


Abstract
The Saccharomyces cerevisiae Tgs1 methyltransferase (MTase) is responsible for conversion of the m7G caps of snRNAs and snoRNAs to a 2,2,7- trimethylguanosine structure. To learn more about the evolutionary origin of Tgs1 and to identify structural features required for its activity, we performed a structure–function study. By using sequence comparison and phylogenetic analysis, we found that Tgs1 shows strongest similarity to Mj0882, a protein related to a family comprised of bacterial rRNA:m2G MTases RsmC and RsmD. The structural information of Mj0882 was used to build a homology model of Tgs1p which allowed us to predict the range of the minimal globular MTase domain and the localization of other residues that may be important for enzyme function. To further characterize functional domains of Tgs1, mutants were constructed and tested for their effects on cell viability, subcellular localization and binding to the small nuclear ribonucleoproteins (snRNPs) and small nucleolar RNPs (snoRNPs). We found that the N-terminal domain of the hypermethylase is dispensable for binding to the common snRNPs and snoRNPs proteins but essential for correct nucleolar localization. Site- directed mutagenesis of Tgs1 allowed also the identification of the residues likely to be involved in the formation of the m7G-binding site and the catalytic center.
INTRODUCTION
Small ribonucleoproteins (RNPs) are complexes required for processing RNA precursors into mature RNA species (reviewed in 1). Based on their intracellular location and function, these RNPs can be classified in two groups, the nucleoplasmic small nuclear RNPs (snRNPs) that play a role in the maturation of pre-mRNAs and the small nucleolar RNPs (snoRNPs) that reside in the cell nucleolus and are required for maturation of pre-rRNA (reviewed in 2–4).
The U1, U2, U4/U6 and U5 snRNPs are essential components of the spliceosome. They contain a set of common proteins also called Sm proteins (B/B′ in mammals, D1, D2, D3, E, F and G) that assemble as a heptameric doughnut-like structure around the Sm site of the snRNAs (5). With the exception of U6, the snRNAs are transcribed by RNA polymerase II, acquire a m7G cap in the nucleus and, after export to the cytoplasm, associate with the Sm proteins, which allows hypermethylation of the m7G cap to a trimethylguanosine (m3G) 5′ cap structure (6,7). In mammals, both the Sm core complex and the m3G cap structure of snRNAs provide signals for subsequent nuclear import of the newly assembled snRNPs (2,8).
The m7G cap of a subset of snoRNAs transcribed by RNA polymerase II is also hypermethylated (9). While a few snoRNAs, such as U3, are known to be involved in the cleavage of primary rRNA transcript, the majority of snoRNAs function as guide RNAs that select 2′-O-methylation sites (snoRNAs of box C/D) and pseudouridylation sites (snoRNAs of box H/ACA) of rRNAs bases. Each family of snoRNAs is complexed with four core proteins to form a stable RNP. The box C/D snoRNAs bind to Snu13, Nop56, Nop58 and Nop1, while the box H/ACA snoRNAs associate with Cbf5, Nhp2, Gar1 and Nop10 (3,4). The biogenesis of vertebrate snoRNPs does not require a cytoplasmic transit suggesting that the snoRNAs acquire the trimethylguanosine cap structure in the nucleus (10). That a hypermethylase acts in this compartment is supported by studies showing that U3, as well as U1 and U2, are efficiently hypermethylated in oocyte nuclear extracts (11,12).
Studies in mammalian systems suggested that the snRNA-(guanosine-N2)-methyltransferase is a non-snRNP protein recognizing the U1 snRNP by binding to the SmB/B′ proteins (13). Moreover, stepwise reconstitution of U1 snRNP showed that the U1 particle is hypermethylated in vitro by a HeLa cytosolic extract while a subcore lacking the SmB/B′ protein is not (14). These observations indicated that the SmB/B′ protein might represent a docking site for the hypermethylase. This is consistent with a recent report showing that the human hypermethylase binds preferentially to the C-terminal extension of the SmB protein (15). Accordingly, the yeast hypermethylase (Tgs1) responsible for m3G cap formation of snRNAs was identified in a two-hybrid screen as binding to the C-terminal tail of the SmB protein (16). The same study also showed that the yeast hypermethylase binds preferentially to the SmB protein in vitro, indicating that the properties of the yeast Tgs1 protein conform to the characteristics of the mammalian enzyme. Remarkably, Tgs1 is also responsible for hypermethylation of yeast snoRNAs (16). By analogy with the snRNPs, where Tgs1 binds to the basic-rich C-tail of yeast SmB, the hypermethylase associates also with the basic-rich domains of the yeast C/D snoRNP core protein Nop58 and the H/ACA snoRNP core protein Cbf5 (16). Taken together, these studies indicate that the enzyme recognizes basic-rich domains of different common core proteins explaining how a unique enzyme can bind to different RNPs.
In yeast, TGS1 is not essential for viability but its deletion produces a cold-sensitive phenotype. The Tgs1 protein is evolutionarily conserved and in higher eukaryotes the hypermethylases possess a large N-terminal domain absent in lower eukaryotes, in which Tgs1 is mainly composed of the conserved catalytic domain (16). While in mammals the hypermethylase locates both in Cajal bodies and in the cytoplasm (15,17), subcellular localization studies revealed that the yeast hypermethylase is localized in the nucleolus, suggesting that yeast snRNAs and snoRNAs cycle through this compartment to undergo cap hypermethylation (16). Given that little is known about the mechanism of small RNA cap hypermethylation, we initiated a structure–function analysis of the yeast Tgs1 protein. In the present report, we found that Tgs1 shows strongest similarity to the structure of Mj0882, a member of a family comprised of bacterial rRNA:m2G methyltransferases (MTases) RsmC and RsmD. The homology model of Tgs1 based on the Mj0882 structure was used to guide the mutagenesis experiments. We identified the structural elements of yeast Tgs1 that are essential for function in vivo. We found that the N-terminal domain of the hypermethylase is dispensable for binding to common snRNPs and snoRNPs core proteins but essential for correct nucleolar localization. Site-directed mutagenesis of Tgs1 allowed the identification of the residues presumably involved in the formation of the m7G-binding site/catalytic center. Interestingly, the alanine mutation of a Ser residue in motif IV, known to be conserved among nearly all amino-MTases, did not abolish cap hypermethylation, which suggests a unique character of the Tgs1 hypermethylase compared with related enzymes acting on different substrates.
MATERIALS AND METHODS
Sequence database searches and phylogenetic analysis
The non-redundant (nr) protein sequence database and putative translations of the expressed sequence tag (EST), sequence-tagged site (STS), high throughput genomic (HTG) and genome survey sequence (GSS) divisions of the GenBank database and publicly available partially sequenced genomes were searched using the PSI-BLAST algorithm (18) using the online services of the National Center for Biotechnology Information (Bethesda, MD; http://www.ncbi.nlm.nih.gov) and the GeneSilico server (Warsaw, Poland; http://genesilico.pl/~blast/). The multiple sequence alignment was generated using CLUSTALX (19) with default parameters. Manual adjustments were introduced based on pairwise alignments reported by PSI-BLAST, results of secondary structure prediction, tertiary fold recognition (FR) and refinement of the structural model (see below).
Phylogenetic inference was carried out using the conserved blocks of the multiple sequence alignment. The matrix of pairwise distances was calculated from sequences according to the JTT model (20) with gaps treated as missing data. The neighbor-joining (NJ) tree was inferred as described in Saitou and Nei (21).
Protein structure prediction
Protein structure prediction was carried via the MetaServer interface (22; http://genesilico.pl/meta/) using publicly available online services for FR (SAM-T02, RAPTOR, 3DPSSM and FUGUE) and secondary structure prediction (PSIPRED). Disordered regions were predicted with PONDR (23). Homology modeling was carried out according to the ‘Frankenstein’s Monster’ approach, which in the hands of J.M.B and coworkers led to successful predictions of the mRNA-cap:m7G (cap 0) MTase Abd1 structure (24) and many other proteins during the recent CASP5 experiment (25). Briefly, preliminary models of Tgs1p were generated based on the alignments returned by the aforementioned FR servers. The sequence–structure fit in these models was evaluated using VERIFY3D (26) (window, five amino acids); polypeptide fragments with scores <0.1 were deleted and the remaining parts were merged to produce a ‘consensus’ model. Final adjustments have been made using ‘patches’ from the known MTase structures. The consensus model was energy-minimized in vacuo using the GROMOS force-field (27) to relieve steric clashes. The coordinates of the final model are freely available online at ftp://genesilico.pl/iamb/models/Tgs1/tgs1.model.pdb.
Yeast strains, media and genetic methods
Strain YB157K (Mata ura3-Δ0 his3-Δ1 leu2-Δ0 met15-Δ0 tgs1::KAN) contains a chromosomal disruption of the YPL157w/TGS1 locus and was derived from strain BY4743 (16). Yeast strains were transformed and grown using standard procedures and media (28).
Recombinant DNA work
Plasmid pGFP-Tgs1 containing the TGS1 coding sequence fused to the GFP reporter protein was constructed as described (16). The N- and C-terminal deletion mutants were constructed by amplifying fragments from pGFP-Tgs1 plasmid using oligonucleotides carrying 5′ XbaI and 3′ HindIII sites. The XbaI–HindIII fragments were then transferred into pGFP-Nfus (URA3 CEN) vector (29), previously cut with XbaI and HindIII. The point mutations were introduced into the TGS1 gene by oligonucleotide-directed mutagenesis using the megaprimer strategy (30). To place the N- and C-terminal deletion mutants under T7 promoter, fragments were PCR-amplified using oligonucleotides carrying an ATG start codon. The PCR fragments were then cloned into XbaI–HindIII-digested pBluescript vector and used for in vitro transcription and translation using rabbit reticulocyte lysate (Promega) in the presence of 35S-methionine. The pGST plasmids containing the SmB gene and the C-terminal tail of Cbf5p in fusion with glutathione S-transferase (GST) were previously described (16).
Protein and RNA analyses
Protein extraction, SDS–PAGE and western blots were carried out as described previously (31,32). Western blot analysis was performed using anti-GFP antibodies at 1/2000 (Molecular Probes) and rabbit secondary antibodies. Immunoprecipitation experiments were performed using whole-cell extracts from indicated strains with 20 µl of protein A–Sepharose beads (Sigma) and 15 µl of anti-m3G antibodies (Calbiochem). The resin was washed five times with 1 ml of buffer and after phenol–chloroform extraction and ethanol precipitation, the RNA samples were subjected to northern analysis. To this end, RNA samples were separated on 6% polyacrylamide/7 M urea denaturing gels and transferred electrophoretically to a Hybond membrane in 20 mM NaPO4 (pH 6.5) at 7 V for 12 h at 4°C. Hybridization was performed as previously described (33).
Fluorescence microscopy
The localization of GFP fusion proteins was examined in living yeast cells. Cells transformed with pGFP constructs were grown in synthetic selective solid or liquid media to early log phase. Cells were washed with phosphate-buffered saline, mounted on slides and examined by fluorescence microscopy. Samples were observed using a DMRA microscope and a 1.25× eyepiece. Digital images were recorded with a C4795-NR CCD camera (Hamamatsu). Acquisitions were performed using the Metamorph (Universal Imaging) software.
RESULTS
Tgs1 is related to other RNA:m2G MTases
To learn more about the evolutionary origin and the sequence–structure–function relationships of Tgs1p, we carried out comprehensive bioinformatics analysis in order to build a structural model, interpret it in the phylogenetic context and use it as a guide for site-directed mutagenesis. The amino acid sequence of Saccharomyces cerevisiae Tgs1 (16) was used in PSI-BLAST database searches to identify homologous proteins. Since we were interested in identification of closely related enzymes with similar functions and not all MTases, a stringent cut-off expectation (e) value (= 10–15) was used to avoid expansion of the profile onto the whole S-adenosylmethionine-dependent MTase superfamily. The eukaryotic orthologs of Tgs1p were detected in the first two iterations; the search converged after the fourth iteration. Interestingly, Tgs1p exhibited significant similarity to an uncharacterized family of putative MTases from Archaea and Gram-positive bacteria (the MM1487 ORF from Methanosarcina mazei Goe1 reported with an e-value of 10–18 in the second iteration). Sequences of Tgs1 and its orthologs were retrieved from the databases; a multiple sequence alignment (Fig. 1) was generated and refined, and a distance-based phylogeny (Fig. 2) was reconstructed (see Materials and Methods for technical details).
Figure 1.

Multiple alignment of the Tgs1 protein family. Conserved motifs are labeled according to the nomenclature described for the ‘classical’, Rossmann-fold AdoMet-dependent MTase superfamily (35). Invariant residues are highlighted in black, conserved residues are highlighted in gray. Residues studied by site-directed mutagenesis (in this work; 16) are indicated by ‘#’. Secondary structure (predicted for Tgs1, observed in Mj0882/1dus) is indicated. For Tgs1, regions predicted to be disordered in the absence of additional binding partners (for instance other proteins), are indicated by ‘x’.
Figure 2.

Unrooted evolutionary tree of the Tgs1 protein family. The names of the taxons are outlined. The numbers at the nodes indicate the statistical support of the branching order by the bootstrap criterion. The bar at the bottom of the phylogram indicates the evolutionary distance to which the branch lengths are scaled based on the estimated divergence.
The evolutionary tree (Fig. 2) reveals that the Tgs1 family is exclusively comprised of eukaryotic proteins, in agreement with the phylogenetic occurrence of the monomethylated cap structures (m7G), which are the substrate for the hypermethylase. The lineage grouping proteins from animals and plants, as well as its internal sub-branches, are relatively well supported by the bootstrap criterion. On the other hand, relationships between the Tgs1 family members from lower eukaryotes are ambiguous, with only the alveolates forming a well resolved branch. All organisms with fully sequenced genomes possess a single copy of the Tgs1 ortholog, the only exception are plants (Arabidopsis thaliana and Oryza sativa), which have two copies. Both copies seem functional, i.e. they possess a complete set of conserved motifs required for the MTase activity. Interestingly, the At1g30550.1 protein from A.thaliana seems to contain two tandemly arranged sets of MTase motifs, of which only the C-terminal one is complete. Our results suggest that a duplication of the Tgs1-encoding gene occurred in the ancestor of modern plants. Subsequently, intragenic duplication occurred in one of the Tgs1 copies in A.thaliana, giving rise to a pseudodimer with the N-terminal copy of the MTase domain inactivated by deletion in the cofactor-binding region. It will be interesting to determine if the Tgs1 duplication in plants was linked to subfunctionalization of the two copies, i.e. if for instance the paralogs exhibit different substrate specificities, and what is the role of the ‘inactivated’ N-terminal repeat in the At1g30550.1 protein.
The multiple sequence alignment was used as a query for the FR metaserver (21; http://genesilico.pl/meta). All algorithms reported a perfect match between the Tgs1 sequence and known MTase structures, with the putative MTase Mj0882 (1dus) always reported with the best score (SAM-T02, 1.92 × 10–24; RAPTOR, 16.4; 3DPSSM, 0.00309; FUGUE, 15.78; the scoring scales of the individual methods are not normalized). Mj0882 is a close relative of rRNA:m2G MTases RsmC and RsmD and most likely shares a similar function, i.e. guanosine-N2 methylation (34). It is remarkable that computational sequence analysis and structure prediction find a strong link between Tgs1, a bona fide m2(2)G MTase, and a family of known and putative m2G MTases, even in the absence of evident sequence similarities other than motifs common to all MTases, including those acting on different substrates. This suggests that the experimentally determined structure of the Mj0882 protein could be used to guide functional annotation of the Tgs1 sequence. The S.cerevisiae Tgs1 sequence was also analyzed using PONDR (23) to identify disordered regions. Together with the information from the multiple sequence alignment and FR analysis, these results allowed the dissection of the primary structure of Tgs1 into three regions: the globular core, corresponding to the catalytic domain, and flexible, presumably disordered terminal tails (amino acids 1–58 and 266–315; Fig. 1).
A three-dimensional structure of the globular core of yeast Tgs1p was homology modeled based on the FR alignment of the Mj0882 protein, according to the protocol described in Materials and Methods. Figure 3 shows the key elements mapped onto the Tgs1 structure. In the MTase superfamily there are nine common motifs conserved at the sequence and structure level (35). All secondary structure elements are conserved in the Tgs1 family, even though only six motifs display pronounced sequence conservation (Fig. 1). Con served residues in motifs I, II and III form the AdoMet-binding site, while motifs X, IV and VI form the substrate-binding/catalytic pocket (Fig. 3). It is worth noting that in addition to the region spanning the conserved MTase motifs (approximately positions 60–265), some members of the Tgs1 family also possess extended N- and C-termini. Indeed, the N-terminal domains of Tgs1 orthologs from animals and plants are much longer than the corresponding termini of Tgs1 from lower eukaryotes (16). Moreover, the yeast Tgs1 protein is one of a few members of the family possessing a small extended C-terminal domain (∼45 residues). While the N-terminal tails are usually rich in positive amino acids, the C-terminal tails contain more acidic residues.
Figure 3.

Homology model of Tgs1 shown in a ‘cartoon’ representation. Docked ligands (AdoMet and the N7-methylated guanosine) and residues studied by mutagenesis are shown in the a ‘wireframe’ representation. Red indicates indispensable side chains (corresponding to inactive alanine mutants); pink indicates positions in which non-conservative substitution with bulky side chains abrogate the MTase activity (see also Bottom); cyan indicates bulky side chains, which are dispensable for the MTase activity. (Bottom) Models of I83R, S175R and S175E mutants predicted to block the active site and abrogate the substrate binding. Mutated residues are shown in green. Steric clashes with m7G are shown as pink broken lines.
Mapping the functional domains of Tgs1
To verify the structural model and to characterize the functional domains of the yeast Tgs1 protein, we constructed a set of deletion alleles of Tgs1. We first generated N- and C-terminal mutants (Fig. 4): the Tgs1ΔN and Tgs1ΔNΔN mutants exhibit N-terminal deletions of 40 and 58 residues, respectively, while the Tgs1ΔC allele exhibits a deletion of the C-terminal 43 residues. The N-terminal deletion constructs were chosen because closer inspection of this region revealed the presence of two basic-rich sequences (residues 16–19 and 42–58) showing similarities to nuclear and nucleolar localization signals (see below) (36,37). Concerning the C-terminal domain, due to its acidic nature, we reasoned that it could represent a substrate-binding surface. To study the functional properties of these domains, the various mutants were fused to the GFP reporter protein.
Figure 4.

Schematic representation of the Tgs1 deletion alleles. The structures of the mutant constructs are schematically shown: the filled bar represents the sequence (positions 42–58) that carries basic amino acids and the gray bars represent conserved MTase motifs. A summary of the subcellular localizations, the growth phenotypes at 16°C and the binding properties of the mutants is shown at the right. Np, nucleoplasm; No, nucleolus; cyt, cytoplasm; NA, not applicable.
The deletion mutants were first tested for their ability to complement the growth defect of the tgs1::KAN strain. Indeed, we previously showed that the GFP–Tgs1 protein complements the cold-sensitive phenotype of the tgs1::KAN strain and restores hypermethylation of the snRNA cap structure (16). After transformation of the different fusions into the tgs1::KAN strain, transformants were plated on medium lacking uracil to maintain the plasmids and the plates were incubated at 28 and 16°C. As shown in Figure 5A, cells bearing the different GFP–Tgs1 deletion fusions grew readily at 28°C indicating that none of the mutations show a dominant phenotype. Cells carrying wild-type GFP–Tgs1 and the GFP–Tgs1ΔC allele also grew readily at 16°C whereas the GFP–Tgs1ΔN and GFP–Tgs1ΔNΔN mutants could not support colony formation at this temperature. These results demonstrate that the N-terminal region (amino acids 1–58) of Tgs1 is critical for Tgs1 function while the C-terminal 43 residues are dispensable.
Figure 5.

Functional properties of Tgs1 deletion mutants. (A) Growth assay. Exponentially growing cultures of yeast cells carrying the indicated constructs were serially diluted, spotted on plates and incubated at 16 and 28°C. In contrast to the wild-type and the Tgs1ΔC fusion proteins, the other constructs were unable to restore growth of the tgs1::KAN deletion strain. (B) Western analysis of GFP–Tgs1 mutant fusion proteins. Equivalent amounts of cell extracts prepared from wild-type strains carrying the indicated GFP–Tgs1 alleles were fractionated by SDS–PAGE and immunoblotted with anti-GFP antibodies. Control extract was made from a wild-type strain carrying the GFP vector alone. The Tgs1BRD allele was fused to a GFP–GFP dimer reporter protein which explains the lower mobility of the fusion protein. The other low mobility bands correspond to anti-GFP antibodies cross-reacting polypeptides. (C) Immunoprecipitation experiments. Whole-cell extracts were prepared from tgs1::KAN cells expressing the indicated GFP fusion proteins grown at 28°C and the snRNPs were immunoprecipitated with anti-m3G antibodies. RNA was extracted from the supernatants (S.), from the pellets (P.) and from equivalent aliquots of the total lysates (T.), separated on denaturing polyacrylamide gels, and subjected to northern analysis. Hybridization was performed with probes specific for the yeast U4, U5 and U6 snRNAs.
To test whether the growth defect of both Tgs1ΔN and Tgs1ΔNΔN might be a consequence of instability of the fusion proteins, whole-cell lysates prepared from yeast cells carrying the different constructs and grown at 28°C, were resolved by SDS–PAGE, transferred to membranes and analyzed by western blotting using anti-GFP antibodies. As shown in Figure 5B, the antibodies recognized the GFP–Tgs1 fusion protein of 62 kDa, the 27 kDa GFP protein as well as the GFP fusion mutants of predicted lengths and at approximately similar levels. Similar results were obtained when cells were shifted to 16°C (data not shown). It is interesting to note that the GFP–Tgs1ΔC allele (43 amino acids deleted) migrates faster than the GFP–Tgs1ΔNΔN allele (58 amino acids deleted). This behavior might be due to the highly acidic nature of the C-terminal domain of the Tgs1 protein. These results indicate that all mutant proteins are correctly expressed in vivo.
The effect of the N- and C-terminal truncations on snRNA cap MTase activity were examined after expressing the wild-type and truncated GFP fusion proteins in the tgs1::KAN background. The presence of a hypermethylated cap was analyzed by immunoprecipitation of snRNPs from yeast cells using K121 antibodies specifically recognizing the m3G cap structure. As shown in Figure 5C, immunoprecipitation experiments on whole-cell extracts, prepared from the indicated strains, followed by northern analysis indicated that the U4 and U5 were present in the pellets from the wild-type strain (lane 14) and GFP–Tgs1ΔC containing strain (lane 8) but not from the N-terminal truncation mutants (lanes 2 and 5). The U6 snRNA was also precipitated from the wild-type strain due to its association with the U4 snRNA. These results demonstrate that the growth defects observed for the Tgs1ΔN and Tgs1ΔNΔN alleles correlate with the absence of the m3G cap structure in the snRNAs.
The N- and C-terminal regions of yeast Tgs1p are not required for binding to common snRNP and snoRNP proteins
We next determined whether Tgs1 mutant proteins are defective in binding to the common proteins found in snRNPs and snoRNPs. Indeed, we have previously shown that the enzyme associates to basic-rich domains of the core protein SmB found in snRNPs and the common proteins Nop58 and Cbf5 found in snoRNPs (16). The interaction between the Tgs1 mutants and the SmB protein was tested using GST pull-down assays. As shown in Figure 6A, 35S-methionine-labeled Tgs1ΔN and Tgs1ΔNΔN mutant proteins quantitatively bound to the GST–SmB protein as efficiently as the 35S-Tgs1 wild-type and the 35S-Tgs1ΔC mutant proteins. Similar pull-down experiments, performed to test whether the mutant proteins are defective in binding with the C-terminal tail of the Cbf5 protein, demonstrate that, like the wild-type Tgs1 protein, all 35S-Tgs1 N- and C-terminal deletion mutants are able to interact with GST–Cbf5/Ctail (Fig. 6B). Taken together, these results demonstrate that the N- and C-terminal regions of Tgs1 are not required for in vitro binding with the common core proteins of snRNPs and snoRNPs. They indicate also that the growth defects observed for the N-terminal deletion mutants might not be a consequence of a defect in binding to the RNP particles.
Figure 6.
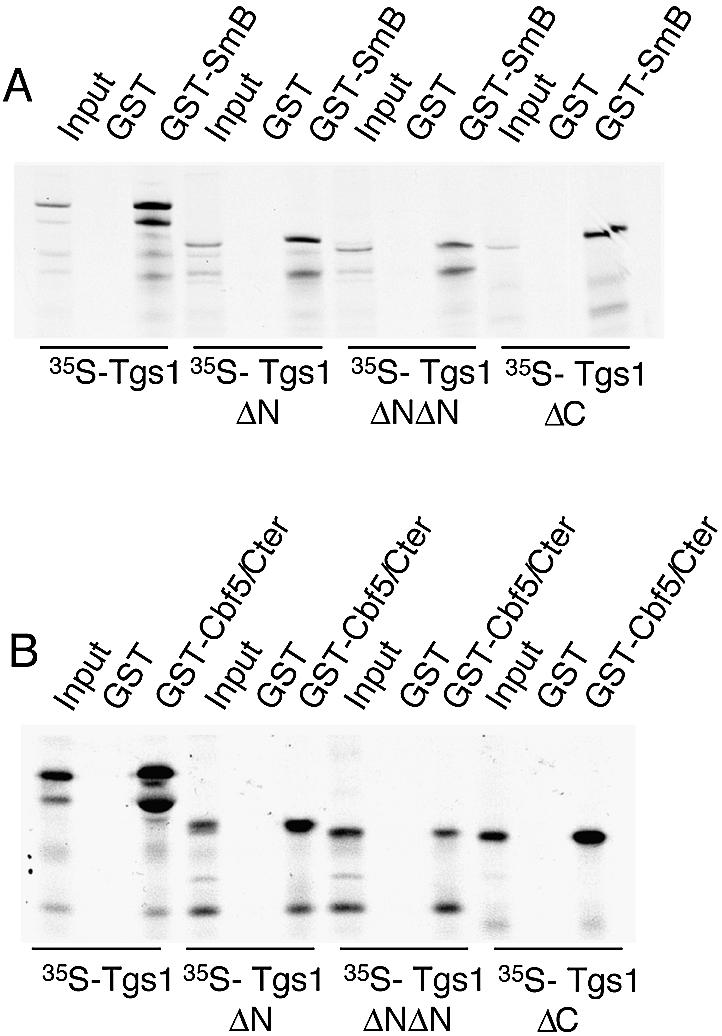
In vitro binding of Tgs1 deletion mutants with SmB and Cbf5 proteins. (A) The indicated 35S-methionine-Tgs1 wild-type and mutant proteins prepared by in vitro transcription and translation were mixed with the GST–SmB fusion protein or GST alone produced in Escherichia coli. Proteins bound to glutathione–Sepharose beads were washed, denatured and separated by SDS–PAGE. Input represents aliquots of radioactive proteins corresponding to 10% of that used in each of the binding reactions. (B) The indicated 35S-methionine-Tgs1 wild-type and mutant proteins were mixed with the GST–Cbf5/Cter fusion protein (carrying the basic amino acid rich C-terminal tail of Cbf5) or with GST alone and treated as described in (A).
The N-terminal region of Tgs1 is required for correct nucleolar localization
Subcellular localization studies showed that Tgs1 is located in the yeast nucleolus (16). The growth defect of the N-terminal deletion mutants could therefore be explained by a mislocalization of the mutant proteins. To test the effect of the mutations on Tgs1 localization, the different GFP–Tgs1 mutants were transformed into a wild-type strain carrying a SRP40–DsRed2 fusion protein and their localizations were observed in living cells. As shown in Figure 7a–c, and as expected, the wild-type GFP–Tgs1 protein was distributed throughout the nucleolus and co-localizes with the SRP40–DsRed2 fusion protein, which allows the visualization of the nucleolus. Indeed, it has been shown that SRP40p is a nucleolar protein that is immunologically and structurally related to rat Nopp140, a snoRNP associated protein of the nucleolus and coiled bodies (38). A similar co-localization was also observed for the GFP–Tgs1ΔC mutant (Fig. 7j–l). In contrast, the GFP–Tgs1ΔNΔN fusion gives rise to a diffuse nuclear and cytoplasmic staining showing that this mutant is unable to localize efficiently to the nucleus (Fig. 7g–i). Concerning the GFP–Tgs1ΔN fusion, it appears that it localizes uniformly throughout the whole nucleus without being clearly concentrated within the nucleolus (Fig. 7d–f). Taken together, these results demonstrate that the N-terminal region (1–58) of Tgs1 is required for correct nucleolar localization. Examination of the sequence of this region indicates the existence of a classical SV40 nuclear localization signal (NLS) (positions 16–19) as well as a sequence rich in basic amino acids (positions 42–58). To more precisely determine which region of Tgs1 allows nucleolar localization, we constructed two additional fusions, namely a GFP–GFP–Tgs1BRD (for basic rich domain) carrying the domain encompassing amino acids 42–58 and a GFP–Nter fusion carrying the entire N-terminal domain of Tgs1 (amino acids 1–58). The BRD domain was fused to a dimeric GFP–GFP reporter protein (32) in order to be over the diffusion limit of the yeast nuclear pore. Western blot analysis indicates that the fusion proteins are correctly expressed in yeast (Fig. 5B and data not shown). After transformation into wild-type yeast cells containing a SRP40–DsRed2 construct, microscopic fluorescence examination showed that the GFP–GFP–Tgs1BRD fusion protein is localized in the nucleus and the nucleolus but not exclusively concentrated into this last compartment (Fig. 7m–o) while a stronger nucleolar staining is observed for the GFP–Nter fusion protein (Fig. 7p–r). Taken together, these results demonstrate that the entire N-terminal region of Tgs1 possesses nucleolar localization properties, being able to locate and retain the reporter fusion protein into the nucleolus.
Figure 7.

Intracellular localizations of Tgs1 deletion mutants. Exponentially growing wild-type cells carrying a SRP40–DsRed2 fusion protein and the indicated GFP fusion proteins were grown at 28°C. The nucleolar SRP40 (red) and the GFP fusion proteins (green) were detected by immunofluorescence. The DNA was stained with DAPI.
Dissection of the putative catalytic center
Based on the homology model proposed in Figure 3 and by analogy to solved structures of MTases that modify exocyclic amino groups in nucleic acids, we predicted that the Tgs1 catalytic center might be represented by residues located in motif X (positions 75–89) and motif IV (positions 171–184) (Fig. 1). In order to test this prediction, we mutated positions W75, E81, I83, S175 and W178 and fused the mutants to the GFP reporter protein. After transformation into the tgs1::KAN strain, cells were tested for function in vivo. To avoid artifacts due to over-expression, the fusions were expressed at low levels from a centromeric vector, with transcription driven at the basal level of the inducible MET25 promoter (39). As shown in Figure 8A, expression of the GFP–Tgs1 point mutants results in the synthesis of fusion proteins of predicted lengths and at approximately similar levels indicating that all mutant proteins are correctly expressed in vivo. Growth assays indicate that the W75A, E81A and S175A alleles complemented the cold-sensitive phenotype of the tgs1::KAN strain while the I83R, S175R, S175E and W178A mutants were unable to restore growth at 16°C (Fig. 8B).
Figure 8.

Functional properties of Tgs1 point mutants. (A) Western analysis of GFP–Tgs1 point mutants. Equivalent amounts of cell extracts prepared from wild-type strains carrying the indicated GFP fusion constructs and grown at 28°C were fractionated by SDS–PAGE and immunoblotted with anti-GFP antibodies. Control extract was made from a wild-type strain carrying the GFP vector alone. The GFP–Tgs1 fusions are indicated by arrows. Additional bands correspond to anti-GFP antibodies cross-reacting polypeptides. (B) Growth assays. Exponentially growing cultures of yeast cells carrying the indicated GFP fusion mutant were serially diluted, spotted on plates and incubated at 16 and 28°C.
Subcellular localization studies showed that all seven GFP–Tgs1 point mutants locate in the yeast nucleolus as the GFP–Tgs1 wild-type fusion protein (data not shown), demonstrating that the mutated residues do not interfere with correct localization. Finally, the presence of a hypermethylated cap structure of the snRNAs and snoRNAs in the tgs1::KAN strain carrying the GFP–Tgs1 point mutants was analyzed by immunoprecipitation of snRNPs and snoRNPs from yeast cells using K121 antibodies. Immunoprecipitation experiments on whole-cell extracts, prepared from the indicated strains, followed by northern analysis show that snRNAs and both C/D and H/ACA snoRNAs were present in the pellets from all viable mutants strains and absent from cells carrying the I83R, S175R, S175E and W178A alleles (Fig. 9). These results demonstrate that alanine substitutions at the highly conserved positions 75, 81 and 175 of Tgs1 do not impede m3G cap formation while the growth defects observed for the I83R, S175R, S175E and W178A alleles correlate with the absence of m3G cap structure in snRNAs and snoRNAs.
Figure 9.

Analysis of m3G cap formation in cells carrying Tgs1 point mutants. The tgs1::KAN cells containing the indicated GFP–Tgs1 mutant protein were grown under conditions maintaining the plasmid. Whole-cell extracts were prepared and snRNPs and snoRNPs were immunoprecipitated with anti-m3G antibodies. RNA was extracted from the supernatents (S.), the pellets (P.) and equivalent aliquots of the total lysates (T.), separated on denaturing polyacrylamide gels and subjected to northern analysis. Hybridization was with probes specific for the yeast U4, U5 and U6 snRNAs as well as for the class C/D (U3) and class H/ACA (snR11) snoRNAs.
DISCUSSION
Structure-guided mutagenesis of Tgs1p
Bioinformatic analysis revealed that Tgs1p is exclusively comprised of eukaryotic proteins and that it is closely related to a family comprised of bacterial rRNA:m2G MTases RsmC and RsmD. In fact, among all proteins of known structure, Tgs1 has shown strongest similarity to Mj0882, which is closely related to RsmC and RsmD (34). The structural information of Mj0882 was used to build a homology model of the yeast Tgs1p, which in turn was used to guide mutagenesis experiments. The model is in agreement with the results of the mutagenesis study of two residues implicated in AdoMet binding and demonstrated to be essential for the activity of Tgs1p (16). According to the model, the residue D103 is involved in water-mediated coordination of the methionine moiety of AdoMet, while D126 hydrogen bonds to the 2′- and 3′-OH groups of the ribose. Based on the model of Tgs1p we predicted the range of the minimal globular MTase domain (amino acids 60–262) and the localization of other residues that may be important for catalysis and/or RNA binding.
In the majority of DNA and RNA MTases, the target base is stabilized by interactions with aromatic or aliphatic residues in motifs IV and VIII (reviewed in 35) or motifs IV and X (34). The catalysis is supported by Asp, Asn or Ser residues in motif IV, which were proposed to promote the hybridization change of the nitrogen in the methylated exocyclic amino group from sp2 towards sp3 (40). By analogy to the template structure of the Mj0882 protein and other structures of MTases that modify exocyclic amino groups in nucleic acids, we predicted that the SPPW motif is involved in positioning of the methylatable base (i.e. of the N7-methylated guanosine cap) and in catalysis of the methyltransfer reaction. In addition to the conserved residues in motif IV, we identified two conserved aromatic residues in motif X, namely W75 and F76, of which the former was invariant in all eukaryotic Tgs1 homologs with the exception of Plasmodium (Fig. 1). The nearly invariant W75, aligned with F35 of Mj0882, is suggested to be the second residue important for stabilization of the target base (34).
According to the model proposed in this report, mutagenesis analysis revealed that alanine mutants at positions W75, E81 and S175 were active, while I83R, S175R, S175E and W178A substitutions turned out to be deleterious for catalysis. It cannot be excluded that alanine substitution at position S175 (as well as in positions W75 and E81) affect the catalysis to some extent but, due to the low efficiency of m3G-capped snRNA binding to the anti-m3G antibodies in the immunoprecipitation experiments (Fig. 9), the inhibitory effect of these mutations may be undetectable with this assay. It is also possible that the W75A, E81A and S175A mutants reduce the catalytic activity of Tgs1 and the rate of cap methylation without affecting the steady state levels of capped RNAs. However, the same assay clearly revealed the functional defects of other mutants studied herein.
Alanine mutants of two conserved residues from motif X (W75 and E81) turned out to be active. We interpret this result as an indication that residues in motif X in Tgs1 are not critical for catalysis, binding or correct folding. However, the I83R was deleterious, in agreement with the model, which suggested that the long side chain of Arg in this position would block the active site (Fig. 3, bottom). We propose that the predicted α-helix structure formed by motif X (Figs 1 and 3) participates in formation of the m7G-binding pocket, but is not directly involved in the methylation reaction.
The results obtained for the mutant W178A confirm that this aromatic residue from motif IV is indeed involved in cap binding, as predicted by our model. On the other hand, the dispensability of the hydroxyl group of S175 (i.e. the viability of the alanine mutant with the side chain truncated beyond the Cβ atom) was surprising. This is in striking contrast to all mutagenesis studies on MTases described previously, in which the Ser, Asn or Asp side chain at the corresponding position in motif IV turned out to be essential for catalysis. Nonetheless, the placement of longer side chains of Glu or Arg in this position totally abrogated the enzymatic activity of Tgs1 (as expected for a residue in the heart of the active site). According to our model, these substitutions (like the above-mentioned I83R mutation) may totally block the m7G-binding site (Fig. 3). Interestingly, the analysis of the multiple sequence alignment reveals that the Tgs1 ortholog from Entamoeba histolytica exhibits a ‘natural’ Ser-Ala substitution in motif IV (APPW; Fig. 1). Taken together, the results of our sequence analysis and mutagenesis study support the prediction that the active site of m7G-N2,N2-dimethyltransferases is similar to that of other amino-MTases, but it is unique in that the Ser residue in the SPPW motif is not essential for the MTase activity.
We propose the following alternative explanations of our finding. (i) The postulated hybridization change of the methylated nitrogen atom from sp2 towards sp3 does not require the side chain-mediated assistance in the m7G-N2,N2-dimethylation reaction, possibly due to a different electron structure of the pre-modified base (m7G) as compared with other substrates (typically, unmethylated bases). In this case, the hybridization would be accomplished solely by the backbone oxygen of P176. (ii) It is also possible that the mechanism of snRNA/snoRNA-cap m7G-N2,N2-dimethylation does not involve the hybridization change, which was postulated for amino-MTases based on the DNA:m6A MTase M.TaqI crystal structure (40). Interestingly, a recent theoretical study of amino-methylation, which employed quantum mechanical calculations, suggested that the hydrogen bonding in the active site of M.TaqI cannot promote the hybridization change of the attacking nitrogen and therefore must fulfill some other role (41). It will be interesting to determine if the cap hypermethylation mechanism is different from other amino-methylations of nucleic acids and why the Ser residue is not essential, while it seems essential for other MTases (for instance for DNA:m4C MTases). However, the detailed comparative analyses of amino-MTases acting on RNA and DNA must await both the development of an in vitro biochemical assay for Tgs1 MTase function and the determination of a high-resolution crystal structure of a member of the Tgs1 family.
Functional domains of Tgs1
Database searches with the yeast Tgs1 sequence revealed that the small RNA cap MTase is evolutionarily conserved, with homologs in metazoans, plants and lower eukaryotes (16). Interestingly, proteins from higher eukaryotes have long N-terminal domains while proteins from lower eukaryotes are only composed of the conserved MTase domain with only small N-terminal extensions. In addition, the yeast protein bears a supplementary highly acidic C-terminal domain. Several eukaryotic nucleolar and ribosomal proteins also contain a stretch of acidic clusters at their C-terminal region (42) and it has been shown for human ribosomal protein L22 that such an acidic region regulates the nucleolar localization and ribosome assembly (43). Although it remains to be determined whether these domains play a general role in nuclear or nucleolar retention, we show here that the deletion of the C-terminal acidic cluster of Tgs1p does not affect its localization and its function.
Our previous data established that Tgs1p is localized in the nucleolar compartment and, under particular growth conditions, is concentrated in a small region of the nucleolus called the nucleolar body (16,17). Given that nucleolar body localization is visible only in ∼20% of cells and depends on particular growth conditions (17), we have not determined whether Tgs1 mutants are localized in this subnucleolar compartment. However, by investigating the subcellular localization of Tgs1 deletion mutants, we were able to show that the N-terminal region serves as a nuclear/nucleolar targeting signal. Indeed, deletion of region 1–40 of Tgs1 hinders exclusive nucleolar localization while deletion of region 1–58 dramatically increases cytoplasmic signal (Fig. 7), suggesting that the entire N-terminal region of Tgs1 serves both as a nuclear localization and nucleolar targeting signal. Accordingly, our results show that the N-terminal domain (amino acids 1–58) of Tgs1 possesses nucleolar localization properties and suggest that Tgs1 might be retained in the nucleolus through the interaction of this region with nucleolar factors. To date, no clear consensus sequence has been reached for nucleolar localization signals but a hallmark of such motifs is a long stretch of basic residues. It appears that nucleolar accumulation of proteins occurs by means of functional domains rather than through linear sequences acting as NoLSs (44). Finally, it is interesting to note that corresponding portions of most Tgs1 orthologs also contain numerous lysine and arginine residues exhibiting resemblance to known nuclear import signals (Fig. 1), but whether these domains also possess nuclear localization properties remains to be determined.
The Tgs1 hypermethylase is distinct from other MTases in that its activity (conversion of the m7G cap to m3G) requires previous binding of the MTase to a correctly assembled Sm core complex (2,6). It has been suggested that the SmB/B′ protein facilitates hypermethylation of snRNAs by providing a docking site for the MTase (13,14). This is consistent with the fact that both yeast and human hypermethylases bind preferentially to the carboxyl extensions of yeast and human SmB proteins (15,16). Our data show that neither the N-terminal (1–58) nor C-terminal (276–335) domains of Tgs1 are necessary for SmB binding in vitro, suggesting that the globular MTase domain (amino acids 58–262) possess the determinants that interact with the SmB target. Our results also demonstrate that the same globular domain is also able to associate with the C-terminal tail of the Cbf5 protein, common to the H/ACA box snoRNPs. Thus, given that the hypermethylase associates with protein domains rich in basic amino acids (16), the globular MTase domain might present a negatively charged surface that could signify the determinant responsible for binding to snRNPs and snoRNPs. In this regard, the crystal structure of the conserved core of protein arginine MTase PRMT3 showed that the top surface of the PRMT core is enriched in acidic residues, representing a substrate binding surface (45). Calculation of the electrostatic potential of the Tgs1 surface showed that its surface is mainly positively charged but segments with a negative charge can also be found (see ftp://genesilico.pl/iamb/Tgs1/tgs1.electro.gif). Identification of the precise surface features of Tgs1 that mediate its interaction with the SmB and Cbf5 proteins will require additional experiments and in the absence of the high-resolution crystal structure, the model of the Tgs1 catalytic domain will serve as a useful platform to carry out such analyses.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Edouard Bertrand and Johann Soret for gift of materials, helpful discussions and critical comments on the manuscript. J.M. was supported by a graduate fellowship from the Ministère de l’Education Nationale, de la Recherche et de la Technologie (MENRT) and a fellowship from the Association pour la Recherche sur le Cancer (ARC). J.M.B. was supported by the EMBO/HHMI Young Investigator award and by a fellowship from the Foundation for Polish Science. This work was supported by the Association Française contre les Myopathies (AFM), the ARC and the Centre National de la Recherche Scientifique (CNRS).
REFERENCES
- 1.Yu Y.T., Scharl,E.C., Smith,C.M. and Steitz,J.A. (1999) The growing world of small nuclear ribonucleoproteins. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 487–524. [Google Scholar]
- 2.Will C. and Lührmann,R. (2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol., 13, 290–301. [DOI] [PubMed] [Google Scholar]
- 3.Kiss T. (2002) Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell, 109, 145–148. [DOI] [PubMed] [Google Scholar]
- 4.Filipowicz W. and Pogacic,V. (2002) Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol., 14, 319–327. [DOI] [PubMed] [Google Scholar]
- 5.Kambach C., Walke,S., Young,R., Avis,J.M., de la Fortelle,E., Raker,V.A., Lührmann,R., Li,J. and Nagai,K. (1999) Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell, 96, 375–387. [DOI] [PubMed] [Google Scholar]
- 6.Mattaj I.W. (1986) Cap trimethylation of U snRNA is cytoplasmic and dependent on U snRNP protein binding. Cell, 46, 905–911. [DOI] [PubMed] [Google Scholar]
- 7.Hamm J., Darzynkiewicz,E., Tahara,S.M. and Mattaj,I.W. (1990) The trimethylguanosine cap structure of U1 snRNA is a component of a bipartite nuclear targeting signal. Cell, 62, 569–577. [DOI] [PubMed] [Google Scholar]
- 8.Fischer U. and Lührmann,R. (1990) An essential signaling role for the m3G cap in the transport of U1 snRNP to the nucleus. Science, 249, 786–790. [DOI] [PubMed] [Google Scholar]
- 9.Maxwell E.S. and Fournier,M.J. (1995) The small nucleolar RNAs. Annu. Rev. Biochem., 64, 897–934. [DOI] [PubMed] [Google Scholar]
- 10.Terns M.P. and Dahlberg,J.E. (1994) Retention and 5′ cap trimethylation of U3 snRNA in the nucleus. Science, 264, 959–961. [DOI] [PubMed] [Google Scholar]
- 11.Terns M.P., Grimm,C., Lund,E. and Dahlberg,J.E. (1995) A common maturation pathway for small nucleolar RNAs. EMBO J., 14, 4860–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Y.T., Shu,M.D. and Steitz,J.A. (1998) Modifications of U2 snRNA are required for snRNP assembly and pre-mRNA splicing. EMBO J., 17, 5783–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plessel G., Fischer,U. and Lührmann,R. (1994) m3G cap hypermethylation of U1 small nuclear ribonucleoprotein (snRNP) in vitro: evidence that the U1 small nuclear RNA-(guanosine-N2)-methyltransferase is a non-snRNP cytoplasmic protein that requires a binding site on the Sm core domain. Mol. Cell. Biol., 14, 4160–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raker V.A., Plessel,G. and Lührmann,R. (1996) The snRNP core assembly pathway: identification of stable core protein heteromeric complexes and a snRNP subcore particle in vitro. EMBO J., 15, 2256–2269. [PMC free article] [PubMed] [Google Scholar]
- 15.Mouaikel J., Narayanan,U., Verheggen,C., Matera,A.G., Bertrand,E., Tazi,J. and Bordonné,R. (2003) Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep., 4, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mouaikel J., Verheggen,C., Bertrand,E., Tazi,J. and Bordonné,R. (2002) Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol. Cell, 9, 891–901. [DOI] [PubMed] [Google Scholar]
- 17.Verheggen C., Lafontaine,D.L., Samarsky,D., Mouaikel,J., Blanchard,J.M., Bordonné,R. and Bertrand,E. (2002) Mammalian and yeast U3 snoRNPs are matured in specific and related nuclear compartments. EMBO J., 21, 2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altshul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones D.T., Taylor,W.R. and Thornton,J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci., 8, 275–282. [DOI] [PubMed] [Google Scholar]
- 21.Saitou N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406–425. [DOI] [PubMed] [Google Scholar]
- 22.Bujnicki J.M., Elofsson,A., Fischer,D. and Rychlewski,L. (2001) Structure prediction meta server. Bioinformatics, 17, 750–751. [DOI] [PubMed] [Google Scholar]
- 23.Garner E., Romero,P., Dunker,A.K., Brown,C. and Obradovic,Z. (1999) Predicting binding regions within disordered proteins. Genome Inform., 10, 41–50. [PubMed] [Google Scholar]
- 24.Bujnicki J.M., Feder,M., Radlinska,M. and Rychlewski,L. (2001) mRNA:guanine-N7 cap methyltransferases: identification of novel members of the family, evolutionary analysis, homology modeling and analysis of sequence–structure–function relationships. BMC Bioinformatics, 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosinski J., Cymerman,I.A., Feder,M., Kurowski,M.A., Sasin,J.M. and Bujnicki,J.M. (2003) A ‘Frankenstein’s monster’ approach to comparative modeling: merging the finest fragments of fold-recognition models and iterative model refinement aided by 3D structure evaluation. Proteins, Suppl. 5, 47–54. [DOI] [PubMed] [Google Scholar]
- 26.Lüthy R., Bowie,J.U. and Eisenberg,D. (1992) Assessment of protein models with three-dimensional profiles. Nature, 356, 83–85. [DOI] [PubMed] [Google Scholar]
- 27.Scott W.R.P., Hunenberger,P.H., Tironi,I.G., Mark,A.E., Billeter,S.R., Fennen,J., Torda,A.E., Huber,T., Kruger,P. and van Gunsteren,W.F. (1999) The GROMOS biomolecular simulation program package. J. Phys. Chem., 103, 3596–3607. [Google Scholar]
- 28.Fink G.R. and Guthrie,C. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol., 194, 3–21. [PubMed] [Google Scholar]
- 29.Niedenthal R.K., Riles,L., Johnston,M. and Hegemann,J.H. (1996) Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast, 12, 773–786. [DOI] [PubMed] [Google Scholar]
- 30.Séraphin B. and Kandels-Lewis,S. (1996) An efficient PCR mutagenesis strategy without gel purification [correction of purificiation] step that is amenable to automation. Nucleic Acids Res., 24, 3276–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camasses A., Bragado-Nilsson,E., Martin,R., Séraphin,B. and Bordonné,R. (1998) Interactions within the yeast Sm core complex: from proteins to amino acids. Mol. Cell. Biol., 18, 1956–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bordonné R. (2000) Functional characterization of nuclear localization signals in yeast Sm proteins. Mol. Cell. Biol., 20, 7943–7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bordonné R., Banroques,J., Abelson,J. and Guthrie,C. (1990) Domains of yeast U4 spliceosomal RNA required for PRP4 protein binding, snRNP–snRNP interactions and pre-mRNA splicing in vivo. Genes Dev., 4, 1185–1196. [DOI] [PubMed] [Google Scholar]
- 34.Bujnicki J.M. and Rychlewski,L., (2002) RNA:(guanine-N2) methyltransferases RsmC/RsmD and their homologs revisited—bioinformatic analysis and prediction of the active site based on the uncharacterized Mj0882 protein structure. BMC Bioinformatics, 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fauman E.B., Blumenthal,R.M and Cheng,X. (1999) In Cheng,X. and Blumenthal,R.M. (eds), S-Adenosylmethionine-Dependent Methyltransferases: Structures and Functions. World Scientific Inc., Singapore, pp. 1–38. [Google Scholar]
- 36.Hatanaka M. (1990) Discovery of the nucleolar targeting signal. Bioessays, 12, 143–148. [DOI] [PubMed] [Google Scholar]
- 37.Boulikas T. (1993) Nuclear localization signals (NLS). Crit. Rev. Eukaryot. Gene Expr., 3, 193–227. [PubMed] [Google Scholar]
- 38.Meier U.T. (1996) Comparison of the rat nucleolar protein nopp140 with its yeast homolog SRP40. Differential phosphorylation in vertebrates and yeast. J. Biol. Chem., 271, 19376–19384. [PubMed] [Google Scholar]
- 39.Mumberg D., Muller,R. and Funk,M. (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res., 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goedecke K., Pignot,M., Goody,R.S., Scheidig,A.J. and Weinhold,E. (2001) Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nature Struct. Biol., 2, 121–125. [DOI] [PubMed] [Google Scholar]
- 41.Newby Z.E., Lau,E.Y. and Bruice,T.C. (2002) A theoretical examination of the factors controlling the catalytic efficiency of the DNA-(adenine-N6)-methyltransferase from Thermus aquaticus. Proc. Natl Acad. Sci. USA, 99, 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wool I.G., Chan,Y.L. and Gluck,A. (1995) Structure and evolution of mammalian ribosomal proteins. Biochem. Cell Biol., 73, 933–947. [DOI] [PubMed] [Google Scholar]
- 43.Shu-Nu C., Lin,C.H. and Lin,A. (2000) An acidic amino acid cluster regulates the nucleolar localization and ribosome assembly of human ribosomal protein L22. FEBS Lett., 484, 22–28. [DOI] [PubMed] [Google Scholar]
- 44.Liu J.L., Lee,L.F., Ye,Y., Qian,Z. and Kung,H.J. (1997) Nucleolar and nuclear localization properties of a herpesvirus bZIP oncoprotein, MEQ. J. Virol., 7, 3188–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X., Zhou,L. and Cheng,X. (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J., 19, 3509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]