

Abstract
Huntington's disease results from a mutation in the HD gene encoding for the protein huntingtin. The function of huntingtin, although beginning to be elucidated, remains largely unclear. To probe the prosurvival function of huntingtin, we modulate levels of wild-type huntingtin in a number of cellular and in vivo models. Huntingtin depletion resulted in caspase-3 activation, and overexpression of huntingtin resulted in caspase-3 inhibition. Additionally, we demonstrate that huntingtin physically interacts with active caspase-3. Interestingly, mutant huntingtin binds active caspase-3 with a lower affinity and lower inhibitory effect on active caspase-3 than does wild-type huntingtin. Although reduction of huntingtin levels resulted in caspase-3 activation in all conditions examined, the cellular response was cell-type specific. Depletion of huntingtin resulted in either overt cell death, or in increased vulnerability to cell death. These data demonstrate that huntingtin inhibits caspase-3 activity, suggesting a mechanism whereby caspase-mediated huntingtin depletion results in a detrimental amplification cascade leading to further caspase-3 activation, resulting in cell dysfunction and cell death.
Keywords: caspases-3, huntingtin, Huntington's disease
Introduction
Huntington's disease (HD) results from an expanded CAG repeat encoding a polyglutamine in exon 1 of the HD gene (Group, 1993). The normal function of huntingtin is beginning to be elucidated, and previous reports have provided evidence that it plays a role in the regulation of BDNF expression, vesicle trafficking, axonal transport, and transcriptional regulation, including dynamic repression of transcription and growth factor genes (DiFiglia et al, 1995; Velier et al, 1998; Steffan et al, 2000; Waelter et al, 2001; Zuccato et al, 2001; Gunawardena et al, 2003; Goehler et al, 2004; Qin et al, 2004; Cattaneo et al, 2005; Woda et al, 2005; Leavitt et al, 2006). Homozygous Hdh-deficient mice die in utero early in embryonic development, demonstrating its key role in development (Duyao et al, 1995; Nasir et al, 1995; Zeitlin et al, 1995). Furthermore, Hdh inactivation, eliminating huntingtin in the postnatal mouse brain, and small interference RNA (siRNA)-mediated reduction of huntingtin in Drosophila, result in neurodegeneration, indicating that huntingtin also plays an important role in neuronal survival in the mature brain (Dragatsis et al, 2000; Gunawardena et al, 2003). However, the mechanisms by which reduction of huntingtin levels leads to neurodegeneration are not known. In this report, we demonstrate that huntingtin binds to active caspase-3, and may have inhibitory effects on this key executioner caspase (Kuida et al, 1996). Reduction of huntingtin levels, which occurs in a variety of neurologic conditions associated with caspase-3 activation, could be not only a consequence but also a cause of a detrimental feedback loop that leads to terminal amplification of caspase-3 activation and cell death (Ona et al, 1999; Chen et al, 2000; Zhang et al, 2003).
Results
Huntingtin expression inversely correlates with caspase-3 activity in N2a cells
To study the mechanisms of huntingtin-depletion-mediated neurodegeneration, we knocked-down huntingtin in mouse neuroblastoma N2a cells by designing an siRNA targeting Hdh mRNA encoding huntingtin. A 21-nucleotide siRNA targeting murine Hdh exon-2, termed I-htt, was transfected into N2a cells (Figure 1A). The reversed sequence of I-htt, termed RI-htt, was used as a control. Immunoblot of cellular protein extracts at 72 h after transiently transfecting cells revealed that huntingtin levels were significantly reduced by I-htt transfection, to approximately 21.7% of levels in N2a cells transfected with RI-htt (Figure 1B). As controls, neurofilament and tubulin protein levels were not altered by I-htt. Analysis of caspase activity in transfected N2a cells indicated that siRNA-mediated huntingtin depletion is associated with a greater than four-fold increase of caspase-3-like activity but not of caspase-1, -2, -6, -8 or -9-like activities (Figure 1C). Caspase-3 activation in the absence of caspase-1, -2, -6, -8 and -9 was confirmed by immunoblot (Figure 1B). Caspase-7 has the same fluorogenic substrate as caspase-3. We therefore performed a Western blot to evaluate whether caspase-7 is activated, and demonstrate that similar to caspase-3, caspase-7 is activated following htt knockdown (Figure 1B). siRNA-mediated reduction of huntingtin resulted in induction of cell death (Figure 1D). These results suggest that huntingtin depletion results in caspase-3/7 activation and N2a neuronal cell death. However, what was most intriguing was that reduction of huntingtin resulted in selective caspase-3/7 activation in N2a cells without evidence of significant activation of other apical caspases such as caspase-1, -2, -6, -8, and -9, which typically mediate caspase-3 activation. We therefore hypothesized that huntingtin might play a role in inhibiting caspase-3.
Figure 1.
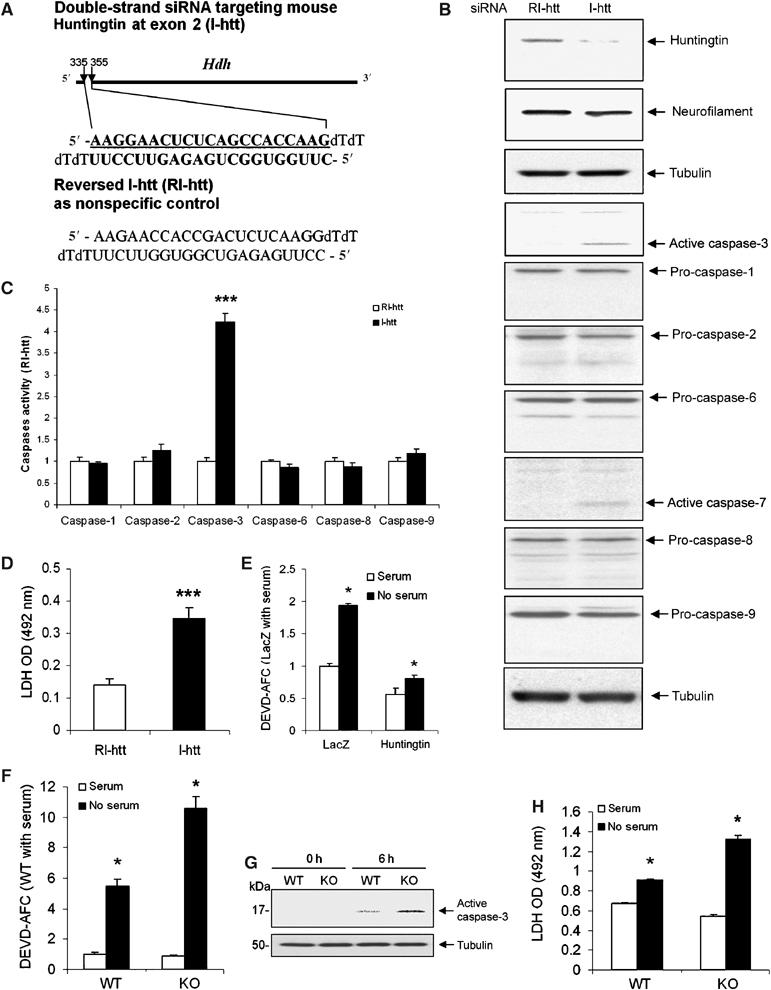
(A) Graphical representation of sites and sequences of double-strand siRNA targeting mouse huntingtin exon 2. I-htt represents the siRNA targeting huntingtin, and RI-htt represents reversed I-htt used as a control. (B) Endogenous huntingtin was depleted 72 h after transfection by I-htt in mouse neuroblastoma N2a cells. The blot was probed with an anti-huntingtin antibody (MAB2166). The same blot was stripped and then probed with antitubulin and antineurofilament H (200 kDa) antibodies to demonstrate equal loading. Densitometry of huntingtin signal was scanned and analyzed as ratio to tubulin. Huntingtin levels were 78.3±6.5% reduced by I-htt transfection when compared with N2a cells transfected with RI-htt. One hundred micrograms of same samples used for huntingtin blot were loaded to do immunoblots for caspase-1, -2, -3, -6, -7, -8, -9, and tubulin. Activation of caspase-3 and -7 was found after huntingtin reduction by siRNA. No activation of caspase-1, -2, -6, -8, and -9 was found. Immunoblots of tubulin demonstrated equal loading. The blots are representative of four independent experiments. (C) Caspase activation was evaluated using fluorogenic substrate assays. Caspase-3-like activity was increased 72 h after transfection by I-htt in N2a cells. Caspase-1, -2, -6, -8, and -9 like activities (YVAD-AFC, VDVAD-AFC, VEID-AFC, IETD-AFC and LEHD-AFC were used, in respectively, fluorogenic substrate assays) were not affected by huntingtin depletion. Data are expressed as the ratio of the I-htt group to the RI-htt group, n=3, ***P<0.001. One representative experiment out of five is shown. (D) Increased cell death was quantified by absorbancy values of LDH activity released into the medium 72 h after RI-htt and I-htt transfection. n=8, ***P<0.001. One representative experiment out of three is shown. (E) N2a cells cotransfected with CD19 and huntingtin or lacZ in a DNA ratio of 1:10. CD19+ cells were selected by magnetic beads coupled with anti-CD19. Overexpressing huntingtin inhibited caspase-3 activation in N2a cells after serum deprivation (n=3). (F) Following serum deprivation, Hdh−/− ES cells have increased caspase-3 activity (F, G) and cell death as detected by LDH release (6 h) (H) (n=6). All the above experiments were repeated at least three times, *P<0.05.
Previous reports have demonstrated a protective role of huntingtin in striatal neuron cell lines. Huntingtin overexpression was associated with reduced caspase-9 and -3 activity (Rigamonti et al, 2000). In vivo, huntingtin overexpression decreases the cellular toxicity of mutant huntingtin and inhibits cerebral ischemia-mediated injury (Leavitt et al, 2001; Zhang et al, 2003; Van Raamsdonk et al, 2005). To evaluate whether huntingtin overexpression can inhibit caspase-3 activation in an alternate cell death model, we transfected N2a cells with a cDNA expression construct encoding full-length mouse huntingtin. Huntingtin overexpression resulted in a reduction of serum-deprivation-mediated caspase-3 activation (Figure 1E), providing further support for the potential inhibitory effect of huntingtin on caspase-3. We predicted that if huntingtin was a direct or indirect inhibitor of caspase-3, huntingtin-deficient cells would be more sensitive to cell death via caspase-3-dependent mechanisms. We therefore evaluated the response of huntingtin-deficient embryonic stem (ES) cells (Hdh−/− ES cells) to serum-deprivation (Duyao et al, 1995). Following serum-deprivation, there is a greater increase of caspase-3 activity in Hdh−/− ES cells, compared to wild-type cells (Figure 1F and G). Consistent with the amplified activation of caspase-3, huntingtin-deficient cells die more readily following serum-deprivation (Figure 1H).
Huntingtin interacts with active caspase-3 and inhibits caspase-3 activation in cell-free systems
We evaluated whether we could detect a direct interaction between endogenous huntingtin and caspase-3. Cell-free N2a extracts were immunoprecipitated with a huntingtin antibody and immunoblotted with two caspase-3 antibodies. We were unable to demonstrate a caspase-3/huntingtin interaction in the extracts (data not shown). As known inhibitors of caspase-3 bind the active form of the enzyme (i.e. XIAP, cIAP1, and cIAP2) (Deveraux et al, 1997; Roy et al, 1997), and as the cell lysates used did not have active caspase-3, we evaluated mixed extracts composed of half untreated extract, and half from serum-deprived cells. In this manner, the extracts endogenously contain full-length huntingtin, pro-caspase-3, and active caspase-3. As demonstrated by immunoblotting, the huntingtin antibody immunoprecipitated processed caspase-3, but not pro-caspase-3 (Figure 2A), demonstrating a physical interaction between huntingtin and active caspase-3. Using purified caspase-3 and in vitro-translated huntingtin, we showed there is indeed a direct interaction between these two proteins (Figure 2B). We then evaluated whether mutant huntingtin was able to bind caspase-3 as well. Mutant huntingtin indeed binds active caspase-3, albeit with a lower affinity (Figure 2B and C).
Figure 2.
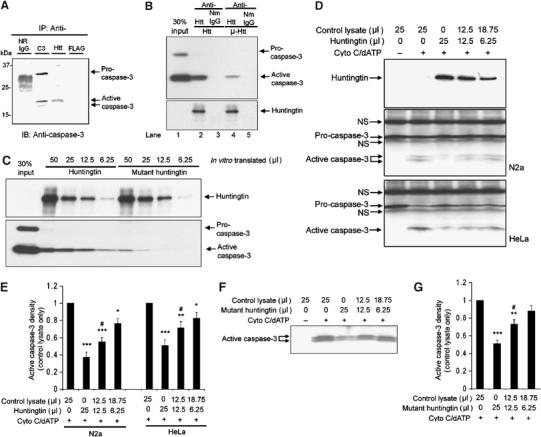
(A) Wild-type huntingtin interact with activated caspase-3. Immunoprecipitation was performed in lysates from N2a cells as described in the Materials and methods section. Caspase-3 antibody (Cell Signal 9662) binds both pro- and active caspase-3. Normal rabbit IgG (N.R.IgG) and mouse anti-FLAG (mouse IgG1) were used as controls. Huntingtin antibody (MAB2166) co-immunoprecipitates active caspase-3. (B) Immunoprecipitation of wild-type huntingtin and mutant huntingtin with active recombinant caspase-3. The full-length huntingtin and mutant huntingtin were first purified from in vitro-translated lysates (50 μl) using conjugated antibodies: anti-huntingtin antibody (MAB 2166) or normal mouse IgG (NmIgG). The purified proteins were then incubated with purified recombinant caspase-3 (2 μg). The bound caspase-3 was detected by Western blot. Blot of anti-huntingtin showed in vitro-translated huntingtin. (C) Western blot of caspase-3 demonstrates different binding affinities of wild-type huntingtin (in an in vitro-translated volume of 50, 25, 12.5, and 6.25 μl) and mutant huntingtin (in an in vitro-translated volume of 50, 25, 12.5, and 6.25 μl) with caspase-3. (D) Western blot for huntingtin and caspase-3 in N2a and HeLa cytosolic extracts following treatment for 1 h with 10 μM cytochrome c and 1 mM dATP at 37°C, with or without in vitro-translated full-length huntingtin. The position of procaspase-3 and active caspase-3 are identified by arrows. Procaspase-3 was mixed with nonspecific bands from in vitro translation reagents (indicated by arrows with ‘NS'. NS=nonspecific band). One representative result of six individual experiments is shown. (E) Densitometric analysis of active caspase-3 signal in Western blot of N2a and HeLa cytosolic extracts with or without in vitro-translated full-length huntingtin as shown in (D). Data are mean±s.e.m. Error bars indicate s.e.m. n=6. ***P<0.001, **P<0.01, *P<0.05 versus control without adding huntingtin. #P<0.05 versus group adding 25 μl of huntingtin. (F) Western blot for caspase-3 in N2a cytosolic extracts following treatment for 1 h with 10 μM cytochrome c and 1 mM dATP at 37°C, with or without in vitro-translated mutant full-length huntingtin. One representative result of six individual experiments is shown. (G) Densitometric analysis of active caspase-3 signal in Western blots as shown in (F). Data are mean±s.e.m. Error bars indicate s.e.m. n=6. ***P<0.001, **P<0.01 versus control without adding mutant huntingtin. #P<0.05 versus group adding 25 μl of mutant huntingtin.
To further explore the biological function of the huntingtin–active caspae-3 binding, using in vitro-translated huntingtin, we evaluated its effect in a cell-free system to determine whether huntingtin could inhibit caspase-3 processing. Addition of cytochrome c and dATP to N2a or to HeLa cell cytosolic extracts results in proteolytic caspase-3 activation (Liu et al, 1996). Recombinant huntingtin inhibited in a dose-dependent fashion cytochrome c/dATP-induced procaspase-3 processing (Figure 2D and E). Additionally, in a concentration-dependent manner, huntingtin also inhibits recombinant caspase-3-mediated cleavage of DEVD-AFC (Supplementary Figure S1). Interestingly, mutant huntingtin is also able to inhibit caspase-3 processing, albeit with a lower degree of efficiency (Figure 2F and G). The inhibitory effect of mutant huntingtin (49% inhibition) is lower when compared to wild-type huntingtin (62% inhibition), using the same amount of in vitro-translated protein (Figure 2G). These results suggest huntingtin inhibits caspase-3 activity, and that the mutant protein, although it retains in part the wild-type function it does so with a lower degree of efficacy.
Cell-specific effects of downregulation of huntingtin by siRNA in HeLa and ST14A cells
We then evaluated in a non-neuronal cell, HeLa cells, whether depletion of huntingtin by siRNA would impact caspase-3 activation (Figure 3). As demonstrated in N2a cells, reduction of huntingtin levels results in significant caspase-3 activation (Figure 3A and B). However, in contrast to the demonstration of cell death in N2a cells, huntingtin depletion did not result in HeLa cell death. Stressing HeLa cells with an apoptotic stimulus (TNFα/CHX (cycloheximide)) results in caspase-3 activation and cell death (Figure 3B and C). We can clearly detect huntingtin cleavage and generation of an N-terminal fragment following TNFα/CHX treatment (Figure 3A). TNFα/CHX when added to cells where huntingtin has been knocked down results in a significant increase in the magnitude of caspase-3 activation (Figure 3A and B). Only when the cell is stressed, does huntingtin depletion in HeLa result in an increased degree of cell death (Figure 3C). From this information, it appears that the consequence of reduction of huntingtin levels is cell-type specific. An unexpected result in the HeLa cells was a dramatic cell shape change following huntingtin knockdown with shrinkage of the cell body and development of long processes (Figure 3D and E). Even though the significance of this change in cell morphology is not evident, this result demonstrates that huntingtin depletion has cell-type-specific consequences.
Figure 3.
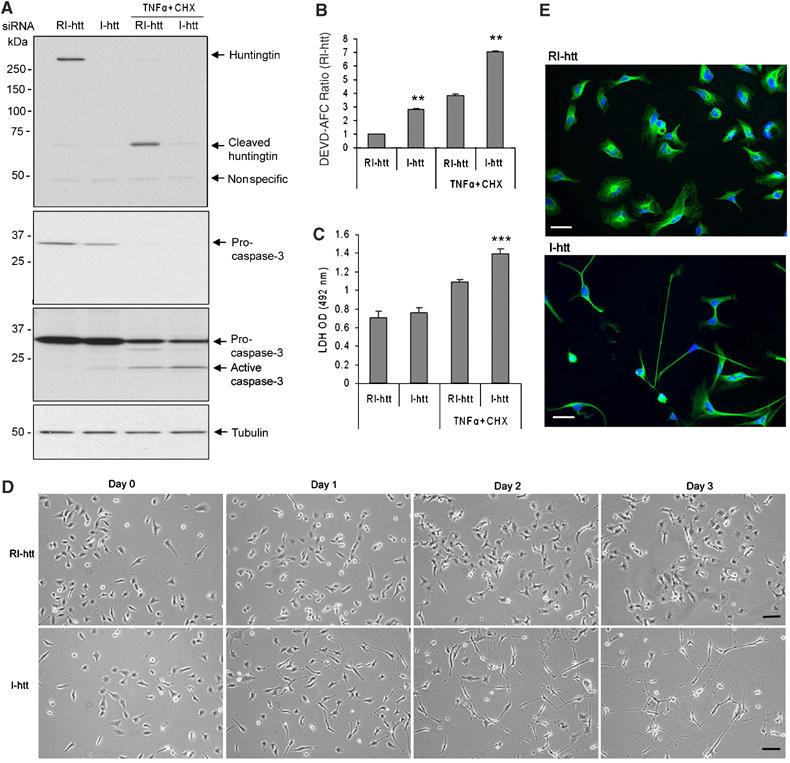
Differential responses of HeLa cells after depletion of huntingtin by siRNA. (A) Huntingtin was significantly reduced by siRNA targeting human huntigntinin in HeLa cells. Sixty hours after siRNA transfection, cells were treated with TNFα/CHX (2000 U/ml of TNFα and 30 μg/ml of CHX) for 12 h. Whole-cell extracts were prepared and run in 7.5% SDS–PAGE (top first blot for huntingtin) and 12% SDS–PAGE (middle second and third blots for caspase-3), second blot is the short exposure one that showed the difference between full-length caspase-3. The third blot is the same blot of second one after longer exposure to show the active caspase-3 when the signal of full-length band is saturated. This blot is striped off and re-blotted with anti-tubulin to show the equal amount of loading. Caspase-3 was slightly cleaved after huntingtin depletion by siRNA. Treatment of TNFα/CHX induced the more activation of caspase-3 and cleavage of huntingtin. (B) Caspase-3-like activity (DEVD-AFC) was detected by fluorometric protease assay after huntingtin depletion by siRNA. TNFα/CHX treatment of 12 h increased the activation of caspase-3 after huntingtin reduction. n=3, **P<0.01. Error bars indicate s.e.m. (C) TNFα/CHX treatment after huntingtin reduction significantly enhances the degree of cell death as detected by LDH assay, whereas cell mortality was not impacted following huntingtin reduction in the absence of apoptotic stimulation. n=18, ***P<0.001. One representative experiment out of three is shown. (D) Morphological changes in HeLa cells after transfection of I-htt compared with transfection of RI-htt for 3 days. Scale bar, 30 μm. (E) Representative photographs of HeLa cells 72 h after transfection of RI-htt and I-htt. Green: anti-tubulin, blue: Hoechst 33342. Scale bar, 35 μm.
As most HD pathology occurs in the striatum, we evaluated the effect of huntingtin depletion on caspase-3 activation and cell death on striatal ST14A cells (Figure 4). ST14A cells were derived from embryonic rat striatum by introducing a temperature-sensitive version of the large-T Antigen. ST14A cells grow at the permissive temperature of 33°C. Mutant huntingtin expressing ST14 cells die once placed at the nonpermissive temperature (37–39°C) (Cattaneo and Conti, 1998; Rigamonti et al, 2000; Wang et al, 2005). We designed siRNA sequence specifically targeting rat endogenous huntingtin, not human huntingtin carrying in mutant ST14A. At the permissive temperature, huntingtin reduction in parental ST14A had a similar effect than that seen in HeLa cells with a two-fold increase in caspase-3 activity, but no increase in cell death (Figure 4A–D). We next evaluated the effect of huntingtin depletion in the context of a cell expressing a human mutant huntingtin N-terminal fragment (N548mu). At the permissive temperature, huntingtin reduction in mutant ST14A cells resulted in a 4.3-fold increase caspase-3 activity and increased cell death when compared with mutant ST14A without huntingtin reduction (Figure 4B and D). Mutant ST14A cells with huntingtin reduction demonstrated a 10.3-fold increase caspase-3 activity when compared to parental ST14A cells without huntingtin reduction (Figure 4B). When parental ST14A cells are placed in nonpermissive conditions (37°C), reduction huntingtin resulted in a greater magnitude of caspase-3 activation (2.5-fold) and cell death when compared to the same conditions without huntingtin reduction (Figure 4B, C and D). Following huntingtin depletion, placing mutant ST14A cells in the nonpermissive temperature, resulted in a significantly greater magnitude of caspase-3 activation (18-fold) and cell death when compared to parental cells without huntingtin reduction at permissive temperature (Figure 4B and D). These data point to the need of multiple factors that are required for the induction of cell death. In this particular cell line, the additive effect of the toxicity of mutant huntingtin, the exposure to the nonpermissive temperature, and the depletion of wild-type huntingtin results in efficient caspase-3 activation and cell death.
Figure 4.
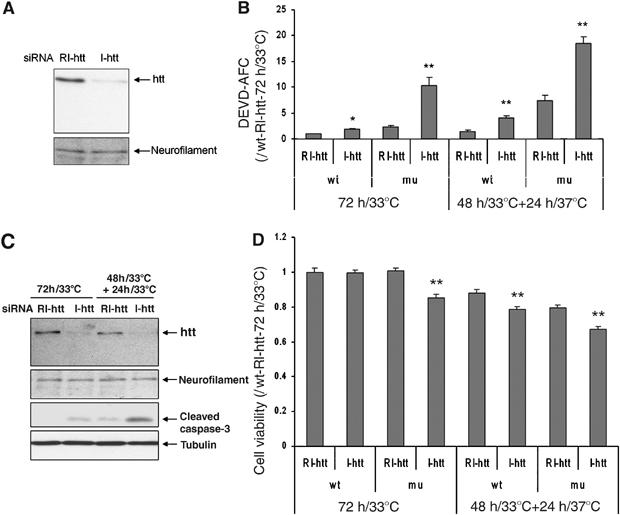
(A) Depletion of huntingtin by siRNA in parental ST14A cells. At 72 h after siRNA transfection, cells were harvested and extracts were separated in 7.5% SDS–PAGE gel and blot with anti-huntingtin (MAB2166). (B–C) Caspase-3-like activation following huntingtin reduction (wt=parental ST14A cells; mu=mutant huntingtin expressing ST14A cells), n=3, *P<0.05, **P<0.01. Western blot confirmed that huntingtin depletion resulted in increased caspase-3 activation in mutant ST14A cells (C). Neurofilament H (for huntingtin) and tubulin (for caspase-3) were used as a loading control. (D) Cell viability as determined by the MTS assay in parental and mutant huntingtin ST14A cells following huntingtin reduction and exposure to nonpermissive temperatures; n=36, **P<0.01. One representative experiment out of three is shown.
Activation of caspase-3 in Hdh−/− neurons in chimeric mice
Although nullizygous (knockout) Hdh mice die early in embryogenesis (Duyao et al, 1995; Nasir et al, 1995; Zeitlin et al, 1995), the survival of neurons lacking huntingtin in chimeric mice in some brain regions indicates that huntingtin is not required for all neurons (Metzler et al, 1999; Reiner et al, 2001). The availability of such chimeric mice provides the opportunity to evaluate the status of caspase-3 activation in adult huntingtin-deficient neurons in vivo. We evaluated caspase-3 activation in the hippocampus, which has been reported to harbor abundant Hdh−/− cells in chimeric mice (Reiner et al, 2001). Complementing the above-described results, huntingtin-deficient neurons stained with X-gal showed significant caspase-3 activation in the hippocampus of chimeric mice (see black arrow in Figure 5F and H, white arrow in Figure 5G). By contrast, less or none caspase-3 staining are showed in hippocampus regions lacking in X-gal labeling (see short red arrow in Figure 5F, G and H). We have counted the number of cells in the hippocampus and evaluated the numbers that are X-gal positive and active caspase-3 positive. The degree of correlation of caspase-3-positive/htt-deficient cells is very high (93.2%, long black arrow in Figure 5F and H, long white arrow in Figure 5G). Neighboring wild-type neurons did not demonstrate caspase-3 activation (long red arrow in Figure 5F, G and H). In other brain regions, huntingtin deficiency and caspase-3 activation were also correlated. It is interesting to note that in these adult chimeric mice, the extent of huntingtin-deficient neurons in regions typically associated with neurodegeneration in HD (striatum and cortex) is much reduced when compared with other brain regions, suggesting that these neuronal populations might be more sensitive to the effects of huntingtin depletion (Reiner et al, 2001). The fact that caspase-3 activation does not lead to cell death in these adult huntingtin-deficient neurons (Figure 5) may be due to the fact that the magnitude of caspase-3 activation might not be sufficient to mediate apoptosis, or that these cells are less sensitive to caspase-3-mediated cell death.
Figure 5.
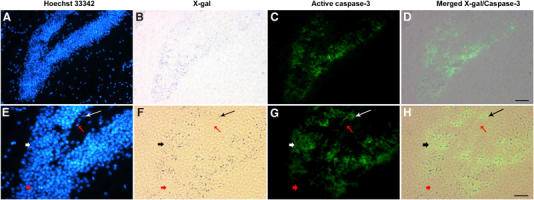
Brain specimens from chimeric mice harboring Hdh−/− cells as well as wild-type cells. These mice have been previously fully described and evaluated (Reiner et al, 2001). In adult mice, X-gal staining cells derived from the Hdh−/− nullizygous ES cells injected at the blastocyst stage. Using adult brain samples, we stained the same section with (A, E) Hoechst 33342 dye (to identify the nuclei), (B, F) X-gal (to identify the huntingtin-deficient cells), and (C, G) an anti-active caspase-3-FITC-conjugated antibody. (D) merged images from (B) and (C), and (H) from (F) and (G), respectively. Hdh−/− cells in regions stained with X-gal labeling are richly labeled active caspase-3 (see short black arrow in (F) and (H), short white arrow in (G)). By contrast, regions in the hippocampus lacking in X-gal labeling are poor in caspase-3 staining (see short red arrow in F, G and H). As showed in E–H, the total cell number is 520, X-gal positive is 221, both X-gal and caspase-3 positive is 206 (93.2%, see long black arrow in (F) and (H), long white arrow in (G)). Neighboring X-gal-negative cells do not show activation of caspase-3 (see long red arrow in (F), (G), and (H)). Scale bar, 50 μm (D), 25 μm (H). This result is representative of four brains evaluated. As a control, when the anti-active caspase-3 antibody is not added, X-gal-positive cells do not fluoresce with the green filter (data not shown).
Overexpressed wild-type huntingtin inhibits caspase-3 activation and neurodegeneration in YAC18 mice
To examine the role of wild-type huntingtin in inhibiting caspase-3 activation in vivo, we evaluated the functional consequence of increasing full-length wild-type huntingtin in a mouse model of excitotoxic neurodegeneration associated with caspase-3 activation. We induced excitotoxic cell death in brains of transgenic mice expressing wild-type human huntingtin (YAC18) at two to three times the levels of endogenous huntingtin by systemic treatment with kainic acid (KA) (Figure 6A). Compared with PBS-treated control mice, both YAC18 mice and wild-type littermates demonstrate a reduction of wild-type huntingtin in the hippocampus 24 h after KA injection. However, KA-treated YAC18 mice still expressed two to three times more huntingtin than did wild-type mice, consistent with the ratio of YAC18 huntingtin to wild-type mouse huntingtin before KA injection (Figure 6A). KA caused prolonged seizures, with no difference in seizure duration, severity, or mortality between wild-type and YAC18 mice. Concomitant with huntingtin depletion in wild-type mice, KA caused acute seizures resulting in neuronal caspase-3 activation and apoptotic neurodegeneration within the CA1 and CA3 regions of hippocampus as demonstrated by immunofluorescence staining of specifically cleaved caspase-3 antibody and Fluoro-Jade labeling (Figure 6B–E). Furthermore, consistent with huntingtin has an inhibitory effect on caspase-3, less hippocampal caspase-3 activation was detected in YAC18 mice compared to wild-type mice following KA-induced seizures (Figure 6F). Following KA-induced seizures, overexpression of huntingtin in YAC18 transgenic mice had a significant protective effect against KA-induced neurodegeneration as demonstrated by less Fluoro-Jade-stained large degenerating neurons (Figure 6G–I) and marked reduction in silver-stained neurons (Figure 6J–L). YAC18 mice averaged approximately 50-fold less degenerating hippocampal CA1 and CA3 neurons than controls (Figure 6M). Previous work showed that huntingtin protects cells from quinolinic acid, an agonist of NMDA receptor, initiated neurodegeneration (Leavitt et al, 2006). The present study suggests that the protective role of huntingtin is, at least in part due to its inhibitory effect on caspase-3, and also provides further in vivo support for a role of wild-type huntingtin inhibiting caspase-3 activation by non-NMDA receptor-mediated excitotoxicity. These investigations suggest that the huntingtin protective mechanism is not restricted to one subtype of glutamate receptor-mediated excitotoxicity, but it results by inhibiting capase-3 activation.
Figure 6.
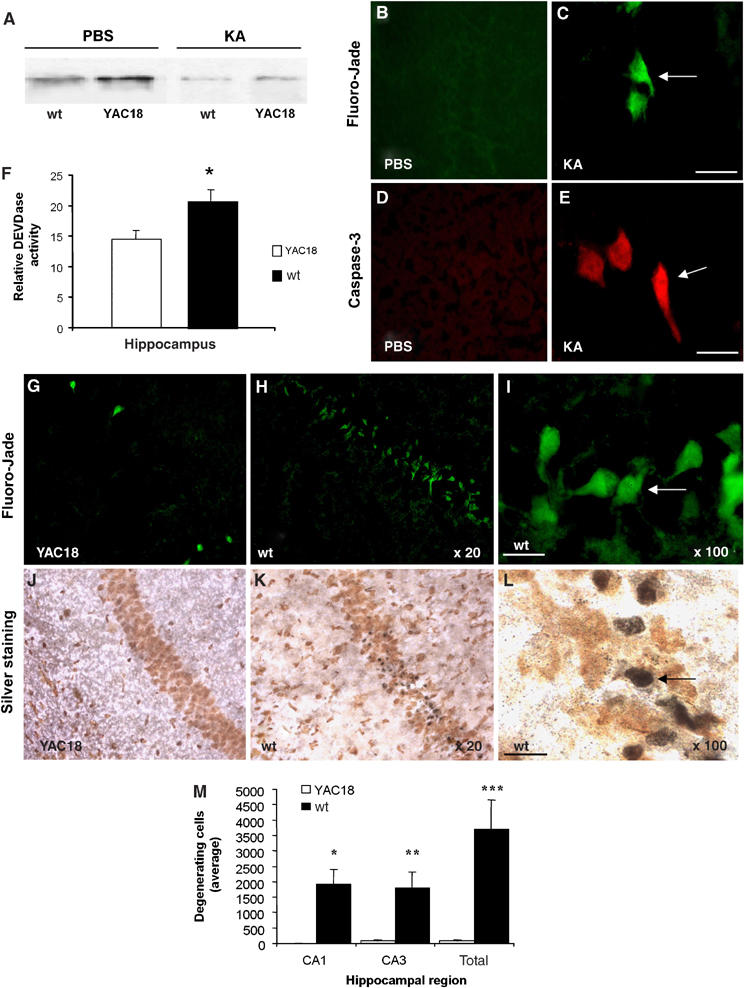
Overexpressed wild-type huntingtin confers significant inhibition of caspase-3 activation and dramatic resistance to neurodegeneration in YAC18 mice. (A) Western blot demonstrates that the amount of full-length huntingtin is approximately two to three times greater in YAC18 mice than in wild-type mice. The blot was probed with anti-huntingtin MAB2166, an antibody recognizing both human and mouse huntingtin. Although KA-mediated seizures result in a significant reduction in full-length huntingtin in wild-type and YAC18 mice (brains evaluated 7 days after seizures), the amount of remaining full-length huntingtin was two- to three-fold greater in YAC18 mice than in wild-type mice. (B–E) Neurodegeneration and caspase-3 activation in hippocampal neurons of wild-type mice 1 week following KA-induced seizures. Degenerating hippocampal neurons (arrows) were identified using Fluoro-Jade labeling in KA-treated mice (C), but none were observed in mice injected with PBS (B). No active caspase-3 was found in hippocampal neurons of mice treated with PBS (D), but in parallel with the degenerating neurons, active caspase-3 was detected within neuronal cell bodies of mice treated with KA (E) with a specific antibody to cleaved caspase-3. (F) Wild-type huntingtin protects neurons from caspase activation. Significantly less hippocampal caspase-3 activity was evident in YAC18 (n=4) than in wild-type mice (n=6) following KA-induced seizures. (G–I) Images of degenerating neurons (arrows) with Fluoro-Jade labeling (G, H, I) and silver staining (J, K, L) within the hippocampus following KA-induced seizures in YAC18-overexpressing wild-type huntingtin and in wild-type littermate controls. (M) Quantification of degenerating hippocampal neurons in YAC18-overexpressing wild-type huntingtin and in wild-type littermate controls (wild-type, n=4; YAC18, n=6). The average number of degenerating neurons per animal identified by Fluoro-Jade labeling is expressed for the CA1, CA3, and total hippocampal regions. Scale bar, 20 μm. *P<0.05, **P<0.01 versus wt of same brain region.
Discussion
This study demonstrates that under certain conditions, and in a cell-specific manner, huntingtin plays an important role in the modulation of caspase-3 activity. We have demonstrated that siRNA-mediated depletion of huntingtin in neuroblastoma N2a cells results in selective caspase-3 activation in the absence of caspase-1, -2, -6, -8, or -9 activation. Given that none of these upstream caspases are activated, activation of caspase-3 associated with huntingtin depletion suggests a potential release resulting in caspase-3 autoactivation (Roy et al, 2001). This is particularly relevant, as procaspase-3 has a low level of proteolytic activity, and is therefore capable of having autocatalytic activity (Roy et al, 2001). Supporting this, huntingtin overexpression results in inhibition of caspase-3 activation in response to apoptotic stimuli. Furthermore, as compared with wild-type ES cells, huntingtin-deficient ES cells are more sensitive to serum-deprivation-mediated cell death and demonstrate elevated caspase-3 activation. Additionally, we demonstrate in a cell-free system that wild-type huntingtin inhibits caspase-3 activation. Adding physiologic relevance to the notion that huntingtin has a relevant in vivo inhibitory effect on caspase-3, we demonstrate that modulation of huntingtin levels, either by chimerism or by transgenic overexpression, modulates the level of caspase-3 activation accordingly. The above-described data provide evidence that huntingtin effectively inhibits caspase-3 in vitro, in cells, and in vivo.
It is of interest to note that similar to wild-type huntingtin, mutant huntingtin also interacts with active caspase-3; however, the interaction of mutant huntingtin is significantly weaker, suggesting a relative loss of function that may be of relevance to the human disease. Mice homozygous with normal levels of mutant huntingtin rescued the characteristic aberrant brain development and perinatal lethality from reduction of wild-type huntingtin (White et al, 1997). Mutant huntingtin probably is protective in early brain development and gradually showing its gain-function side of effect when endogenous cellular environment changes with progression of the pathobiology. It is also important to mention that in animals or cells overexpressing mutant full-length huntingtin or fragments resulted in cytotoxicity and activation of multiple caspases, even the reduction of endogenous huntingtin was evaluated (Rigamonti et al, 2001; Zhang et al, 2003).
It is of interest to note, that reduction of huntingtin has different consequences depending on the cellular context. One universal finding in our study is that huntingtin depletion, both in vivo and in cells, is always associated with caspase-3 activation, and that huntingtin overexpression is associated with lower levels of caspase-3 activation. However, even though huntingtin depletion always resulted in caspase-3 activation, the effect on cell viability was cell-type- and cell condition-dependent. In unstressed cells, huntingtin depletion resulted in cell death only in N2a cells. Huntingtin depletion in unstressed ES, HeLa, and ST14a cells was not associated with increased cell death. However, exposure to a stressful stimulus such as serum withdrawal in ES cells, TNFα/CHX in HeLa cells, or expression of mutant huntingtin or exposure to a nonpermissive temperature in striatal ST14A cells results in enhanced caspase-3 activation and cell death. Therefore, it appears that reduction of huntingtin has effects that are cell-type-specific, somewhat akin to the selective vulnerability of medium spiny neurons in HD.
Huntingtin-mediated caspase-3 inhibition holds significant relevance for neurologic diseases (Friedlander, 2003). We have previously demonstrated depletion of huntingtin in several acute models of neurological diseases (i.e. HD, stroke, traumatic brain and spinal cord injury) (Ona et al, 1999; Zhang et al, 2003). Reduction of endogenous huntingtin may be a consequence of the disease process that may be a component of a detrimental feedback loop resulting in amplification of the caspase cascade. Furthermore, the fact that huntingtin inhibits caspase-3 activation, and that huntingtin is progressively depleted in R6/2 mice (Zhang et al, 2003) suggests that whereas the disease process in HD is first initiated by mutant huntingtin, a loss of huntingtin protective function likely contributes to the ultimate neuronal loss. Additionally, huntingtin loss appears to play a role in the human disease as well (unpublished data). We have demonstrated progressive transcriptional upregulation of caspase-3 in a mouse model of HD (Chen et al, 2000), and loss of full-length huntingtin associated specifically with cleavage by caspase-3 in mouse and human brain (Leavitt et al, 2001; Wellington et al, 2002). Therefore, compounded upon the toxicity resulting from mutant huntingtin, increased generation of caspase-3 results in huntingtin cleavage (Wellington et al, 2002), resulting in depletion of wild-type huntingtin. In fact, huntingtin depletion results in runoff caspase-3 activation and further amplifies the neurodegenerative process. Additionally, the fact that huntingtin deficiency, either during embryonic development or during adulthood, results in significant neurodegeneration and lethality provides additional support for the key role of huntingtin for cell function and cell survival.
Materials and methods
Double-strand siRNA transfection
The siRNA (I-htt) specifically targeting mouse (N2a) and rat (ST14A) huntingtin mRNA was designed as shown in Figure 1. The inverted double-strand oligo-RNA of I-htt, RI-htt, was used as a control. For human huntingtin mRNA (HeLa): target sequence, I-htt: AACAUAGUGGCACAGUCUGUC; RI-htt: AACUGUCUGACACGGUGAUAC. Double-strand I-huntingtin and RI-htt were chemically synthesized, purified, and annealed by Dharmacon Research Inc (Lafayette, CO). Twenty-four hours before transfection, N2a cells were seeded to 30% density in 10% FBS without antibiotics. Double-strand oligo-RNAs were delivered into cells by OligofectAMINE (Invitrogen, Carlsbad, CA) following protocols provided by Invitrogen.
Tissue culture
N2a cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. ES cell were cultured in DMEM with 15% FBS, 2-mercaptoethanol (3.5 μl/500 ml), 1000 U/ml ESGRO (Chemicon, CA), 1 × pen/strep/glutamine, and 1 × nonessential amino acids on gelatinized 75 cm2 flask. Cells were collected and lysed with an NP-40 buffer (150 mM NaCl, 1% NP-40, 50 mM Tris (pH 8.0), protease inhibitor cocktail set I (Calbiochem, CA)).
Caspase activity assay
A protocol provided by Chemicon for caspase-1-like (YVAD-AFC), caspase-2-like (VDVAD-AFC), Caspase-3-like (DEVD-AFC), caspase-6-like (VEID-AFC), caspase-8-like (IETD-AFC), and caspase-9-like (LEHD-AFC) fluorometric protease assay kit was used to assay caspase activity. Collected cell pellets were homogenized in lysis buffer and incubated with an equal volume of 2 × reaction buffer (with 0.01 M dithiothreotol (DTT)) for an additional 1 h at 37°C with caspase-1, -2, -3, -6, -8, and -9 substrates (YVAD-AFC, VDVAD-AFC, DEVD-AFC, VEID-AFC, IETD-AFC, and LEHD-AFC) at a final concentration of 50 μM. For the inhibition assays, two concentrations of in vitro-translated huntingtin were mixed with the indicated concentration of DEVD-AFC for 60 min at 37°C, and 0.25 U active caspase-3 recombinant protein (Chemicon, CA). Activity was measured by release of AFC from DEVD-AFC over 60 min. The fluorescence was measured by a VersaFluor Fluorometer (Bio-Rad) with an excitation filter of 390±22 nm and an emission filter of 510±10 nm.
Detection of cytotoxicity by measurement of LDH activity
Released LDH activity was measured according to the protocol provided by the manufacturer (Roche, Germany). Reaction mixture (100 μl) was added to conditioned media (100 μl) removed from 24-well plates after centrifugation at 250 g for 10 min. For siRNA transfection, seeded N2a cell density is ⩽30%. For ES cells, seeded cell density is 90%. Absorbance of samples at 492 nm was measured in an ELISA reader after 15 min of incubation at room temperature. The same volume of medium was used as the background control.
Western blot
As described previously (Zhang et al, 2003), the transferred PVDF membrane blotted with the following antibodies: mouse anti-huntingtin (MAB2166, Chemicon, Temecula, CA), rabbit anti-caspase-3 (#9662, # 9664, Cell Signal, MA), mouse anti-caspase-2 (#ab18737, Abcam Inc., Cambridge, MA), rabbit anti-caspase-6 (#9762, Cell Signal), rabbit anti-caspase-7 (#9491, Cell Signal), rabbit anti-caspase-8 (#4927, Cell Signal), rabbit anti-caspase-9 (#9504, Cell Signal), and rabbit anti-Neurofilament 200 kDa (AB1989, Chemicon). Blots were striped and reblotted with a mouse anti-tubulin (mouse monoclonal antibody, 1:5000) (Sigma). The signal in Western blot was scanned and its densitometry was analyzed by Quantity One (Bio-Rad, CA). Each lane was normalized to the tubulin signal.
Immunoprecipitation
Huntingtin antibody (MAB2166), caspase-3 antibodies (Cell Signal #9662), FLAG antibody (Sigma), and normal mouse and rabbit IgG (Santa Cruz, CA) were conjugated to protein A, or A/G plus agarose beads with 40 mM dimethylpimelimidate dihydrochloride dissolved in 0.1 M borate buffer (pH 9.0), and the conjugation reaction was stopped with 40 mM ethanolamine dissolved in 0.1 M borate buffer pH 8.0. In order to maximize both the levels of full-length huntingtin and active caspase-3, a mixed one-to-one lysate was prepared using naïve N2a cells as well as ones treated with TNFα/CHX for 12 h. Cells were lysed on ice by adding NP-40 buffer in presence of protease inhibitor cocktail set I (CalBiochem, CA) and precleaned with conjugated normal rabbit or mouse IgG at 4°C for 0.5–1 h. The precleaned lysates were incubated with the conjugated beads at 4°C for 2–6 h. Immune complexes were collected and washed with NP-40 buffer and then eluted with sample-loading buffer. For in vitro direct binding assay, the full-length huntingtin and mutant huntingtin were first purified from in vitro-translated lysates using the conjugated antibodies (MAB2166 or normal mouse IgG as control) overnight in 4°C. The purified proteins were then incubated with 2 μg purified recombinant active caspase-3 (Sigma) at 4°C for 2 h. The bound caspase-3 was collected by washing the beads with NP-40 buffer and then eluted with sample loading buffer.
In vitro transcription/translation
Adding 1 μl of 1 mM methionine with 2 μg DNA of wild-type huntingtin or mutant construct with 40 μl TNT T7 Quick Master Mix based on the reticulocyte lysate system (Promega, Madison, WI) up to a final volume of 50 μl. The reaction was incubated at 30°C for 90 min. In vitro huntingtin and mutant huntingtin production was confirmed by Western blot.
Preparation and induction of the cell-free system
Cell extracts were prepared from N2a or HeLa cells according to the method described in Deveraux et al (1997) and Roy et al (1997). Cells were washed by buffer A (20 mM HEPES, 10 mM KCL, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, and 0.1 mM Phenylmethylsulfonyl fluoride) and gently homogenized with a Kontes Dounce Tissue Grinder in 1 volume buffer A. After being centrifuged at 16 000 g for 30 min, supernatants of homogenates were collected and added NaCl to reach 50 mM and stored at −80°C. For cell-free experiments, 8 μl of N2a or HeLa cytosolic extracts were preincubated with in vitro-translated huntingtin before adding 10 μM Cytochrome c/1 mM dATP for 1 h at 37°C.
Immunocytochemistry
Generation of Hdh−/−chimeras and fixed brain sections were prepared as described previously (Dragatsis et al, 1998; Reiner et al, 2001). For β-galactosidase histochemistry, brain sections were washed in PBS, and incubated with a mixture of X-Gal solution and iron buffer (‘β-Gal Staining Set' from Boehringer Mannheim, Indianapolis, IN) with fluorescein-conjugated anticleaved caspase-3 antibody (1:200; # 9667, Cell Signal) overnight at 4°C. Slides were washed and counterstained with Hoechst 33342 (1:100 000; Molecular Probes, OR). For KA-treated YAC mice: 1 week after KA injection, the brains from one group of the mice were removed and immediately frozen in isopentane on dry ice. Serial 30-μm coronal cryostat sections were cut through the entire hippocampus. After every fifth section, two sections were removed for quantitative analysis and fixed in 3% paraformaldehyde for 30 min in preparation for histochemical staining. Degenerating neurons were identified in hippocampal sections by Fluoro-Jade histochemistry (Histo-Chem) and silver staining (FD Neurotechnologies). The total number of degenerated hippocampal neurons labeled with Fluoro-Jade was recorded for the CA1, CA3, and total hippocampus regions of each section selected for quantitative analysis. Slide-mounted sections were viewed with a Zeiss (Axiovert) fluorescent microscope, digital photomicrographs were captured with a cooled CCD camera (Princeton), and degenerating hippocampal neurons were counted manually in a blinded fashion.
Statistical analysis
Data are presented as the mean±s.e.m. Statistical comparisons were made by Student's t-test.
Supplementary Material
Supplementary Figure S1
Acknowledgments
This work was supported by grants from the Huntington's Disease Society of America (RMF and MRH), NIH/NINDS (RMF, MEM NS 32765 and NS 16367), the Hereditary Disease Foundation/High-Q Foundation (MRH, BRL, DG, and ID), the Canadian Genetic Diseases Network and the Canadian Institutes of Health Research (MRH and BRL). Dr Michael Hayden is the holder of a Canada Research Chair in Human Genetics.
References
- Cattaneo E, Conti L (1998) Generation and characterization of embryonic striatal conditionally immortalized ST14A cells. J Neurosci Res 53: 223–234 [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M (2005) Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci 6: 919–930 [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6: 797–801 [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304 [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM, Aronin N (1995) Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14: 1075–1081 [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Efstratiadis A, Zeitlin S (1998) Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development 125: 1529–1539 [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S (2000) Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet 26: 300–306 [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME (1995) Inactivation of the mouse Huntington's disease gene homolog Hdh. Science 269: 407–410 [DOI] [PubMed] [Google Scholar]
- Friedlander RM (2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348: 1365–1375 [DOI] [PubMed] [Google Scholar]
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Bussow K, Coleman SH, Gutekunst CA, Landwehrmeyer BG, Lehrach H, Wanker EE (2004) A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell 15: 853–865 [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983 [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40: 25–40 [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384: 368–372 [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR (2001) Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet 68: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR (2006) Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem 96: 1121–1129 [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157 [DOI] [PubMed] [Google Scholar]
- Metzler M, Chen N, Helgason CD, Graham RK, Nichol K, McCutcheon K, Nasir J, Humphries RK, Raymond LA, Hayden MR (1999) Life without huntingtin: normal differentiation into functional neurons. J Neurochem 72: 1009–1018 [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR (1995) Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81: 811–823 [DOI] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM (1999) Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399: 263–267 [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M (2004) Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci 24: 269–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Del Mar N, Meade CA, Yang H, Dragatsis I, Zeitlin S, Goldowitz D (2001) Neurons lacking huntingtin differentially colonize brain and survive in chimeric mice. J Neurosci 21: 7608–7619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E (2000) Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci 20: 3705–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Sipione S, Goffredo D, Zuccato C, Fossale E, Cattaneo E (2001) Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J Biol Chem 276: 14545–14548 [DOI] [PubMed] [Google Scholar]
- Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J 16: 6914–6925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Bayly CI, Gareau Y, Houtzager VM, Kargman S, Keen SL, Rowland K, Seiden IM, Thornberry NA, Nicholson DW (2001) Maintenance of caspase-3 proenzyme dormancy by an intrinsic ‘safety catch' regulatory tripeptide. Proc Natl Acad Sci USA 98: 6132–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97: 6763–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Rogers DA, Bissada N, Vogl AW, Hayden MR, Leavitt BR (2005) Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet 14: 1379–1392 [DOI] [PubMed] [Google Scholar]
- Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M (1998) Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol 152: 34–40 [DOI] [PubMed] [Google Scholar]
- Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE (2001) The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet 10: 1807–1817 [DOI] [PubMed] [Google Scholar]
- Wang X, Wang H, Figueroa BE, Zhang WH, Huo C, Guan Y, Zhang Y, Bruey JM, Reed JC, Friedlander RM (2005) Dysregulation of receptor interacting protein-2 and caspase recruitment domain only protein mediates aberrant caspase-1 activation in Huntington's disease. J Neurosci 25: 11645–11654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J Neurosci 22: 7862–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME (1997) Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat Genet 17: 404–410 [DOI] [PubMed] [Google Scholar]
- Woda JM, Calzonetti T, Hilditch-Maquire P, Duyao MP, Conlon RA, MacDonald ME (2005) Inactivation of the Huntington's disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev Biol 5: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A (1995) Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet 11: 155–163 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li M, Drozda M, Chen M, Ren S, Mejia Sanchez RO, Leavitt BR, Cattaneo E, Ferrante RJ, Hayden MR, Friedlander RM (2003) Depletion of wild-type huntingtin in mouse models of neurologic diseases. J Neurochem 87: 101–106 [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293: 493–498 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1