

Abstract
The dihydrolipoamide acyltransferase (E2b) component of the branched-chain α-ketoacid dehydrogenase complex forms a cubic scaffold that catalyzes acyltransfer from S-acyldihydrolipoamide to CoA to produce acyl-CoA. We have determined the first crystal structures of a mammalian (bovine) E2b core domain with and without a bound CoA or acyl-CoA. These structures reveal both hydrophobic and the previously unreported ionic interactions between two-fold-related trimers that build up the cubic core. The entrance of the dihydrolipoamide-binding site in a 30-Å long active-site channel is closed in the apo and acyl-CoA-bound structures. CoA binding to one entrance of the channel promotes a conformational change in the channel, resulting in the opening of the opposite dihydrolipoamide gate. Binding experiments show that the affinity of the E2b core for dihydrolipoamide is markedly increased in the presence of CoA. The result buttresses the model that CoA binding is responsible for the opening of the dihydrolipoamide gate. We suggest that this gating mechanism synchronizes the binding of the two substrates to the active-site channel, which serves as a feed-forward switch to coordinate the E2b-catalyzed acyltransfer reaction.
Keywords: branched-chain α-ketoacid dehydrogenase complex, cubic-core assembly, ionic clamping interaction, maple syrup urine disease, synchronized substrate gating
Introduction
The mitochondrial branched-chain α-ketoacid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC) and 2-oxoglutarate dehydrogenase complex (OGDC) are a family of highly conserved macromolecular catalytic machines (Reed and Hackert, 1990; Reed, 2001). These complexes catalyze multistep reactions (Reaction 1) that lead to the oxidative decarboxylation of α-ketoacids giving rise to acetyl-CoA, acyl-CoA or succinyl-CoA. These products are subsequently utilized in the Krebs cycle.
Inherited defects in these complexes produce severe metabolic diseases in humans; for example, maple syrup urine disease (MSUD) involves defective BCKDC (Chuang and Shih, 2001) and lactic acidemia results from mutated PDC (Robinson and Sherwood, 1984). Both metabolic disorders are manifested by often-fatal α-ketoacidosis, neurological derangement and mental retardation. In addition, the association of OGDC with Alzheimer's disease and Parkinson's disease has been reported (Mizuno et al, 1994; Kish, 1997). The core components of α-ketoacid dehydrogenase complexes are the major autoantigens in primary biliary cirrhosis (Yeaman et al, 2000).
The BCKDC is a 4-MDa catalytic machine organized around a core scaffold consisting of dihydrolipoamide acyltransferase (E2b, with ‘b' for branched-chain) subunits, to which multiple copies of branched-chain α-ketoacid dehydrogenase complex (E1b) and dihydrolipoamide dehydrogenase (E3) are noncovalently tethered (Pettit et al, 1978). This architectural design is conserved in PDC and OGDC with specific pyruvate dehydrogenase (E1p) and acetyltransferase (E2p) for the former complex and 2-oxoglutarate dehydrogenase (E1o) and succinyltransferase (E2o) for the latter (Reed, 2001). When examined by electron microscopy, the E2b of BCKDC and the E2o of OGDC from all species, and the E2p of PDC from Gram-negative bacteria exhibit a 24-meric cubic core, whereas the E2p's of PDC from Gram-positive bacteria, fungi and mammals show a 60-meric dodecahedral scaffold (Oliver and Reed, 1982). In the eukaryotic BCKDC, two regulatory enzymes, that is, the BCKD kinase (Chuang et al, 2002) and presumably the BCKD phosphatase also bind to the E2b core, which control BCKDC activity by reversible phosphorylation (Harris et al, 1997).
The E2b subunit of BCKDC consists of an N-terminal lipoyl-bearing domain (LBD), an internal subunit-binding domain (SBD) and a C-terminal core (catalytic) domain (E2bCD) (Griffin et al, 1988; Lau et al, 1991). The E2bCD possesses a catalytic site for the acyltransfer reaction and is responsible for the formation of the cubic core. The LBD and SBD are tethered to the E2bCD through flexible linker regions with E1b and E3 binding to the same SBD in a mutually exclusive manner (Lessard et al, 1996). In BCKDC, as in PDC and OGDC, LBD plays an important role in connecting distinct reaction steps catalyzed by the E1b, E2b and E3 components. A lipoylamide moiety covalently attached to LBD (Lip-LBD) of E2b serves as a ‘swinging arm' to extract an acyl group from the E1b active site. The resultant S-acyldihydrolipoamide moiety on LBD is transferred to the E2b active site (the ‘swinging arm' active-site coupling mechanism) (Koike et al, 1963; Perham, 2000), where the acyl group is conjugated with CoA, producing acyl-CoA by the reversible E2bCD-catalyzed acyltransfer reaction (in the forward direction of Reaction 2).
Finally, the reduced Lip-LBD visits the E3 active site, where the dihydrolipoamide [Lip(SH)2] moiety on LBD is reoxidized, with NAD+ as the ultimate electron acceptor.
High-resolution crystal structures of full-length E2's have not been deciphered presumably owing to the flexibility of the linker segments. However, a significant amount of structural information has been obtained for individual domains of E2. The NMR structures of LBD and SBD have been reported (Perham, 2000; Reed, 2001). The crystal structures of cubic E2CD cores from Azotobacter vinelandii PDC (E2pCD) and Escherichia coli OGDC (E2oCD) as well as dodecahedral E2pCD cores from Bacillus stearothermophilus and Enterococcous faecalis PDCs have been determined (Mattevi et al, 1992; Knapp et al, 1998; Izard et al, 1999). These E2CD structures corroborated the proposition, based on sequence similarities, that the topology of E2CD is analogous to that of E. coli chloramphenicol acetyltransferase (CAT) (Guest, 1987; Leslie et al, 1988). E2CD forms a disk-shaped homotrimer with a pseudo three-fold axis in the center. Each of the eight or 20 trimers of E2CDs occupies one vertex of a hollow cube (24-mer) with octahedral 432 symmetry, or a dodecahedron (60-mer) with icosahedral 532 symmetry.
In the forward acyltransfer reaction catalyzed by E2bCD (Reaction 2), the two substrates CoA and the S-acyldihydrolipoamide moiety on LBD bind to the active site of E2bCD. The A. vinelandii E2pCD structure has shown that the CoA substrate binds to one entrance of the 30 Å long active-site channel formed by two three-fold-related subunits within a trimer. The second substrate, LBD-free dihydrolipoamide, binds to the opposing entrance of the channel facing the outside of the cubic core (Mattevi et al, 1993). The invariant histidine residue (His391′ in bovine E2bCD) located in the center of the active-site channel functions as a general-base catalyst to abstract a proton from CoA for activation of the substrate (Russell and Guest, 1990; Meng and Chuang, 1994; Hendle et al, 1995). However, the regulation of this bimolecular reaction in the active-site channel of the E2CDs has not been elucidated.
The mammalian BCKDC degrades branched-chain α-ketoacids derived from the branched-chain amino acids leucine, isoleucine and valine with the concomitant conversion of CoA to isovaleryl-CoA, α-methylbutyryl-CoA and isobutyryl-CoA (IB-CoA), respectively (Pettit et al, 1978). MSUD mutations have been identified in the E1b-α, E1b-β, E2b and E3 subunits of the human BCKDC (Chuang and Shih, 2001). The lack of a mammalian E2bCD structure has hampered the understanding of how MSUD mutations impede the function and assembly of the E2b core of human BCKDC. The available structures of cognate E2pCD and E2oCD are from bacteria, which share less than 30% sequence identity with their human counterpart.
In the present study, we determined the crystal structure of the cubic core of bovine E2bCD, which shares 93% sequence identity with the human homolog. Our structural and biochemical data describe, for the first time, adverse effects of certain MSUD mutations on the assembly and catalysis of E2bCD. Significantly, the structures of bovine E2bCD reveal a novel gating mechanism in which the binding of CoA to one entrance of the active-site channel synchronizes the opening of the otherwise closed gate on the opposite end for S-acyldihydrolipoamide binding. The CoA-induced gate opening results in markedly increased affinity of E2bCD for dihydrolipoamide as measured by isothermal titration calorimetry (ITC). To our knowledge, this synchronized substrate-gating mechanism is the first example of regulating a bimolecular reaction through distant communication between the two-substrate entrances of the same active-site channel. We propose that this synchronized gating mechanism serves as a feed-forward switch to coordinate and maintain the catalytic efficiency of the E2b-catalyzed acyltransfer reaction. This molecular switch is likely a common feature in the catalytic cores of mammalian α-ketoacid dehydrogenase complexes.
Results and discussion
Monomer and oligomer structures of bovine E2bCD
We determined four structures of bovine E2bCD (residues 162–421): an apo form at 2.7 Å resolution, two CoA-bound forms (produced with both a short 15-min and a long overnight soak) at 2.7 and 2.17 Å, respectively, and a product isobutyryl-CoA (IB-CoA)-bound form at 2.5 Å (Supplementary Table SI). In all structures, the linker region between SBD and E2bCD (residues 162–187) is disordered. Eight monomers of E2bCD were found in the asymmetric unit of the crystal: two trimers and two monomers, each of the latter from one of the two additional trimers that are related by crystallographic symmetry. The structures of these eight monomers in the same crystal are essentially identical (the r.m.s. deviations are less than 0.4 Å for all Cα atoms), except the monomers in the E2bCD-CoA (long soak) structure.
The overall structure of bovine E2bCD monomer is similar to those of E. coli E2oCD (r.m.s.d=1.3 Å for the corresponding 217 Cα atoms) and A. vinelandii E2pCD (r.m.s.d=1.2 Å for the corresponding 223 Cα atoms) (Figure 1A and B) (Mattevi et al, 1992; Knapp et al, 1998), despite the less than 30% sequence identities between these core domains (Figure 2). Chloramphenicol acetyltransferase (CAT) has a similar topology to those of E2CDs (Leslie et al, 1988), and the r.m.s.d between E2bCD and CAT is 1.7 Å for the corresponding 153 Cα atoms (Figure 1B).
Figure 1.

Monomer and oligomer structures of bovine E2bCD. (A) Monomer structure of bovine E2bCD. The bound CoA is shown as a stick model. (B) Stereo diagram of the monomer structures of E. coli E2oCD (blue, PDB code 1E2O) (Knapp et al, 1998), A. vinelandii E2pCD (green, 1EAA) (Mattevi et al, 1992) and E. coli CAT (gray, 3CLA) (Leslie et al, 1988) superimposed on bovine E2bCD (red). The bound CoA of E2bCD is shown as a stick model in red. (C) Trimer structure of bovine E2bCD. Each subunit is differently colored. A dashed line (left panel) and a small filled triangle (right panel) indicate a pseudo three-fold axis of the trimer. The bound CoA molecules are depicted as space-filling models. (D) The 24-mer cubic structures of bovine E2bCD (left), E. coli E2oCD (center) and A. vinelandii E2pCD (right) shown as space-filling models. For clarity, one of the eight trimers in the cube is colored the same as (C). White ovals highlight the trimer–trimer interactions that dictate the cubic core assembly.
Figure 2.

Sequence alignments and secondary structure assignments of the three E2CD's. The sequence alignments of bovine E2bCD, E. coli E2oCD and A. vinelandii E2pCD were performed with Clustal W (Thompson et al, 1994) and modified with ESPript (Gouet et al, 1999); red box: identical residues, yellow box: conserved, open box: semi-conserved. Secondary structures and residue numbers for each protein are indicated above and below the alignments, respectively, with the same color coordination as in the protein nomenclatures. Triangles immediately above aligned residues depict the following functions or effects in bovine E2bCD: black, CoA binding; red, MSUD mutations; blue, active-site residues; orange, hydrophobic-socket residues for the trimer–trimer interaction; magenta, gatekeepers for the dihydrolipoamide-binding site.
Like bacterial E2CDs, three monomers of bovine E2bCD assemble into a trimer of 61 Å in height and 63 Å in diameter, whose monomers are related by a single three-fold rotational axis (Figure 1C). The subunit interfaces between one E2bCD monomer and the other two are 2900 Å2, including 47 hydrogen bonds (H-bonds). These values are comparable to those of E. coli E2oCD (subunit interface: 2740 Å2, 52 H-bonds and six salt bridges) (Knapp et al, 1998). A. vinelandii E2pCD has a more extended N-terminal region that crosses over a neighboring subunit from the edge to the center of the trimer, thereby providing the largest subunit interface for trimer formation (3900 Å2, 48 H-bonds involved) among the three E2 core structures (Mattevi et al, 1992).
The cubic core of bovine E2bCD generated with crystallographic symmetry-related molecules is similar to those of bacterial E2CDs in both the shape and dimensions (∼130 Å edge) (Figure 1D). Eight trimers of E2bCD, which are building blocks of the cube, are located at corners of the cube with octahedral 432 symmetry. The trimer–trimer interactions are accomplished solely through C-terminal contacts between the 2-fold-related subunits, each from a separate neighboring trimer. Each octahedral face of these cubes has a large rectangular hole, which provides the passage for CoA to approach the CoA-binding site inside the cubic core (Mattevi et al, 1992).
Distinct trimer–trimer interactions in bovine E2bCD
In the two-fold trimer–trimer interface of bovine E2bCD, the two C-terminal helices (H7 and H7′) from respective monomers of the two interacting trimers line up with each other in the center of the interface (Figure 3A and B). The side chain of Leu418 (or Leu418′) from each helix projects, like a ‘knob', into a hydrophobic ‘socket' of the opposing subunit, as observed in E. coli E2oCD and A. vinelandii E2pCD (Figure 3 and Supplementary Figure S1) (Mattevi et al, 1992; Knapp et al, 1998). This ‘socket' in bovine E2bCD is formed by hydrophobic residues Leu227′, Leu233′, Ile236′, Leu244′, Phe249′ and Phe307′. Residues Leu417 or Leu417′ confer additional hydrophobic interactions between the two C-terminal helices. Removal of the two C-terminal leucine residues in porcine E2o corresponding to Leu417 and Leu418 of bovine E2b completely prevents cubic-core formation (Koike et al, 2000). Many of the residues in the hydrophobic ‘knob-and-socket' interactions are conserved in bacterial E2oCD and E2pCD (Figure 2). However, among the three E2CDs, bovine E2bCD possesses the largest interface area between trimers (1161 Å2).
Figure 3.

Trimer–trimer interactions in the bovine E2bCD cubic core. (A) Hydrophobic contacts in the ‘knob-and-socket' interactions between two-fold-related subunits (blue and green) from each neighboring trimer in the bovine E2bCD core. The residues in the hydrophobic sockets are colored in orange. (B) The same trimer–trimer interface shown in (A) with the residues forming H-bonds and ion pairs as stick models. (C) Pattern diagrams showing trimer–trimer interactions in the cubic cores of the bovine E2bCD (left), E. coli E2oCD (middle) and A. vinelandii E2pCD (right). The protruding C-terminal helix is labeled with a white letter C. The hydrophobic ‘knob-and-socket' interface is colored in orange. White lines represent H-bonds and ion pairs. (D) Sedimentation coefficients of wild-type and mutant bovine E2bCDs. The sedimentation coefficients (s20,w) of the wild-type (red) and R240C mutant (blue) are 17.1S and 17.3S, respectively, which are close to the calculated coefficient (17.5S) from the theoretical molecular mass of the 24-mer E2bCD cubic core. The sedimentation coefficients of the K252N (green) and ΔC2 (orange) mutants are both 4.4S. When modeled as discrete species, the calculated masses of the mutants agree well with the theoretical mass of a E2bCD trimer.
A distinct feature in bovine E2bCD is that many H-bonds and salt bridges contribute to additional trimer–trimer interactions (Figure 3B). Notably, the C-terminal residue Lys421 makes H-bonds with Lys421′ as well as Lys252′ and Asn302′ from the other subunit, with the latter residues forming a ‘clamp' holding the backbone of Lys421. As a result of the symmetrical interactions, the two clamps provided by Lys421 and Lys421′ practically embrace each other. Thus, in addition to the hydrophobic ‘knob-and-socket' interactions, bovine E2bCD employs the second ionic clamping mechanism to augment the trimer–trimer contacts, which we term ‘clamp-and-socket' interactions (Figure 3C).
The significance of the clamping mechanism is corroborated by the K252N MSUD mutation (Chuang and Shih, 2001) that eliminates one H-bond for Lys421 in bovine E2bCD (Figure 3B). This mutation prevents formation of the cubic E2bCD core, resulting in an exclusively trimeric species, as indicated by a sedimentation coefficient of 4.4S (Figure 3D). Similarly, when the last two C-terminal residues (Leu420 and Lys421) are truncated, E2bCD (ΔC2) is no longer able to form the cubic core and shows the same sedimentation coefficient. In contrast, the R240C MSUD mutation (Chuang and Shih, 2001) that abolishes a salt bridge with Asp419′ in the trimer–trimer interface (Figure 3B) does not affect the cubic assembly of E2bCD, judging from its sedimentation coefficient of 17.3S, which is similar to the wild type (17.1S) (Figure 3D). The presence of a His-tag at the C-terminus of bovine E2bCD also does not impede the formation of the cubic core (unpublished data). In contrast, the introduction of a C-terminal His-tag in E. coli E2oCD produces exclusively trimers (Knapp et al, 2000). Through an apparent steric hindrance, the cubic core of the A. vinelandii E2p dissociates upon binding of either E1p or E3 (Bosma et al, 1984). These results suggest that the clamp interactions between the two C-terminal lysine residues are critical to the increased stability of the bovine E2bCD core, compared to its bacterial counterparts.
Two different conformations between the bound CoA and the bound acyl-CoA
In the bovine E2bCD-CoA (short soak) structure, all eight monomers in the asymmetric unit bind an intact CoA with its pantetheine tail extended in the active-site channel (Figure 4A). The bound CoA is held in place by as many as 12 direct and five water-mediated (indirect) H-bonds. Arg230 is highly conserved in the E2 family (Figure 2); this residue forms an H-bond with the 3′-phosphate group of the bound CoA. The R230G MSUD mutation (Chuang et al, 1997) increases the Kd of E2bCD for CoA by 10-fold, and reduces the kcat/Km by six-fold over the wild type (Table I). Asn339 makes three direct H-bonds to the adenine ring and pyrophosphate of the bound CoA. In the majority of E2bCDs and E2pCDs from Gram-negative bacteria, this residue is replaced by a serine, whose side chain is not able to form an H-bond with CoA, as is the case with Ser559 in A. vinelandii E2pCD (Figure 2) (Mattevi et al, 1993). Furthermore, Gln317, which is present only in the mammalian E2bCD (Figure 2), forms direct and indirect H-bonds with the pyrophosphate moiety of the bound CoA. This position in all other E2CDs is occupied by a glycine, alanine or serine residue, whose side chains are too short to form H-bonds with CoA. These structural properties suggest that the bovine E2bCD may have evolved the increased specificity and affinity for CoA over the prokaryotic E2 cores.
Figure 4.
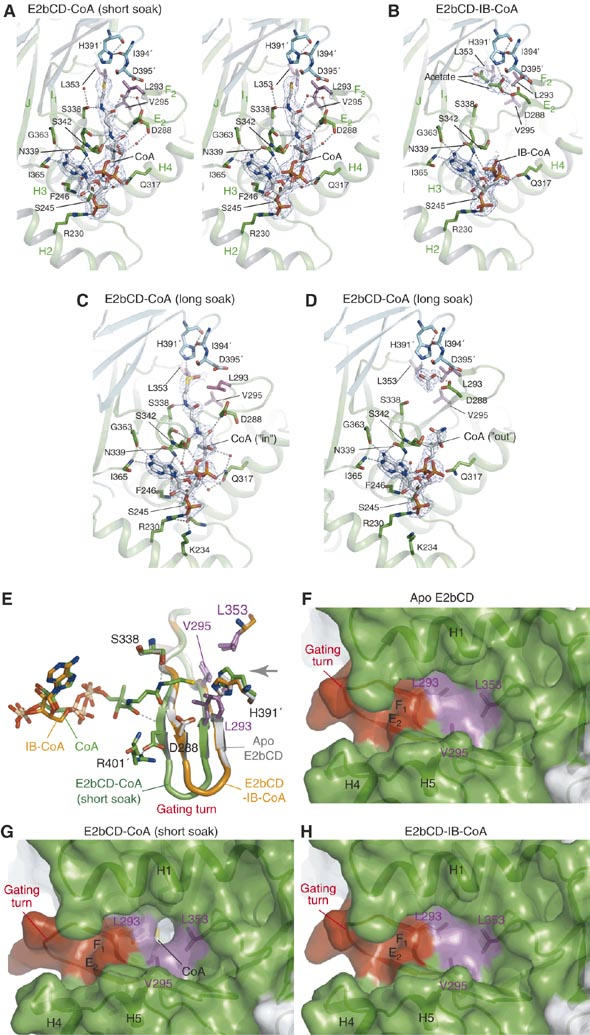
A CoA-mediated gating mechanism for synchronized dihydrolipoamide binding in the active-site channel of bovine E2bCD. (A) Stereo view of the CoA-binding site of E2bCD-CoA (short soak). The bound CoA in the ‘in' conformation and its surrounding residues are shown in stick models. Residues from the three-fold-related subunit are shown in cyan. The residues in magenta are the gatekeepers at the dihydrolipoamide entrance of the active-site channel. Water molecules are shown as small spheres. The 2Fo-Fc density of the bound CoA is superimposed at a 1-σ contour level. (B) The bound IB-CoA in the ‘out' conformation in E2bCD-IB-CoA. The 2Fo-Fc densities are at the 1-σ level. (C) The oxidized CoA in the ‘in' conformation in E2bCD-CoA (long soak). Three of the eight E2bCD monomers in the asymmetric unit of this crystal show this structure. The omit density of the oxidized CoA was at the 1-σ level. (D) The disordered pantetheine tail of the CoA (the ‘out' conformation) bound to the remaining five monomers in E2bCD-CoA (long soak). The 2Fo-Fc density is contoured at the 1-σ level. (E) Superimposed gating turns and bound cofactors; white: apo, green: E2bCD-CoA (short soak), orange: E2bCD-IB-CoA. The bound CoA (carbon atoms in green) in E2bCD-CoA (short soak) is in the ‘in' conformation, whereas the pantetheine tail of the bound IB-CoA (carbons in orange) in E2bCD-IB-CoA is disordered (the ‘out' conformation). The gray arrow indicates the direction of the dihydrolipoamide-binding site. The gatekeeper residues are in magenta. (F) Surface model of the closed dihydrolipoamide gate in the apo E2bCD structure. Three gatekeeper residues (Leu293, Val295 and Leu353) are colored in magenta and the gating turn in red. (G) The open gate for the dihydrolipoamide-binding site in E2bCD-CoA (short soak). The thiol group of the bound CoA in the ‘in' conformation is shown in yellow. (H) The closed dihydrolipoamide gate in E2bCD-IB-CoA.
Table 1. CoA-binding and kinetic constants of wild-type and mutant E2bCDs.
| CoA bindinga | IV-CoA bindinga | Kineticsb | Oligomeric statec | Mutant category | |||||
|---|---|---|---|---|---|---|---|---|---|
| Protein |
Kd (μM) |
ΔG (kcal/mol) |
Kd (μM) |
ΔG (kcal/mol) |
kcat (s−1) |
Km (μM) |
kcat/Km (s−1 μM−1) |
|
|
| Wild type | 3.0±0.35 | −7.5 | 6.8±0.35 | −7.1 | 1874±122 | 219±14 | 8.6 | 24 | — |
| R230Gd | 29±2.5 | −6.1 | — | — | 1294±56 | 1349±58 | 0.95 | 24 | CoA binding |
| R240Cd | 9.1±1.6 | −6.7 | — | — | 1713±89 | 222±11.5 | 7.7 | 24 | Trimer–trimer |
| K252Nd | 11±1.2 | −6.6 | — | — | 818±58 | 340±24 | 2.4 | 3 | Trimer–trimer |
| ΔC2 | 5.0±0.71 | −7.3 | — | — | 2019±161 | 211±16 | 9.5 | 3 | Trimer–trimer |
| L293A | 25±1.4 | −6.6 | — | — | 347±23 | 1005±61 | 0.35 | 24 | Gating |
| D288A | 13±1.1 | −6.5 | — | — | 828±68 | 386±18 | 2.1 | 24 | Gating |
| All measurements for CoA or IV-CoA binding and kinetics were in triplicate. | |||||||||
| Kd and ΔG for CoA or IV-CoA were measured by ITC. | |||||||||
| Kinetics was measured in the reverse direction of the acyltransfer reaction (Reaction 2) with dihydrolipoamide and IV-CoA as substrates. Km is for IV-CoA. | |||||||||
| Oligomeric states were judged by gel filtration and designated as follows: 24: 24-mer, 3: trimer. | |||||||||
| MSUD mutations. | |||||||||
The extended conformation of the bound CoA in bovine E2bCD-CoA (short soak) (Figure 4A) is similar to the ‘in' conformation previously reported in the A. vinelandii E2pCD structure (Mattevi et al, 1993). In this conformation, the thiol group of the bound CoA is poised for activation by the catalytic residue His391′. A conserved serine residue Ser338 forms an H-bond with a peptide nitrogen atom in the pantetheine tail. Ser338 was shown to be involved in the acyltransfer reaction presumably through stabilization of the tetrahedral intermediate (Meng and Chuang, 1994). On the other hand, in the structure of bovine E2bCD-IB-CoA, the pantetheine tail of the bound IB-CoA, which is a substrate for the reverse acyltransfer reaction (Reaction 2), is largely disordered, with several acetate molecules from the crystallization buffer occupying the active-site channel (Figure 4B). Acetate at the concentration present in the crystallization buffer (250 mM) has minimal effects on the binding of IV-CoA to bovine E2bCD as determined by ITC (data not shown). This suggests that the disordering of the pantetheine tail is not caused by the presence of acetate ions in the crystal. We refer to the partially disordered conformation of the bound IB-CoA as the ‘out' conformation, which is reminiscent of the compact conformation of the bound CoA with the folded pantetheine tail observed outside the active-site channel of A. vinelandii E2pCD (Mattevi et al, 1993).
A novel CoA-mediated gating mechanism for the dihydrolipoamide-binding site
The entrance of the dihydrolipoamide-binding site in the active-site channel opposing the CoA-binding site is open in all reported structures of A. vinelandii E2pCD (apo, CoA-bound and dihydrolipoamide-bound forms) as well as in the apo structure of E. coli E2oCD (Supplementary Figure S2) (Mattevi et al, 1993; Knapp et al, 1998). Surprisingly, in the structures of the apo and IB-CoA-bound bovine E2bCD, the entrance for dihydrolipoamide is completely closed (Figure 4F and H), whereas the same entrance is open in the E2bCD-CoA (short soak) structure (Figure 4G). The gate at the dihydrolipoamide entrance is formed by Leu293, Val295 and Leu353 as well as part of the helix H1. In the closed-gate state, there are hydrophobic interactions between the side chains of these gatekeeper residues clustered at the channel entrance (Figure 4F and H). Gate opening is brought about by the conformational change on a β-hairpin turn that runs from strands E2 to F1 (designated as the gating turn). This gating turn is part of the active-site channel separating the two opposite entrances for CoA and dihydrolipoamide. When the dihydrolipoamide gate is open, the gating turn moves toward the CoA-binding site by maximally ∼2.5 Å with a concomitant shift and flip for the Leu293 and Asp288 side chains, respectively, which are located on this turn (Figure 4E).
The open- and closed-gate conformations at the dihydrolipoamide-binding site are correlated with the conformation of the bound CoA. All monomers in bovine E2bCD-CoA (short soak) with the open gate bind CoA in the ‘in' conformation (Figure 4A and E). The gating turn in the open-gate conformation is stabilized by several direct and indirect H-bonds between the pantetheine tail of the bound CoA and Asp288 on the gating turn, as well as by a salt bridge between the side chains of Asp288 and Arg401′. In contrast, E2bCD monomers with a closed-gate conformation are either in the apo form or with the bound IB-CoA in the ‘out' conformation (Figure 4E). The absence of interactions between the pantetheine tail and the gating turn apparently locks the gate in the closed conformation.
Two conformations of the bound CoA in the E2bCD-CoA structure produced by long soak
Among the eight monomers in the asymmetric unit of bovine E2bCD-CoA (long soak), two monomers have an open gate for dihydrolipoamide with the bound CoA in the ‘in' conformation (Figure 4C and Supplementary Figure S3B). The remaining five monomers have a closed gate with the bound CoA in the ‘out' conformation (Figure 4D and Supplementary Figure S3C). These results further support the correlation between the conformations of the gating turn and the bound CoA. In addition, the presence of two different conformations for the bound CoA in the same crystal provides insight into the reaction mechanism of E2bCD. In the structure of E2bCD-CoA (short soak), all of the bound CoAs are intact and assume the ‘in' conformation (Figure 4A). The bound CoA in E2bCD-CoA (long soak) is oxidized, similar to the bound CoA in the A. vinelandii E2pCD structure (Figure 4C and Supplementary Figure S2C) (Mattevi et al, 1993). We hypothesize that, during the prolonged soaking, the thiol group of the bound CoA is activated by the catalytic His391′ residue, resulting in the oxidation of the sulfur atom. As a consequence, the bulk of the bound CoAs are transformed from the ‘in' conformation to the ‘out' conformation in the structure of E2bCD-CoA (long soak) (Figure 4D and Supplementary Figure S3A). These results suggest that once the thiol group of the bound CoA is oxidized, the lengthened pantetheine tail imparts steric collisions between the modified thiol group and residues in the active-site channel, leading to a weaker binding of the oxidized CoA to the active-site channel. This hypothesis is reinforced by the E2bCD-IB-CoA structure, in which the bound IB-CoA with its thiol group modified by the bulky acyl side-chain is maintained in the ‘out' conformation (Figure 4B). The relatively high B-factors for the atoms in the bound IB-CoA in this structure (94.7 Å2, Supplementary Table SI) may be due to low occupancy of the bound ligand, resulting from the low affinity of E2bCD for acyl-CoA (Table I). Different conformations observed with oxidized CoA in E2bCD-CoA (long soak) are further discussed in Supplementary Figure S3.
CoA binding markedly increases affinity of E2bCD for dihydrolipoamide
The model of dihydrolipoamide-bound bovine E2bCD in the open-gate conformation, based on the structure of the dihydrolipoamide-bound A. vinelandii E2pCD, shows that the gate opening is large enough to accommodate the alkyl chain of dihydrolipoamide (Supplementary Figure S2E). Together with the above results, we propose that the bound CoA in the ‘in' conformation promotes gate opening to facilitate the dihydrolipoamide entry into the active-site channel. To validate this gating mechanism, the binding affinity of bovine E2bCD for dihydrolipoamide, an LBD-free substrate analog for the reverse acyltransfer reaction, was measured by ITC in both the absence and presence of CoA (Table II). In the absence of CoA, the binding of dihydrolipoamide to the wild-type E2bCD is not detected, but in the presence of 0.5 mM CoA, the binding affinity for dihydrolipoamide is markedly increased (Kd=48 μM) (Supplementary Figure S4A). We also measured the binding affinity for Lip-LBD, which is an LBD-containing analog of dihydrolipoamide. Similar to the above results, Lip-LBD showed no detectable binding to E2bCD without CoA, and the presence of CoA strikingly increases the binding affinity of E2bCD for Lip-LBD (Kd=11 μM) (Supplementary Figure S4B).
Table 2. Binding affinity of E2CDs for dihydrolipoamide and Lip-LBD in the absence or presence of cofactors.
| Protein | Cofactor | Kd (μM) for dihydrolipoamide | ΔG (kcal/mol) | Kd (μM) for Lip-LBD | ΔG (kcal/mol) |
|---|---|---|---|---|---|
| Bovine E2bCD | |||||
| Wild type | — | NMa | — | N.M.a | — |
| CoA | 48±7.2 | −5.8 | 11±1.3 | −6.3 | |
| D288A | — | NMa | — | — | — |
| CoA | NMa | — | — | — | |
| L293A | — | 6±1.5 | −7.0 | — | — |
| CoA | 61±6.5 | −5.6 | — | — | |
| H391A | — | NMa | — | — | — |
| CoA | 12±2.5 | −6.6 | — | — | |
| IV-CoAb | 65±6.6 | −5.6 | — | — | |
| Human E2pCD | |||||
| Wild type | — | NMa | — | — | — |
| CoA | 21±5.3 | −5.2 | — | — | |
| All measurements were in triplicate. | |||||
| NM, not measurable. | |||||
| Isovaleryl-CoA. | |||||
As described above, Asp288 on the gating turn is an important residue that stabilizes the open-gate conformation by interacting with the pantetheine tail of the bound CoA in the ‘in' conformation (Figure 4A and E). Dihydrolipoamide binding to the D288A mutant cannot be detected in either the absence or presence of CoA (Table II). Although the binding affinity of the D288A mutant for CoA (Kd=13 μM) is three-fold weaker than that of the wild type (Kd=3 μM) (Table I), under the ITC conditions (0.5 mM CoA, 100 μM protein) for measuring dihydrolipoamide binding, >95% of the mutant protein is expected to bind CoA. Therefore, undetectable binding of the D288A mutant for dihydrolipoamide in the presence of CoA implies that the gate is closed, even though CoA binds to the mutant protein. It is conceivable that without proper interactions between the pantetheine tail and the gating turn, the bound CoA cannot cause the movement of the latter.
In contrast to the D288A mutant, the gatekeeper variant L293A binds to dihydrolipoamide both in the absence (Kd=6 μM) and presence (Kd=61 μM) of CoA (Table II). The differing binding constants suggest that this mutant has two open-gate conformations for dihydrolipoamide binding. The one in the absence of CoA results from the deletion of the L293 side chain. Because the affinity of dihydrolipoamide for this conformation is eight times greater than that for the wild-type E2bCD, this mutant may feature structural rearrangements that enhance dihydrolipoamide binding in addition to opening the gate. The other open-gate conformation induced by CoA binding may be mediated by the shift of the gating turn, similar to the wild type, because Asp288 is not perturbed in this mutant. In the presence of CoA, both the mutant and the wild type (Kd=48 μM) show comparable binding affinities for dihydrolipoamide. However, the kcat of the L293A variant for the reverse acyltransfer reaction is significantly reduced to a level equal to only 19% of the wild type (Table I). The kinetic data strongly suggest that the open-gate conformation of the L293A mutant, in the presence of CoA, is significantly different from the corresponding conformation of the wild type. These observations will be explored by additional biochemical and structural studies of the L293A mutant. Taken together, results of these binding experiments buttress the concept that the closed-gate conformation blocks the entry of dihydrolipoamide into the active-site channel of bovine E2bCD; conversely, the CoA-induced gate opening promotes the binding of dihydrolipoamide to E2bCD.
The affinity of bovine E2bCD for dihydrolipoamide in the presence of IV-CoA, a homolog of IB-CoA and an alternate substrate for the reverse acyltransfer reaction, was determined using the H391A mutant. The inactivation of the catalytic histidine residue in this variant nullifies both the forward and the reverse acyltransfer reaction (Reaction 2) so as to allow the accurate determination of binding constants for dihydrolipoamide by ITC. Similar to the wild type, the H391A mutant has no detectable dihydrolipoamide binding in the absence of a cofactor. The addition of CoA imparts observable dihydrolipoamide binding to the mutant (Kd=12 μM) (Table II). The H391A mutant also binds dihydrolipoamide with nearly wild-type affinity when IV-CoA is present (Kd=65 μM). This occurs despite the finding that the E2bCD-IB-CoA structure shows a closed-gate conformation for dihydrolipoamide (Figure 4H). It is plausible that the active-site channel in E2bCD moves to accommodate the extra atoms in bound IB-CoA, therefore allowing the dihydrolipoamide gate to oscillate between the open and closed conformations. However, the binding of dihydrolipoamide to the H391A mutant is 5.4-fold tighter in the presence of CoA than IV-CoA. The data suggest that the equilibrium in the reversible acyltransfer reaction favors the forward direction.
Synchronized substrate gating regulates the E2bCD-catalyzed acyltransfer reaction cycle
Based on the above structural and binding data, we propose the following reaction cycle for the acyltransfer reaction (Reaction 2) catalyzed by bovine E2bCD (Figure 5). At the onset of the cycle (Step 1), the dihydrolipoamide gate in the active-site channel is closed as captured in the apo E2bCD structure (Figure 4F). When a substrate CoA binds to E2bCD (Step 2), the pantetheine tail of the bound CoA is imbedded inside the active-site channel (the ‘in' conformation), which induces a shift in the gating turn by forming H-bonds between the pantetheine tail and the gating turn (Figure 4A and E). The shift of the gating turn results in the opening of the dihydrolipoamide gate (Figure 4G) that allows the second substrate, S-acyldihydrolipoamide on LBD, to enter the active-site channel (Step 3). The catalytic His391′ abstracts a proton from the thiol group of the bound CoA, and the activated thiol group subsequently attacks the acyl moiety on the S-acyldihydrolipoamide to promote acyltransfer (Russell and Guest, 1990; Meng and Chuang, 1994; Hendle et al, 1995). Once the acyl group is conjugated with CoA, deacylated Lip-LBD dissociates. The acylated pantetheine tail of the product interacts poorly with the enzyme, as evidenced by the E2bCD-IB-CoA structure (Figure 4E and H) and our ITC solution data that indicate weaker binding for IV-CoA than for CoA (Table I). The weaker binding of acyl-CoA may cause the pantetheine tail to shuttle between the ‘in' and ‘out' conformations. This in turn renders the gating turn to oscillate between the open- and closed-gate configurations (Step 4). Under these conditions, the acyl-CoA is released from the active-site channel. The loss of interactions with the tail upon acyl-CoA dissociation results in gate closure, returning the cubic core to the ground state (Step 1) of the reaction cycle.
Figure 5.

The proposed scheme for the bovine E2bCD-catalyzed acyltransfer reaction cycle. The active-site channel of bovine E2bCD is depicted with the CoA-binding site on the left side and the dihydrolipoamide-binding site on the right. The catalytic resides in the channel (His391′ and Ser338) and two important residues on the gating turn (Leu293 and Asp288) are shown. Gray dotted lines: H-bonds, a green oval with ‘Ac': acyl group, yellow dots: thiol groups and red dots: water molecules. See the text for details of each reaction step.
Gating mechanisms in certain enzymes metabolizing small substrates, for example, phosphoenolpyruvate mutase (Liu et al, 2004), D-aminoacylase (Liaw et al, 2003) and cytosine deaminase (Ko et al, 2003), are designed to sequester the bound substrate in a catalytic site away from bulk solvent so as to foster an enzymatic reaction. Other enzymes utilize various gating mechanisms to select for a specific substrate, as exemplified by oxidosqualen cyclase (Oliaro-Bosso et al, 2005), polyunsaturated fatty acid isomerase (Liavonchanka et al, 2006) and monoamine oxidase B (Hubalek et al, 2005). In the present study, we show that bovine E2bCD has evolved a novel gating mechanism that is distinct from those of the enzymes mentioned above. The binding of the first substrate, CoA, to one entrance of the long active-site channel synchronizes the opening of the gate for the second substrate, S-acyldihydrolipoamide, at the opposite end (Figure 5, Step 3). The concerted binding of two substrates may be important for the catalytic efficiency of the bimolecular acyltransfer reaction of E2bCD. Bovine E2bCD slowly hydrolyzes acyl-CoA in the absence of dihydrolipoamide (data not shown). It is therefore conceivable that the E2bCD would deacylate S-acyldihydrolipoamide if it binds to the active-site channel in the absence of CoA. The net result would be the wasteful loss of the acyl group in the physiological forward acyltransfer reaction. Thus, the synchronized binding of CoA and S-acyldihydrolipoamide prevents the loss of the latter reaction intermediate in the active-site coupling between E1b and the E2b catalytic core.
In parallel to the results with bovine E2bCD, the binding of dihydrolipoamide in the absence of CoA to the E2p core from human PDC (E2pCD) cannot be detected by ITC (Table II). At 0.5 mM CoA concentration, human E2pCD binds dihydrolipoamide with a Kd of 21 μM, which is similar to that of the wild-type bovine E2bCD. These results strongly suggest that human E2pCD also employs the CoA-mediated synchronized gating mechanism in the cognate acetyltransfer reaction. On the other hand, the E. coli E2oCD and A. vinelandii E2pCD structures show a persistent open conformation at the entrance for dihydrolipoamide (Supplementary Figure S2) (Mattevi et al, 1992; Knapp et al, 1998). It is possible that these bacterial E2s do not use the synchronized gating system to regulate the acyltransfer reaction in the active-site channel. The prokaryotes may instead adjust the amounts of E2s in response to physiological demands as a means to regulate acyltransferase activities on the global level. Supporting this model is the fact that the concerted expression of enzyme components for E. coli PDC and OGDC is under tight transcriptional control of the respective operons (Quail et al, 1994; Cunningham and Guest, 1998). It is obvious that additional structural and biochemical studies with bacterial E2CDs, like those undertaken here, are needed to address these questions.
Feed-forward switches for Lip-LBD binding: a common regulatory scheme in BCKDC
We have recently reported that the Tyr113-α residue in the human E1b active site serves as a feed-forward switch to coordinate the two E1b-catalyzed half reactions, that is, decarboxylation and reductive acylation (Machius et al, 2006). The binding and decarboxylation of an α-ketoacid in the E1b active site produces a ThDP-carbanion intermediate that induces a conformational change in the Tyr113-α residue. This information is transmitted to the putative LBD-binding region through a ‘switch-turn' that harbors Tyr113-α. This communication results in an enhanced binding of Lip-LBD to foster the subsequent reductive acylation by the lipoyl group of the bound Lip-LBD. In parallel, the present study shows that the gating mechanism in bovine E2bCD functions as a feed-forward switch of its catalytic events. The binding of CoA to one side of the active-site channel of E2bCD promotes the open conformation of the gating turn on the other end to increase binding of dihydrolipoamide or Lip-LBD (Table II and Supplementary Figure S4), augmenting the efficiency of the E2bCD-catalyzed acyltransfer reaction. Thus, both the E1b and E2b components employ a turn region in the respective active site to regulate Lip-LBD binding. In this regard, these two feedforward mechanisms for E1b and E2b catalysis may function as a means to coordinate active-site coupling through Lip-LBD (Koike et al, 1963) that effects substrate channeling in the BCKDC. These molecular switches are likely a common feature in mammalian α-ketoacid dehydrogenase complexes, and may have implications for understanding how other macromolecular catalytic machines function.
Materials and methods
See Supplementary data for ‘Materials', ‘Purification of proteins' and ‘Activity assay'.
Crystallization and structure determination
Wild-type E2bCD was crystallized at 20°C in 1:1 mixture drops between the protein and the well solutions, with the latter containing 100 mM Na-acetate (pH 4.6), 28% (w/v) polyethylene glycol 4000 and 150 mM NH4-acetate. Crystals grew up to 0.15 × 0.15 × 0.2 mm3 in 2 weeks. Crystals were soaked in a harvest buffer containing the well solution and 20 mM DTT, transferred in a cryo-buffer made with the harvest buffer including 12% (v/v) glycerol, and subjected to flash cooling in liquid propane. For long soak with CoA or IB-CoA, crystals were immersed for overnight in the harvest buffer containing 5 mM CoA or 7 mM IB-CoA, respectively, and transferred to the cryo-buffer, followed by flash cooling. For short soak with CoA, crystals were harvested in a buffer containing 100 mM Na-citrate (pH 4.6), 28% (w/v) PEG 4000, 150 mM Na-citrate and 20 mM DTT, and soaked for 15 min in the same buffer containing 12% (v/v) glycerol and 5 mM CoA, followed by flash cooling. Diffraction data were collected at beamline 19ID or 19BM at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). The data were processed with HKL2000 (Otwinowski and Minor, 1997).
The bovine E2bCD structure was solved by the molecular replacement method using Phaser (Storoni et al, 2004). The search template was a trimer model of bovine E2bCD that was built using the homology-modeling program SwissModel (Schwede et al, 2003) based on the published A. vinelandii E2pCD structure (PDB code: 1EAA) (Mattevi et al, 1992). Initially, two trimers of E2bCD were found in the asymmetric unit. After rigid-body refinements with REFMAC5 (Murshudov et al, 1997), the electron-density map was improved using DM (Cowtan and Main, 1996) combined with noncrystallographic symmetry averaging. This improved map clearly showed extra densities for two additional monomers in the asymmetric unit. Models for these two monomers were manually fitted into these extra densities and a rigid-body refinement was again applied. The E2bCD models were manually rebuilt with Coot (Emsley and Cowtan, 2004). During subsequent refinement cycles, CoA, chloride ion, acetate and water molecules were gradually added to the model. In the final models, the N-terminal 28 residues of the protein (residues 162–187) were disordered and not included. Data processing and refinement statistics are summarized in Supplementary Table SI. Molecular graphics for structural representations were generated with PyMOL (DeLano Scientific LLC.).
Binding measurements by ITC
Both MBP-fused wild-type and mutant bovine E2bCD and human E2pCD were dialyzed exhaustively against the ITC buffer containing 50 mM K-phosphate (pH 7.5) and 50 mM KCl. To measure CoA or IV-CoA binding, the solution of 2 mM CoA or IV-CoA in a syringe was injected in 8-μl increments into the reaction cell containing 1.8 ml of 100 μM E2bCD (based on monomer) at 20°C in a VP-ITC microcalorimeter (MicroCal, Northampton, MA). Titrations for dihydrolipoamide or Lip-LBD binding were carried out by consecutive 8-μl injections of reduced dihydrolipoamide (10–20 mM) or Lip-LBD (1.2 mM) into the reaction cell containing 1.8 ml of 100 μM bovine E2bCD or human E2pCD. To measure dihydrolipoamide or Lip-LBD binding in the presence of CoA or IV-CoA, both the syringe and cell include 0.5 mM CoA or IV-CoA. Binding constants (Kd) and free energy (ΔG) were calculated using ORIGIN v7.0 (MicroCal). The concentration of Lip-LBD was determined by absorbance at 280 nm using the calculated extinction coefficient of 1.07 mg/ml.
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out in a Beckman XL-1 ultracentrifuge using an An60-Ti rotor. Sedimentation (40 000 r.p.m.) of the proteins (0.3 mg/ml) in the ITC buffer at 20°C was monitored using absorbance at 280 nm. The data were analyzed by calculating sedimentation coefficient (S20,w) distributions c(s) using SEDFIT and SEDPHAT (Schuck, 2000). The partial specific volume of bovine E2bCD, buffer viscosity and buffer density were calculated using SEDNTERP (www.rasmb.bbri.org).
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 2IHW, 2II3, 2II4 and 2II5.
Supplementary Material
Supplementary Materials and Methods
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Acknowledgments
We are indebted to Drs Mischa Machius and Diana Tomchick in the Structural Biology Laboratory for the collection of synchrotron data. Use of the Argonne National Laboratory Structural Biology Center beam lines at the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research under Contract No. W-31-109-ENG-38. This work was supported by Grants DK26758 and DK62306 from the National Institutes of Health and Grant I-1286 from the Welch Foundation. We declare that there is no financial conflict of interest related to this work.
References
- Bosma HJ, De Kok A, Van Markwijk BW, Veeger C (1984) The size of the pyruvate dehydrogenase complex of Azotobacter vinelandii. Association phenomena. Eur J Biochem 140: 273–280 [DOI] [PubMed] [Google Scholar]
- Chuang DT, Shih VE (2001) Maple syrup urine disease (branched-chain ketoaciduria). In The Metabolic and Molecular Basis of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, Childs B (eds), pp 1971–2006. New York, NY: McGraw-Hill [Google Scholar]
- Chuang JL, Cox RP, Chuang DT (1997) E2 transacylase-deficient (type II) maple syrup urine disease. Aberrant splicing of E2 mRNA caused by internal intronic deletions and association with thiamine-responsive phenotype. J Clin Invest 100: 736–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JL, Wynn RM, Chuang DT (2002) The C-terminal hinge region of lipoic acid-bearing domain of E2b is essential for domain interaction with branched-chain alpha-keto acid dehydrogenase kinase. J Biol Chem 277: 36905–36908 [DOI] [PubMed] [Google Scholar]
- Cowtan KD, Main P (1996) Phase combination and cross validation in iterated density-modification calculations. Acta Crystrallogr D 52: 43–48 [DOI] [PubMed] [Google Scholar]
- Cunningham L, Guest JR (1998) Transcription and transcript processing in the sdhCDAB-sucABCD operon of Escherichia coli. Microbiology 144 (Part 8): 2113–2123 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15: 305–308 [DOI] [PubMed] [Google Scholar]
- Griffin TA, Lau KS, Chuang DT (1988) Characterization and conservation of the inner E2 core domain structure of branched-chain alpha-keto acid dehydrogenase complex from bovine liver. Construction of a cDNA encoding the entire transacylase (E2b) precursor. J Biol Chem 263: 14008–14014 [PubMed] [Google Scholar]
- Guest JR (1987) Functional implications of structural homologies between chloramphenicol acetyltransferase and dihydrolipoamide acetyltransferase. FEMS Microbiol Lett 44: 417–422 [Google Scholar]
- Harris RA, Hawes JW, Popov KM, Zhao Y, Shimomura Y, Sato J, Jaskiewicz J, Hurley TD (1997) Studies on the regulation of the mitochondrial alpha-ketoacid dehydrogenase complexes and their kinases. Adv Enzyme Regul 37: 271–293 [DOI] [PubMed] [Google Scholar]
- Hendle J, Mattevi A, Westphal AH, Spee J, de Kok A, Teplyakov A, Hol WG (1995) Crystallographic and enzymatic investigations on the role of Ser558, His610, and Asn614 in the catalytic mechanism of Azotobacter vinelandii dihydrolipoamide acetyltransferase (E2p). Biochemistry 34: 4287–4298 [DOI] [PubMed] [Google Scholar]
- Hubalek F, Binda C, Khalil A, Li M, Mattevi A, Castagnoli N, Edmondson DE (2005) Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J Biol Chem 280: 15761–15766 [DOI] [PubMed] [Google Scholar]
- Izard T, Aevarsson A, Allen MD, Westphal AH, Perham RN, de Kok A, Hol WG (1999) Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 96: 1240–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ (1997) Brain energy metabolizing enzymes in Alzheimer's disease: alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann N Y Acad Sci 826: 218–228 [DOI] [PubMed] [Google Scholar]
- Knapp JE, Carroll D, Lawson JE, Ernst SR, Reed LJ, Hackert ML (2000) Expression, purification, and structural analysis of the trimeric form of the catalytic domain of the Escherichia coli dihydrolipoamide succinyltransferase. Protein Sci 9: 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp JE, Mitchell DT, Yazdi MA, Ernst SR, Reed LJ, Hackert ML (1998) Crystal structure of the truncated cubic core component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J Mol Biol 280: 655–668 [DOI] [PubMed] [Google Scholar]
- Ko TP, Lin JJ, Hu CY, Hsu YH, Wang AH, Liaw SH (2003) Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J Biol Chem 278: 19111–19117 [DOI] [PubMed] [Google Scholar]
- Koike M, Reed LJ, Carroll WR (1963) alpha-Keto acid dehydrogenation complexes. IV. Resolution and reconstitution of the Escherichia coli pyruvate dehydrogenation complex. J Biol Chem 238: 30–39 [PubMed] [Google Scholar]
- Koike K, Suematsu T, Ehara M (2000) Cloning, overexpression and mutagenesis of cDNA encoding dihydrolipoamide succinyltransferase component of the porcine 2-oxoglutarate dehydrogenase complex. Eur J Biochem 267: 3005–3016 [DOI] [PubMed] [Google Scholar]
- Lau KS, Lee J, Fisher CW, Cox RP, Chuang DT (1991) Premature termination of transcription and alternative splicing in the human transacylase (E2) gene of the branched-chain alpha-ketoacid dehydrogenase complex. FEBS Lett 279: 229–232 [DOI] [PubMed] [Google Scholar]
- Leslie AG, Moody PC, Shaw WV (1988) Structure of chloramphenicol acetyltransferase at 1.75-A resolution. Proc Natl Acad Sci USA 85: 4133–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard IA, Fuller C, Perham RN (1996) Competitive interaction of component enzymes with the peripheral subunit-binding domain of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus: kinetic analysis using surface plasmon resonance detection. Biochemistry 35: 16863–16870 [DOI] [PubMed] [Google Scholar]
- Liavonchanka A, Hornung E, Feussner I, Rudolph MG (2006) Structure and mechanism of the Propionibacterium acnes polyunsaturated fatty acid isomerase. Proc Natl Acad Sci USA 103: 2576–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw SH, Chen SJ, Ko TP, Hsu CS, Chen CJ, Wang AH, Tsai YC (2003) Crystal structure of D-aminoacylase from Alcaligenes faecalis DA1. A novel subset of amidohydrolases and insights into the enzyme mechanism. J Biol Chem 278: 4957–4962 [DOI] [PubMed] [Google Scholar]
- Liu S, Lu Z, Han Y, Jia Y, Howard A, Dunaway-Mariano D, Herzberg O (2004) Conformational flexibility of PEP mutase. Biochemistry 43: 4447–4453 [DOI] [PubMed] [Google Scholar]
- Machius M, Wynn RM, Chuang JL, Li J, Kluger R, Yu D, Tomchick DR, Brautigam CA, Chuang DT (2006) A versatile conformational switch regulates reactivity in human branched-chain alpha-ketoacid dehydrogenase. Structure 14: 287–298 [DOI] [PubMed] [Google Scholar]
- Mattevi A, Obmolova G, Kalk KH, Teplyakov A, Hol WG (1993) Crystallographic analysis of substrate binding and catalysis in dihydrolipoyl transacetylase (E2p). Biochemistry 32: 3887–3901 [DOI] [PubMed] [Google Scholar]
- Mattevi A, Obmolova G, Schulze E, Kalk KH, Westphal AH, de Kok A, Hol WG (1992) Atomic structure of the cubic core of the pyruvate dehydrogenase multienzyme complex. Science 255: 1544–1550 [DOI] [PubMed] [Google Scholar]
- Meng M, Chuang DT (1994) Site-directed mutagenesis and functional analysis of the active-site residues of the E2 component of bovine branched-chain alpha-keto acid dehydrogenase complex. Biochemistry 33: 12879–12885 [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ikebe S, Hattori N, Mochizuki H, Nakagawa-Hattori Y, Kondo T (1994) Studies on the pathogenesis of Parkinson's disease in Japan. Arch Gerontol Geriatr 19: 105–121 [DOI] [PubMed] [Google Scholar]
- Murshudov G, Vagin A, Dodson E (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Oliaro-Bosso S, Schulz-Gasch T, Taramino S, Scaldaferri M, Viola F, Balliano G (2005) Access of the substrate to the active site of squalene and oxidosqualene cyclases: comparative inhibition, site-directed mutagenesis and homology-modelling studies. Biochem Soc Trans 33: 1202–1205 [DOI] [PubMed] [Google Scholar]
- Oliver RM, Reed LJ (1982) In Electron Microscopy of Proteins, Harris JR (ed), Vol. 2, pp 1–48. London: Academic [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of x-ray diffraction data collected in oscillation mode. In Methods in Enzymology, CW Carter JRMS (ed), Vol. 276, pp 307–326. London: Academic Press [DOI] [PubMed] [Google Scholar]
- Perham RN (2000) Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 69: 961–1004 [DOI] [PubMed] [Google Scholar]
- Pettit FH, Yeaman SJ, Reed LJ (1978) Purification and characterization of branched chain alpha-keto acid dehydrogenase complex of bovine kidney. Proc Natl Acad Sci USA 75: 4881–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Haydon DJ, Guest JR (1994) The pdhR-aceEF-lpd operon of Escherichia coli expresses the pyruvate dehydrogenase complex. Mol Microbiol 12: 95–104 [DOI] [PubMed] [Google Scholar]
- Reed LJ (2001) A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem 276: 38329–38336 [DOI] [PubMed] [Google Scholar]
- Reed LJ, Hackert ML (1990) Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem 265: 8971–8974 [PubMed] [Google Scholar]
- Robinson BH, Sherwood WG (1984) Lactic acidaemia. J Inherit Metab Dis 7 (Suppl 1): 69–73 [DOI] [PubMed] [Google Scholar]
- Russell GC, Guest JR (1990) Overexpression of restructured pyruvate dehydrogenase complexes and site-directed mutagenesis of a potential active-site histidine residue. Biochem J 269: 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31: 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D 60: 432–438 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman SJ, Kirby JA, Jones DE (2000) Autoreactive responses to pyruvate dehydrogenase complex in the pathogenesis of primary biliary cirrhosis. Immunol Rev 174: 238–249 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and Methods
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4