Y270F thus obtained was referred to as TUB2tax. The yeast strain expressing TUB1 and TUB2tax was used as the wild-type in the following analyses.
To examine the function of β-tubulin H12 in kinesin motility, we generated a set of charged-to-alanine point mutations in the sequence coding for H12 in β-tubulin (Figure 1). Among these four mutants, E410A and D417A were haploid lethal. Therefore, we attempted to isolate these mutated tubulins by expressing two species of tub2tax genes in a strain, one of which, tub2tax-plusE (tub2tax-440GDFGEEEEGEEEEGEEEEGEEEEGEEEEA), contains numerous negatively charged amino acids at the C-terminus and the other, tub2tax-E410A or tub2tax-D417A, which has point mutation in H12; the latter is under the control of the inducible galactose promoter (Burke et al, 1989). The haploid cells were first grown in YPD medium, expressing only tub2tax-plusE. When the growth reached the late-log phase, the cells were transferred to YPG medium containing galactose for inducing the coexpression of tub2tax-E410A (or tub2tax-D417A). The cells were cultured for another 8 h in YPG medium, and then harvested for tubulin purification. The cell lysate contains two species of tubulin dimers, including either Tub2taxp-plusE or Tub2taxp-E410A (or Tub2taxp-D417A) as β-tubulin subunit, but these two species can be separated using their charge differences during purification (see Supplementary Figure S1). For the other two mutants, E412A and E421A, tubulin could be readily purified from the cultured haploid cells as these cells containing only this mutated tubulin were viable under normal growth conditions (YPD medium).
Figure 1.
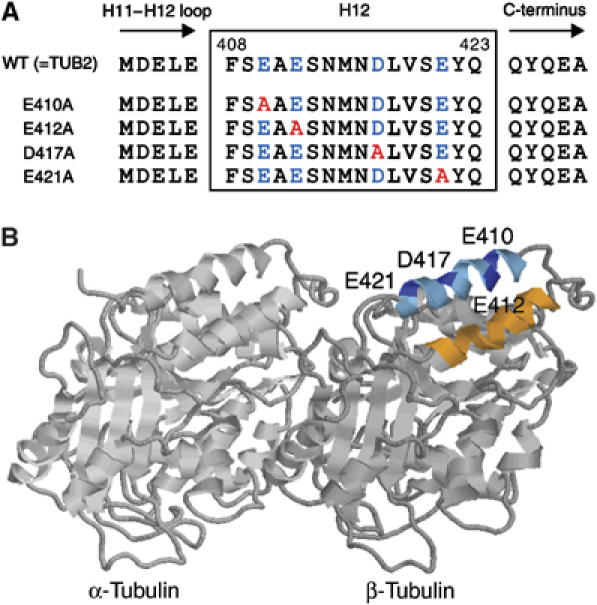
Design of the H12 mutants of β-tubulin in Saccharomyces cerevisiae. (A) Sequences of the H12 region are shown with negatively charged residues indicated by blue and the residues substituted by alanines are indicated by red. (B) A ribbon diagram of the tubulin dimer viewed from the side of the microtubule with its minus end to the left (Nogales et al, 1998). Image analysis of the kinesin–microtubule complex revealed that in both nucleotide free and AMP-PNP state, kinesin motor domain is associated in close proximity to H11 (orange), H12 (cyan), and the COOH terminus (undefined in crystal structure) of β-tubulin (Kikkawa et al, 2000; Hoenger et al, 2000). The acidic residues in H12 mutagenized to alanine are indicated in blue.
To purify tubulin from the cell lysate, a couple of anion exchange column chromatographies were successively used (Davis et al, 1993), and the crude tubulin fraction eluted from the second column was further purified by polymerization and depolymerization (see Supplementary Methods and Figure S2). This procedure generated approximately 30 μg of assembly-competent tubulin from 6L of culture with purity higher than 95% on SDS gel electrophoresis (Figure 2A). To examine if these mutated tubulins were post-translationally modified at their C-terminus (Luduena, 1998), both α- and β-polypeptides were analyzed by electrospray ionization/ion trap and quadrupole-TOF mass spectrometry. The MS and MS/MS data suggested that the isolated tubulin dimers predominantly contained α- and β-polypeptides lacking any post-translational modifications at the C-terminal region (Supplementary Figure S3).
Figure 2.
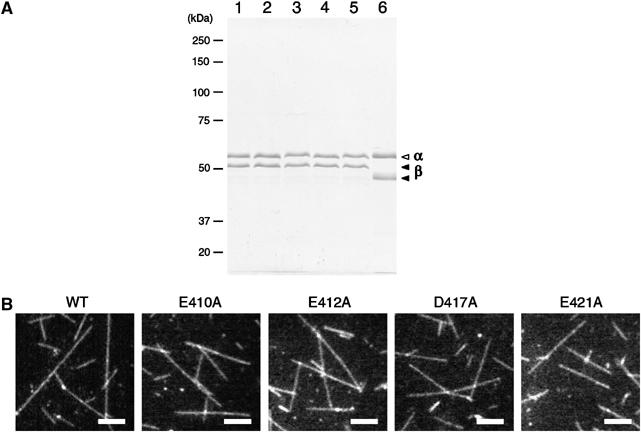
Purity of yeast tubulin and images of polymerized microtubules. (A) SDS–PAGE analysis of purified tubulin. Lane 1, yeast wild-type tubulin; lane 2, E410A; lane 3, E412A; lane 4, D417A; lane 5, E421A; and lane 6, porcine brain tubulin. In each lane, 1 μg of sample was loaded and stained by Coomassie blue. In SDS gel containing Sigma SDS (L-5750), α- and β-polypeptide of porcine brain tubulin were separated more as compared to these peptides purified from yeast cells (Best et al, 1981; Bode et al, 2003). (B) Dark-field images of the microtubules polymerized from yeast wild-type and mutated tubulins in the presence of 1 μM Taxol. Bar=5 μm.
When these isolated tubulins (0.5–1.0 mg/ml) were incubated at 30°C in the presence of 1 μM Taxol, all species of tubulins could polymerize into long filaments having length up to ∼20 μm (Figure 2B).
In vitro motility assay
To examine the influence of the mutations on kinesin–microtubule interaction, these mutated microtubules were first tested in the microtubule gliding assay on the conventional two-headed kinesin. When the density of kinesin on the glass surfaces was >1000 μm−2, both wild-type and mutated microtubules showed smooth gliding movement at similar velocities (Table I). When the direction of the movement was examined using polarity marked microtubules (Tanaka-Takiguchi et al, 1998), the movement of kinesin was plus-end-directed in all cases. However, at kinesin density of ∼100 μm−2, the wild-type and the E412A microtubules showed smooth gliding movement, whereas the E410A, D417A, and E421A microtubules often rotated erratically about a roughly vertical axis, resulting in wobbly gliding motions. This indicates that the latter three mutants may have lower affinity for kinesin as compared to the wild-type.
Table 1. Velocity of microtubule movement in the gliding assaya.
| Construct | Velocity (μm/s)b | n |
|---|---|---|
| WT | 0.75±0.08 | 120 |
| E410A | 0.78±0.06 | 120 |
| E412A | 0.76±0.06 | 103 |
| D417A | 0.73±0.08 | 123 |
| E421A | 0.74±0.10 | 102 |
| a Gliding assay was performed at a kinesin density of ∼2000/μm2. | ||
| b Mean±s.e.m. | ||
To quantify the affinity of these mutated microtubules for kinesin, we measured the fraction of the microtubules that moved a distance greater than their own lengths, f, over a range of kinesin densities (Figure 3A and B; Howard et al, 1989). Here, to compare between each mutant and wild-type, the length of the microtubules was adjusted to approximately 3 μm in all strains (see Materials and methods). The result showed that in the wild-type, virtually all the microtubules moved more than their own length at kinesin density >300 μm−2, and the fraction of the microtubules that covered distances greater than their own length gradually decreased with the decrease in kinesin density. At kinesin density <10 μm−2, no microtubules covered distances greater than their own lengths. For continuous movement of a microtubule over distance greater than its own length, if a minimum number, n, of kinesin molecules are required to interact with the microtubule, then the data points can be fit to the equation
Figure 3.
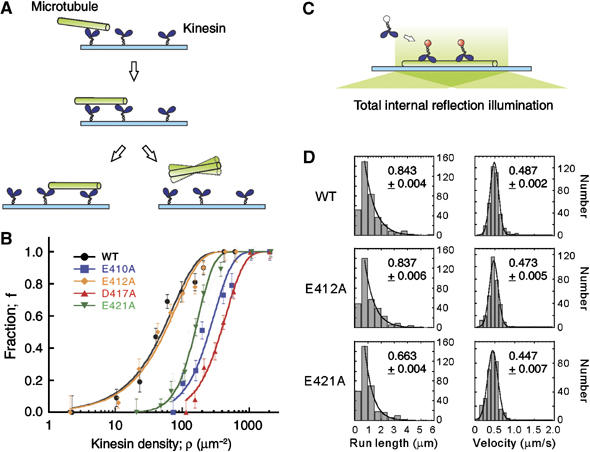
Motility assays using mutated microtubules. (A) In microtubule gliding assay, the fraction of microtubules that moved a distance greater than their own length (∼3 μm), f, was measured, and (B) plotted as a function of kinesin density, ρ. Total number of microtubules counted for wild-type (black), E410A (blue), E412A (orange), D417A (red), and E421A (green) were 495, 328, 657, 496, and 388, respectively. Continuous curves are the plot of equation (1). Error bars represent the statistical counting errors, calculated according to Materials and methods. (C) In single molecule motility assay using TIRFM, the motility of HK560-Cy3 was examined, and (D) the distribution of the kinesin run length and the velocity was analyzed for both the wild-type and the mutated microtubules. HK560-Cy3 did not interact with the E410A and D417A microtubules. Total number of the events counted for wild-type, E412A, and E421A were 374, 329, and 333, respectively.The mean run length and the mean velocity, calculated according to Supplementary Methods, are shown in each panel with the errors of the curve fits.
where ρ is the kinesin density (variable), ρ0 is a fit parameter (Materials and methods; Howard et al, 1989; Hancock and Howard, 1998). The best fit was obtained with n=1 and ρ0=39.8 μm−2 (P=0.041; χ2 test), indicating that the wild-type microtubule can move processively along a single kinesin molecule randomly located on the glass surface. The result is similar to the previous observation on brain microtubules.
In contrast, whereas the data set for E412A showed a dependence on kinesin density similar to the wild-type (n=1, ρ0=43.6 μm−2, P=0.032; χ2 test), E410A, D417A, and E421A required significantly higher concentrations of kinesin to move a distance greater than their own lengths (Figure 3B). For the latter three mutants, the fraction curves were steeper and shifted to the right along the log density axis. When each of the data set was fit to the same equation, the best fit was obtained with n=2, ρ0=100.9 μm−2 for E410A, n=2, ρ0=152.1 μm−2 for D417A, and n=3, ρ0=42.9 μm−2 for E421A, and the corresponding P-values were 0.047, 0.012, and 0.010, respectively (χ2 test). These results indicate that alanine substitution at E410, D417, and E421 might have reduced the affinity of microtubules for kinesin either by lowering the efficiency of the initial interaction of the microtubules with kinesin molecules and/or by reducing the extent of microtubules processivity along the kinesin molecules.
ρ0 for E410A and D417A was higher than that for the wild-type. This implies that at a given kinesin density, although the collision frequency was constant for all the species of microtubules, the initial interaction of the E410A/D417A microtubules with kinesin molecules might have occurred at a relatively low frequency as compared to the frequency observed with the wild-type microtubules. Contrastingly, ρ0 for E421A was comparable to that of the wild-type, indicating that in E421A mutants, the initial interaction might have occurred at a frequency similar to that of the wild-type. The parameter ρ0 may reflect the activation energy of the microtubule necessary for its initial interaction with kinesin on the glass surface (Fersht, 1984; Hackney, 1995).
Conversely, the parameter n is a measure of multivalency in the kinesin–microtubule interaction occurring at the microtubule's interface with the kinesin-coated surface. For E410A, D417A, and E421A mutants, n was >1. This result may indicate that either a simultaneous, collective action of multiple motor molecules is required for the stable gliding movement of mutated microtubules (Hancock and Howard, 1998; Shima et al, 2006) or that a run length of a single kinesin along the mutated microtubule is too short to support the gliding movement of microtubule over a distance greater than its own length.
To understand which the operative mechanism is, we conducted a single molecule motility assay (Figure 3C); the motility of fluorescently labeled two-headed kinesin HK560-Cy3 (0.29 nM) was examined along these mutated microtubules using total internal reflection fluorescence microscopy (TIRFM). The result showed that whereas kinesin moved processively along the wild-type and E412A microtubules, it scarcely interacted with the E410A and D417A microtubules (Figure 3D). For E421A, kinesin could move along the microtubule, but its processivity was reduced as compared to that of the wild-type. The mean run length of kinesin for the wild-type, E412A, and E421A microtubules was 0.843±0.004 μm, 0.837±0.006 μm, and 0.663±0.004 μm, respectively. These results are consistent with the values of ρ0 and n derived from the fraction curves in Figure 3B. The estimated value of n=3 for E421A was due to the reduced processivity of single kinesin.
It is reasonable that only the E410A, D417A, and E421A mutations affected the kinesin–microtubule interaction. Docking experiments of the crystal structure of kinesin into cryoelectron-microscopy maps of kinesin–microtubule complex revealed that H12 of β-tubulin is located on the surface of microtubules and aligned diagonally to the longitudinal axis of protofilament (Hoenger et al, 2000; Kikkawa et al, 2000). In its coiled structure, the residues E410, D417, and E421 are facing kinesin, whereas E412 is positioned on the opposite side, facing the microtubule core.
Unbinding force
To clarify the chemical state at which the binding affinity of the microtubules for kinesin was modulated in the mutants, we measured the force required to dissociate kinesin from these mutated microtubules (unbinding force) under two nucleotide conditions—in the presence of ADP and AMP-PNP (Kawaguchi and Ishiwata, 2001; Uemura et al, 2002).
One-headed kinesin heterodimers were used for the measurement because our previous work demonstrated that the two distinct binding modes (weak and strong), each inherent to the nucleotide condition, can be unambiguously characterized with the single-headed kinesin (Uemura et al, 2002). Using optical tweezers, a polystyrene bead attached with a single-headed kinesin (Kojima et al, 1997) was made to interact with a microtubule in the presence of either 1 mM ADP or 1 mM AMP-PNP. An external load was gradually applied to the kinesin–microtubule complex by moving the stage of the microscope towards the plus end or the minus end of the microtubule until the bead dissociated from it. The unbinding force was calculated by multiplying the magnitude of abrupt bead displacement during detachment with the stiffness of the optical tweezers.
The yeast wild-type microtubules showed properties similar to those observed previously for brain microtubules (Figure 4; Kawaguchi and Ishiwata, 2001; Uemura et al, 2002). Unbinding force in the AMP-PNP state was significantly higher than that in the ADP state, and in each nucleotide state, the unbinding force for the minus-end loading was higher than that for the plus-end loading. The dependence on loading direction in the ADP state, however, was not as significant as in the AMP-PNP state.
Figure 4.
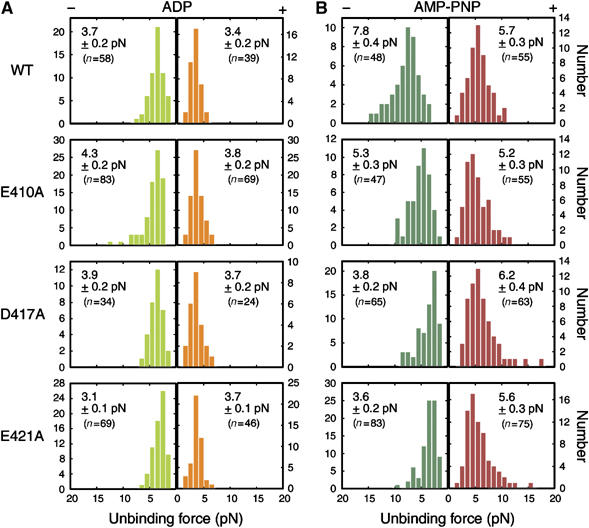
Unbinding force distribution of one-headed kinesin in (A) ADP and (B) AMP-PNP state. An external load was applied towards either the plus end (orange for (A) and red for (B)) or the minus end of the microtubule (light and dark green for (A) and (B), respectively). The stiffness of the trap was 0.038 pN/nm (ADP) and 0.076 pN/nm (AMP-PNP). The average unbinding force (pN) with s.e.m. is shown in each panel.
Whereas the unbinding force in ADP state for mutated microtubules was almost similar to that measured for wild-type (Figure 4A), the unbinding force in AMP-PNP state was significantly altered by mutations (Figure 4B). In the presence of AMP-PNP, the unbinding force for minus-end loading was considerably reduced in all three mutants (7.8±0.4, 5.3±0.3, 3.8±0.2, and 3.6±0.2 pN for wild-type, E410A, D417A, and E421A, respectively; mean±s.e.m.), yet the unbinding force for plus-end loading was scarcely affected (5.7±0.3, 5.2±0.3, 6.2±0.4, and 5.6±0.3 pN for wild-type, E410A, D417A, and E421A, respectively). Unexpectedly, the mutations rendered the kinesin–microtubule interaction less stable only for minus-end loading. In the presence of ADP, the unbinding force measured for mutants was fairly similar to that of the wild-type except in the case of E421A, where the asymmetry for loading direction was reversed.
The residues E410, D417, and E421 appeared to be crucial for the strong binding of kinesin to the microtubules, and this may underlie the reduced affinity of these mutated microtubules for kinesin during movement (Figure 3).
Stall force
Previous measurements of the mechanical properties of two-headed kinesin indicated that the kinesin stall force might be determined by (1) the binding affinity of the head to the microtubule in the strongly bound state and (2) the ATP-binding kinetics to the nucleotide-free head (Visscher et al, 1999; Nishiyama et al, 2002; Lakämper and Meyhöfer, 2005; Shao and Gao, 2006). Thus, the kinesin stall force exerted on the mutated microtubules with reduced affinity for kinesin in AMP-PNP state is expected to be smaller than that exerted on the wild-type microtubules. To examine this hypothesis, we next attempted to measure the stall force of conventional two-headed kinesin along these mutated microtubules.
When the displacements and forces caused by single kinesin molecules were measured in the presence of 1 mM ATP using optical tweezers (trap stiffness=0.076 pN/nm), the stall force measured for E410A, D417A, and E421A was 3.5±0.1, 3.0±0.1, and 3.1±0.1 pN, respectively (mean±s.e.m.). These values were significantly lower than that measured for the wild-type (5.6±0.2 pN) (Figure 5A and B). Although we observed the processive movement of kinesin only for the wild-type, E412A, and E421A microtubules in the single molecule motility assay (Figure 3D), in the experiment using optical tweezers, a single-kinesin-bound bead is also observed to move along the E410A and D417A microtubules, probably because their initial interaction/successive interaction with the microtubule was enforced by the laser trap.
Figure 5.
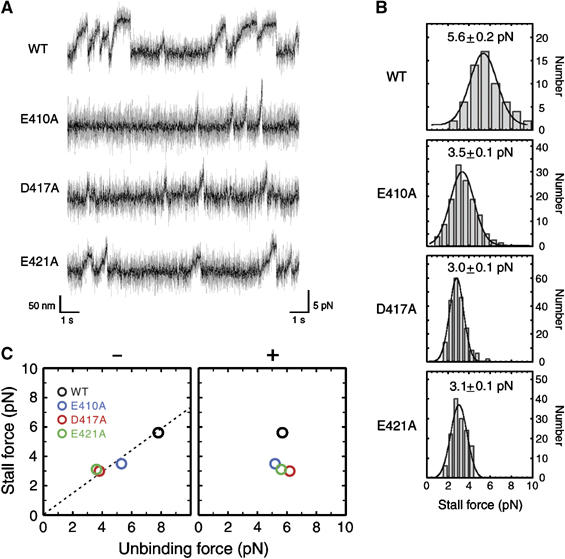
Stall force measurement. (A) Representative tracing of a trapped bead powered by a conventional two-headed kinesin along the wild-type, E410A, D417A, and E421A microtubules, measured at the trap stiffness of 0.076 pN/nm. Light shaded, unfiltered; solid, filtered at 100 Hz. (B) Distribution of the stall force for wild-type and mutated microtubules. The average stall force with s.e.m. is shown in each panel. Total number of events counted are (from top to bottom) 61, 104, 102, and 69, respectively. (C) The stall force plotted against the unbinding force for minus- (left) and plus-end loading (right). The stall force was linearly related to the unbinding force for minus-end loading (linear coefficient; 0.73).
For wild-type and E421A mutant, the stall forces were independent of the trap stiffness, ranging from 0.019 to 0.076 pN/nm (Kojima et al, 1997). Contrastingly, for E410A and D417A microtubules, the stall force was almost constant down to the trap stiffness of 0.038 pN/nm. However, below this trap stiffness value, the stall force gradually declined with the decrease of the stiffness (data not shown), indicating that in these mutants, the duration of the kinesin–microtubule interaction might be limited not only by the limit in the maximum force but also by the limit in the run length (∼200 nm).
When the stall force measured at the trap stiffness of 0.076 pN/nm was plotted against the unbinding force measured in the presence of AMP-PNP (Figure 5C), it became clear that the stall force is a linear function of the unbinding force for minus end loading. Our result clearly demonstrates that the size of the stall force depends on the force the head can bear against a load imposed towards the minus end in its strong binding state (Visscher et al, 1999; Nishiyama et al, 2002; Lakämper and Meyhöfer, 2005; Shao and Gao, 2006).
Discussion
Phenotype of yeast mutants and motility of the mutated microtubules in vitro
Utilizing yeast expression system, we have produced a set of charged-to-alanine mutations in β-tubulin H12, which has been implicated as a major constituent of kinesin interface in previous structural studies (Hirose et al, 1999; Hoenger et al, 2000; Kikkawa et al, 2000; Skiniotis et al, 2004). Among the four charged amino acids in H12, two amino-acid residues E410 and D417 appeared to be critical for the function of microtubules in living cells because substitution of these residues with alanine caused the yeast cells expressing only this mutated tubulin to become haploid lethal. It is unlikely that the lethality is due to the unsuccessful protein folding or global change in the tubulin structure because the mutated tubulins could polymerize into microtubules (Figure 2B). The most likely explanation for the observed lethality is that the alanine substitution of these charged residues might have deteriorated the intermolecular interaction of microtubules with various proteins (Al-Bassam et al, 2002; Mizuno et al, 2004), leading to the aberrant cellular transport and/or abnormal assembly of cellular architecture. All four acidic residues in H12 are highly conserved across the species (Little and Seehaus, 1988), indicating that the structure of H12 is refined through evolution to play a key role in the intermolecular interactions of microtubules.
Microtubule gliding assay using these mutated microtubules showed that the reduced charges in E410, D417, and E421 led to a decrease in their affinity for kinesin during movement, whereas the reduced charge in E412 had no influence (Figure 3B). Ineffectiveness of the mutation at residue E412 is consistent with the 3D model of kinesin–microtubule complex showing that E412 faces the microtubule core and is situated in a direction opposite the kinesin interface (Figure 1B; Hoenger et al, 2000; Kikkawa et al, 2000). Although the other three mutants showed reduced affinity for kinesin, the profile of their fraction curves indicates that the mechanism underlying the reduction of affinity might be different among the mutants; in E410A and D417A mutants, both initial interaction of kinesin with the microtubule and subsequent processive movement might have deteriorated, whereas in the E421A mutant, only the processivity might have been modulated by mutation. This interpretation was confirmed by the behavior of two-headed kinesin observed under TIRFM (Figure 3C and D). At the kinesin concentration of 0.29 nM, we could observe the processive movement of single kinesin molecules along the wild-type and E421A microtubules, whereas no interaction was observed for E410A and D417A microtubules. In the E421A mutant, the mean run length achieved by single kinesin was slightly reduced as compared to the wild-type.
It is possible that these differences between E410A/D417A and E421A mutants might explain the phenotypic differences between these two groups. The yeast cells of the former group were haploid lethal probably because the organelle vesicles with merely few motor proteins attached could not interact with the E410A/D417A microtubules, causing serious deterioration in organelle transport. As kinesin shares the same binding site on microtubules with the motors having opposite directionality (Lockhart et al, 1995; Mizuno et al, 2004), these mutations of microtubules might also disrupt the organelle transport directed towards the microtubule minus end. Conversely, the yeast E421A strain was viable because vesicles could still move along the E421A microtubules, although the efficiency of transport was lowered by the mutation.
The reduced stability of the kinesin–microtubule interaction in H12 mutants is consistent with the 3D model of the kinesin–microtubule complex showing that several positively charged amino acids in L8 and L12 of kinesin are located in close proximity to the negatively charged residues of H12 in β-tubulin (Hoenger et al, 2000; Kikkawa et al, 2000). Our result is also complementary to the result of the previous mutagenesis study on kinesin, demonstrating that the positively charged residues in L7/8, L11, and a4/L12/a5 of kinesin are crucial for kinesin–microtubule interaction during movement (Woehlke et al, 1997). These positively charged residues in kinesin L8 and L12 may electrostatically interact with the negatively charged residues of H12, playing a key role in the mechanism of motility.
Charge reduction in H12 decreased the stability of the strong binding state
Consistent with the reduced affinity measured in the motility assays, the measurement of the unbinding force has revealed that in these mutants, the stability of the kinesin–microtubule interaction in the AMP-PNP state was reduced for minus-end loading, but the stability in ADP state remained constant (Figure 4). This result contrasts with the previous observation that the removal of the negatively charged C-terminus of tubulin (E-hook) by protease treatment decreased the stability of the kinesin–microtubule interaction only in the ADP state, but not in the AMP-PNP state (Okada and Hirokawa, 2000; Skiniotis et al, 2004; Lakämper and Meyhöfer, 2005). E-hook and β-tubulin H12 apparently serve as an interface for kinesin specific for ADP and AMP-PNP state, respectively. These independent interfaces might be the structural basis for the physicochemical properties distinct for each chemical state. In the ADP state, mobility freedom of the motor has to be ensured to a certain extent while it is loosely anchored to the microtubule surface (Okada and Hirokawa, 2000; Wang and Sheetz, 2000; Sosa et al, 2001; Skiniotis et al, 2004; Lakämper and Meyhöfer, 2005), whereas in the AMP-PNP state, the motor has to be tightly attached to the microtubule so that it can sustain load (Visscher et al, 1999; Kawaguchi and Ishiwata, 2001; Nishiyama et al, 2002; Uemura et al, 2002; Shao and Gao, 2006).
As suggested by the structural studies (Hoenger et al, 2000; Kikkawa et al, 2000) and the mutational analyses of kinesin (Woehlke et al, 1997), the binding site for kinesin in the strong binding state may not be confined in H12, but is rather spanned over an extensive area of tubulin. In this respect, it is noteworthy that the stability of the interaction in AMP-PNP state was modulated by mutations in H12 only for minus-end loading. For plus-end loading, some unidentified structure in tubulin, independent of β-tubulin H12, might be responsible for the stability of the interaction.
Although the structural elements that determine the shape of the binding potential for strong binding have not been fully identified, the measurement of the stall force demonstrated that the size of the stall force was linearly related to the unbinding force for minus-end loading, but it was independent of the unbinding force for plus-end loading (Figure 5C). As predicted from the analyses of the force–velocity relationship of single kinesin molecule (Visscher et al, 1999; Nishiyama et al, 2002; Carter and Cross, 2005; Lakämper and Meyhöfer, 2005; Shao and Gao, 2006), for two-headed kinesin to produce force, it might be crucial for the trailing head in the strong binding state to sustain against the load imposed towards the minus-end direction. Our result is the first direct demonstration that the stall force is directly related to the stability of the strong binding state.
Stability of the strong binding state is not directly related to kinesin processivity
Now, if we review the results obtained in two motility assays (Figure 3B and D) with the understanding that the affinity of the kinesin–microtubule interaction in the strong binding state was modulated in all three mutants (E410A, D417A, and E421A), we can have further insights into the mechanism of kinesin processivity. Despite the reduced affinity in the strong binding state, in the single molecule motility assay, processive movement of kinesin was reduced only slightly in the E421A mutant, whereas kinesin could scarcely interact with the E410A and D417A microtubules (Table II). In the latter two mutants, even when the initial interaction of kinesin with the microtubule was enforced using optical tweezers, kinesin showed only a poor processive movement, with its longest mean run length being ∼200 nm (Figure 5). These results indicate that the stability of the strong binding state is not the only factor that determines the processivity of the two-headed kinesin.
Table 2. Summary of single molecule assay.
| Motility assay |
Mechanical measurement |
||||
|---|---|---|---|---|---|
| Velocity (μm/s) | Run length (μm) | Stall force (pN) | Unbinding force (pN)a |
||
| (−) | (+) | ||||
| WT | 0.49 | 0.84 | 5.6 | 7.8 | 5.7 |
| E410A | —b | —b | 3.5 | 5.3 | 5.2 |
| D417A | —b | —b | 3.0 | 3.8 | 6.2 |
| E421A | 0.45 | 0.66 | 3.1 | 3.6 | 5.6 |
| a Unbinding force obtained in AMP-PNP state in each minus- and plus-end loading direction. | |||||
| b Data not available because single kinesin molecules did not spontaneously bind to these microtubules. | |||||
The differences between E410A/D417A and E421A mutants might depend on whether or not the leading head can readily attach to the microtubule before the trailing head dissociates from the microtubule. As proposed in the walking model of the kinesin movement, the processive movement of the two-headed kinesin might be a result of mechanical and chemical coordination between the two heads (Hackney, 1994; Ma and Taylor, 1997; Rice et al, 1999; Kawaguchi and Ishiwata, 2001; Uemura et al, 2002; Uemura and Ishiwata, 2003; Schief et al, 2004; Shao and Gao, 2006). If the ADP release from the leading head and the strong binding of this head to the microtubule occurs before the phosphate release from the trailing head and concomitant dissociation of the trailing head from the microtubule, kinesin might succeed to make a forward step. Such a coordination of the two heads might be somehow distracted in E410A/D417A mutants.
The 3D model of the kinesin–microtubule complex indicates that the basic residue in kinesin L8 faces the acidic residues E410 and D417 of β-tubulin, whereas some of the basic residues in kinesin L12 face the residue E421 of β-tubulin (Hoenger et al, 2000; Kikkawa et al, 2000; Skiniotis et al, 2004). Considering this structure, one can hypothesize that the interaction via kinesin L8 might be critical for the leading head to tightly bind the microtubule (Alonso et al, 1998; Klumpp et al, 2003; Ogawa et al, 2004). Currently, such an idea remains highly speculative, and to understand the exact mechanism, kinetical steps in the enzymatic cycle that are affected by each of the mutations should be addressed in future studies. Mutational analysis of tubulin developed in this study has opened a route for future studies to thoroughly identify the structural elements on kinesin interface in both strong and weak binding states, and to relate how each element is involved in the mechanochemical cycle of kinesin.
Materials and methods
Preparation of the H12 mutants
The plasmids and the yeast mutant strains were constructed as described in Supplementary Methods and are listed in Table SII.
Purification of yeast tubulin from mutants
Tubulin was purified from the yeast cell lysate via the successive use of ion exchange column chromatographies (DEAE-Sepharose and Mono-Q; Davis et al, 1993), followed by a cycle of tubulin polymerization and depolymerization. For details, see Supplementary Methods, Figures S1 and S2.
Microtubule gliding assay
We adapted a previously reported method (Howard et al, 1989) for our microtubule gliding assay, except that the standard buffer solution in the original assay was replaced by motility assay buffer (MA buffer) comprising 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 5 mM K-acetate, 2 mM MgSO4, 1 mM EGTA and 0.1 mM EDTA (pH 6.8) (see Supplementary Methods for detail). To maintain the ionic condition of the solution constant throughout the study, MA buffer was used in the microtubule gliding assay, single molecule motility assay, and unbinding force and stall force measurements.
In an experiment that measured the fraction of microtubules that moved for a distance greater than their own length (Figure 3A and B; Howard et al, 1989; Hancock and Howard, 1998), the mean lengths of the microtubules was adjusted to ∼3 μm by controlling the concentration of tubulin and Taxol at the initial stage of polymerization. Based on the video images, microtubule lengths and distances that moved were measured using the public domain program, Image J. The data were collected at variable kinesin densities, and at each density, the fraction of microtubules that moved for a distance greater than their own length, f, was calculated. The data points were fit by equation (1), where ρ is the kinesin density (variable), ρ0 is a fit parameter, and n is the number of kinesin molecules required for a microtubule to move for a distance greater than its own length. The error bars in the curves (Figure 3B) represent the statistical counting errors defined as follows, s.e.m.=(f(1−f)/N)1/2, where f≠0 or 1; s.e.m.=1/N if f=0 or 1. Although the lengths of the microtubules were adjusted to ∼3 μm in both mutated and wild-type microtubules, their mean lengths were slightly different among the preparation of microtubules and among the populations of microtubules subjected to the analyses at each kinesin density. To compensate for this slight difference and to compare the ρ dependence of f between mutants, ρ in Figure 3B was corrected for each data point, assuming that the increase in the microtubules length is equivalent to the increase in the kinesin density in its effect, which increases the probability of the kinesin–microtubule encounter.
Single molecule motility assay
The protocol for single molecule motility assay originally designed for brain microtubules (Inoue et al, 2001) was modified for yeast microtubules. A flow chamber made of two coverslips (dimensions 9 mm × 9 mm × 80 μm) was first incubated with 6 μl of cytochrome c (C-2506, Sigma; 5 mg/ml in MA buffer) for 3 min, extensively washed with 50 μl of MA buffer, and loaded with 10 μl of yeast-microtubule solution (10 μg/ml in MA buffer containing 10 μM Taxol (Taxol without fluorescent label; T-7402, Sigma)). The coverslips were coated with cytochrome c to facilitate the adhesion of yeast microtubules, which had lower affinity for the glass surfaces than the brain microtubules, to the wall of chamber. The chamber was subsequently incubated with 15 μl of BSA (1 mg/ml in MA buffer) to block nonspecific binding of kinesin to the glass surface. Then, 15 μl of MA buffer containing 1 μM BODIPY-FL Taxol (P-7500, Molecular Probes) and an oxygen scavenger (Harada et al, 1990) was introduced into the chamber, and the microtubules were observed under TIRFM. After the position of a BODIPY-FL-labeled microtubule was recorded (Digital video recorder DR20; Sony), 15 μl of HK560-Cy3 solution (0.29 nM in MA buffer) containing 0.1 mg/ml casein, 1 mM ATP, 10 μM Taxol (Taxol without fluorescent label), and an oxygen scavenger was introduced into the chamber, and the interaction of HK560-Cy3 with the microtubule was recorded. For image analysis of kinesin movement and experimental setup of the TIRFM microscope, see Supplementary Methods.
Measurement of unbinding force and stall force
For measurement of the unbinding force, kinesin-coated beads were prepared as described previously (Kojima et al, 1997), except that the fluorescent polystyrene beads with a diameter of 1 μm (F-8823, Molecular Probes) were used. A flow chamber was first incubated with 15 μl of cytochrome c (5 mg/ml in MA buffer) for 5 min, and then washed with 50 μl of MA buffer. Subsequently, 10 μl of polarity-marked yeast microtubule solution including 10 μM Taxol (Taxol without fluorescent label) was infused into the chamber and microtubules were allowed to adsorb on the surface for 10 min. The chamber was then incubated with 15 μl of BSA (1 mg/ml in MA buffer) to block the nonspecific binding of kinesin-bound beads to the glass surface. Finally, 15 μl of kinesin-bound beads solution (25 fM of beads in MA buffer) containing an oxygen scavenger system (Harada et al, 1990), 1 μM BODIPY-564/570-Taxol (P-7501, Molecular Probes), 1 mM AMP-PNP and 1 U/ml apyrase (AMP-PNP state) or 1 mM ADP and 1 U/ml hexokinase (ADP-state) was introduced into the chamber.
The chamber was set under the fluorescence microscope (Nishizaka et al, 1995), and using optical tweezers, a single kinesin-bound bead was made to interact with a microtubule. An external load was applied to this bead–microtubule complex by moving the stage of the microscope at a constant speed (100 nm/s) by a piezoelectric transducer (P-611 NanoCubeTM, Physic Instrument, Germany). The stiffness of the optical trap was 0.038 and 0.076 pN/nm for the experiment in the presence of ADP and AMP-PNP, respectively. All experiments were carried out at 24±1°C.
For the stall force measurement, the same protocol was used, except that the beads were coated with conventional two-headed kinesin and the force was measured in the presence of 1 mM ATP in a range of trap stiffness 0.019–0.076 pN/nm.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr RD Vale for the kind gift of the HK560cys kinesin construct, Dr K Kitamura for technical advice in establishing TIRFM, and Dr T Hashikawa for the assistance with electron microscopy. We are indebted to the Research Resources Centre in Brain Science Institute (RIKEN) for DNA sequencing and mass spectroscopy. This research was partly supported by the Grants-in-Aid for Specially Promoted Research and for the 21st Century COE Program (Physics of Self-Organization Systems) at Waseda University from the Ministry of Education, Sports, Culture, Science and Technology of Japan (SI).
References
- Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA (2002) MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol 157: 1187–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso MC, Damme J, Vandekerckhove J, Cross RA (1998) Proteolytic mapping of kinesin/ncd-microtubule interface: nucleotide dependent conformational changes in the loops L8 and L12. EMBO J 17: 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best D, Warr PJ, Gull K (1981) Influence of the composition of commercial sodium dodecyl sulphate preparations on the separation of α- and β-tubulin during polyacrylamide gel electrophoresis. Anal Biochem 114: 281–284 [DOI] [PubMed] [Google Scholar]
- Block SM, Goldstein LSB, Schnapp BJ (1990) Bead movement by single kinesin molecules studied with optical tweezers. Nature 348: 348–352 [DOI] [PubMed] [Google Scholar]
- Bode CJ, Gupta ML, Suprenant KA, Himes RH (2003) The two alpha-tubulin isotypes in budding yeast have opposing effects on microtubule dynamics in vitro. EMBO Rep 4: 94–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Gasdaska P, Hartwell L (1989) Dominant effects of tubulin overexpression in Saccharomyces cerevisiae. Mol Cell Biol 9: 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter NJ, Cross RA (2005) Mechanics of the kinesin step. Nature 435: 308–312 [DOI] [PubMed] [Google Scholar]
- Davis A, Sage CR, Wilson L, Farrell KW (1993) Purification and biochemical characterization of tubulin from the budding yeast Saccharomyces cerevisiae. Biochemistry 32: 8823–8835 [DOI] [PubMed] [Google Scholar]
- Fersht A (1984) Measurement and magnitude of enzymatic rate constants. In Enzyme Structure and Mechanism, pp 121–154. New York, USA: WH Freeman and Company Press [Google Scholar]
- Goldstein LS, Yang Z (2000) Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu Rev Neurosci 23: 39–71 [DOI] [PubMed] [Google Scholar]
- Gupta ML Jr, Bode CJ, Georg GI, Himes RH (2003) Understanding tubulin–Taxol interactions: mutations that impart Taxol binding to yeast tubulin. Proc Natl Acad Sci USA 100: 6394–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD (1994) Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc Natl Acad Sci USA 91: 6865–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD (1995) Implications of diffusion-controlled limit for processivity of dimeric kinesin head domains. Biophys J 68: 267s–270s [PMC free article] [PubMed] [Google Scholar]
- Hancock WO, Howard J (1998) Processivity of the motor protein kinesin requires two heads. J Cell Biol 140: 1395–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y, Sakurada K, Aoki T, Thomas DD, Yanagida T (1990) Mechanochemical coupling in actomyosin energy transduction studied by in vitro movement assay. J Mol Biol 216: 49–68 [DOI] [PubMed] [Google Scholar]
- Hirose K, Löwe J, Alonso M, Cross RA, Amos LA (1999) 3D electron microscopy of the interaction of kinesin with tubulin. Cell Struct Funct 24: 277–284 [DOI] [PubMed] [Google Scholar]
- Hoenger A, Thormählen M, Diaz-Avalos R, Doerhoefer M, Goldie KM, Muller J, Mandelkow E (2000) A new look at the microtubule binding patterns of dimeric kinesins. J Mol Biol 297: 1087–1103 [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD (1989) Movement of microtubules by single kinesin molecules. Nature 342: 154–158 [DOI] [PubMed] [Google Scholar]
- Hua W, Young EC, Fleming ML, Gelles J (1997) Coupling of kinesin steps to ATP hydrolysis. Nature 388: 390–393 [DOI] [PubMed] [Google Scholar]
- Inoue Y, Hikikoshi-Iwane A, Miyai T, Muto E, Yanagida T (2001) Motility of single one-headed kinesin molecule along microtubules. Biophys J 81: 2838–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelachvili JN (1992) Intermolecular and Surface Forces. San Diego, USA: Academic Press [Google Scholar]
- Kawaguchi K, Ishiwata S (2001) Nucleotide-dependent single to double-headed binding of kinesin. Science 291: 667–669 [DOI] [PubMed] [Google Scholar]
- Kikkawa M, Okada Y, Hirokawa N (2000) 15 Å resolution model of the monomeric kinesin motor, KIF1A. Cell 100: 241–252 [DOI] [PubMed] [Google Scholar]
- Klumpp LM, Brendza KM, Rosenberg JM, Hoenger A, Gilbert SP (2003) Motor domain mutation traps kinesin as a microtubule rigor complex. Biochemistry 42: 2595–2606 [DOI] [PubMed] [Google Scholar]
- Kojima H, Muto E, Higuchi H, Yanagida T (1997) Mechanics of single kinesin molecules measured by optical trapping nanometry. Biophys J 73: 2012–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakämper S, Meyhöfer E (2005) The E-hook of tubulin interacts with kinesin's head to increase processivity and speed. Biophys J 89: 3223–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little M, Seehaus T (1988) Comparative analysis of tubulin sequences. Comp Biochem Physiol 90B: 655–670 [DOI] [PubMed] [Google Scholar]
- Lockhart A, Crevel IM, Cross RA (1995) Kinesin and ncd bind through a single head to microtubules and compete for a shared MT binding site. J Mol Biol 249: 763–771 [DOI] [PubMed] [Google Scholar]
- Luduena RF (1998) Multiple forms of tubulin: different gene products and covalent modifications. Int Rev Cytol 178: 207–275 [DOI] [PubMed] [Google Scholar]
- Ma YZ, Taylor EW (1997) Interacting head mechanism of microtubule-kinesin ATPase. J Biol Chem 272: 724–730 [DOI] [PubMed] [Google Scholar]
- Mizuno N, Toba S, Edamatsu M, Watai-Nishii J, Hirokawa N, Toyoshima YY, Kikkawa M (2004) Dynein and kinesin share an overlapping microtubule-binding site. EMBO J 23: 2459–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Higuchi H, Yanagida T (2002) Chemomechanical coupling of the forward and backward steps of single kinesin molecules. Nat Cell Biol 4: 790–797 [DOI] [PubMed] [Google Scholar]
- Nishizaka T, Miyata H, Yoshikawa H, Ishiwata S, Kinosita K Jr (1995) Unbinding force of a single motor molecule of muscle measured using optical tweezers. Nature 377: 251–254 [DOI] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH (1998) Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391: 199–203 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Nitta R, Okada Y, Hirokawa N (2004) A common mechanism for microtubule destabilizers—M type kinesins stabilize curling of the protofilament using the class-specific neck loops. Cell 116: 591–602 [DOI] [PubMed] [Google Scholar]
- Okada Y, Hirokawa N (2000) Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc Natl Acad Sci USA 97: 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Willson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD (1999) A structural change in the kinesin motor protein that drives motility. Nature 402: 778–784 [DOI] [PubMed] [Google Scholar]
- Schief WR, Clark RH, Crevenna AH, Howard J (2004) Inhibition of kinesin motility by ADP and phosphate supports a hand-over-hand mechanism. Proc Natl Acad Sci USA 101: 1183–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer MJ, Block SM (1997) Kinesin hydrolyses one ATP per 8-nm step. Nature 388: 386–390 [DOI] [PubMed] [Google Scholar]
- Shao Q, Gao YQ (2006) On the hand-over-hand mechanism of kinesin. Proc Natl Acad Sci USA 103: 8072–8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima T, Imamula K, Kon T, Ohkura R, Sutoh K (2006) Head-head coordination is required for the processive motion of cytoplasmic dynein, an AAA+ molecular motor. J Struct Biol 156: 182–189 [DOI] [PubMed] [Google Scholar]
- Skiniotis G, Cochran JC, Muller J, Mandelkow E, Gilbert SP, Hoenger A (2004) Modulation of kinesin binding by the C-termini of tubulin. EMBO J 23: 989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa H, Peterman EJG, Moerner WE, Goldstein LSB (2001) ADP-induced rocking of the kinesin motor domain revealed by single-molecule fluorescence polarization microscopy. Nat Struct Biol 8: 540–544 [DOI] [PubMed] [Google Scholar]
- Tanaka-Takiguchi Y, Itoh TJ, Hotani H (1998) Visualization of the GDP-dependent switching in the growth polarity of microtubules. J Mol Biol 280: 365–373 [DOI] [PubMed] [Google Scholar]
- Thorn KS, Ubersax JA, Vale RD (2000) Engineering the processive run length of the kinesin motor. J Cell Biol 151: 1093–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura S, Ishiwata S (2003) Loading direction regulates the affinity of ADP for kinesin. Nat Struct Biol 10: 308–311 [DOI] [PubMed] [Google Scholar]
- Uemura S, Kawaguchi K, Yajima J, Edamatsu M, Toyoshima YY, Ishiwata S (2002) Kinesin–microtubule binding depends on both nucleotide state and loading direction. Proc Natl Acad Sci USA 99: 5977–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernos I, Karsenti E (1996) Motors involved in spindle assembly and chromosome segregation. Curr Opin Cell Biol 8: 4–9 [DOI] [PubMed] [Google Scholar]
- Visscher K, Schnitzer MJ, Block SM (1999) Single kinesin molecules studied with a molecular force clamp. Nature 400: 184–189 [DOI] [PubMed] [Google Scholar]
- Wang Z, Sheetz MP (2000) The C-terminus of tubulin increases cytoplasmic dynein and kinesin processivity. Biophys J 78: 1955–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD (1997) Microtubule interaction site of the kinesin motor. Cell 90: 207–216 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information