

Abstract
The phosphatidyl inositol 3-kinase-like kinases (PIKKs), ataxia-telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) regulate parallel damage response signalling pathways. ATM is reported to be activated by DNA double-strand breaks (DSBs), whereas ATR is recruited to single-stranded regions of DNA. Although the two pathways were considered to function independently, recent studies have demonstrated that ATM functions upstream of ATR following exposure to ionising radiation (IR) in S/G2. Here, we show that ATM phosphorylation at Ser1981, a characterised autophosphorylation site, is ATR-dependent and ATM-independent following replication fork stalling or UV treatment. In contrast to IR-induced ATM-S1981 phosphorylation, UV-induced ATM-S1981 phosphorylation does not require the Nbs1 C-terminus or Mre11. ATR-dependent phosphorylation of ATM activates ATM phosphorylation of Chk2, which has an overlapping function with Chk1 in regulating G2/M checkpoint arrest. Our findings provide insight into the interplay between the PIKK damage response pathways.
Keywords: ataxia telangiectasia-mutated protein, DNA damage responses, phosphorylation, PIKKs
Introduction
ATM and ATM- and Rad3-related (ATR) are phosphatidyl inositol 3-kinase-like kinases (PIKKs) that play key roles in the response to DNA damage (Shiloh, 2003; Abraham, 2004). ATM is activated by DNA double-strand breaks (DSBs), whereas ATR is recruited to single-stranded DNA regions, which arise at stalled replication forks or during the processing of bulky lesions such as UV photoproducts (Zou and Elledge, 2003; Zou et al, 2003). ATM is recruited to DNA DSBs by the Mre11–Rad50–Nbs1 (MRN) complex via an interaction between ATM and the C-terminus of Nbs1 (Falck et al, 2005). Mre11, which also interacts with the C-terminus of Nbs1, is additionally required for ATM activation (Uziel et al, 2003). ATM exists as an inactive dimer but undergoes autophosphorylation triggering monomerisation and activation (Bakkenist and Kastan, 2003). In contrast, ATR is recruited to RPA-coated single-stranded DNA regions via its interacting partner, ATR-interacting protein (ATRIP) (Cortez et al, 2001; Costanzo et al, 2003; Zou and Elledge, 2003). Via phosphorylation of damage response proteins, ATM and ATR activate cell cycle checkpoint arrest, aspects of DNA repair and, in some cell types, apoptosis (Shiloh, 2003). ATM and ATR share overlapping phosphorylation targets but also have distinct substrates. Most notably, ATM specifically phosphorylates and activates the transducer kinase, Chk2, whereas ATR preferentially phosphorylates Chk1 (Jazayeri et al, 2006). Ataxia telangiectasia (A-T) is a disorder conferred by mutations in ATM; ATR-Seckel Syndrome is associated with hypomorphic mutations in ATR (O'Driscoll et al, 2003; Chun and Gatti, 2004). The distinct clinical and cellular characteristics of A-T and ATR-Seckel patients and derived cell lines consolidate the notion that the two PIKKs regulate parallel damage response pathways that respond to distinct lesions. Recent studies have shown that after IR in S/G2, activation of the MRN complex by an ATM and CDK-dependent process creates single-stranded regions of DNA, which subsequently activate ATR (Jazayeri et al, 2006). Thus, in this situation, ATM functions upstream of ATR. Here, we provide evidence for a reverse situation by demonstrating that ATR functions upstream of ATM after UV treatment or replication fork stalling. In this situation, ATR can phosphorylate and activate ATM, demonstrating that ATM activation can occur without autophosphorylation.
Results and discussion
ATM-S1981 phosphorylation following hydroxyurea (HU) or UV irradiation is ATR dependent
Following UV irradiation (5 J m−2, 1 h post-treatment), phosphorylation of ATM on Ser1981 and H2AX on Ser139 (γ-H2AX) is observed by immunofluorescence in hTERT-immortalised human fibroblasts (Figure 1A). ATR-Seckel hTERT fibroblasts show much reduced phosphorylation of both substrates (Figure 1A and Supplementary Figure S1A). As phosphorylation is observed in ∼90% of cells, it most likely represents ATR activation during processing of UV photoproducts rather than following replication fork stalling (see also O'Driscoll et al, 2003). Nijmegen Breakage Syndrome (NBS) is another disorder caused by hypomorphic mutations in Nbs1. NBS114hTERT is an hTERT-immortalised fibroblast with a homozygous 1089C>A mutation which gives a clinically severe phenotype (Gennery et al, 2004). NBS114hTERT cells also show reduced but residual ATM-S1981 and H2AX phosphorylation (Figure 1A and Supplementary Figure S1A). The dependency of ATM-S1981 phosphorylation on ATR was surprising as Ser1981 is an established autophosphorylation site following IR treatment (Bakkenist and Kastan, 2003). To determine whether this represents autophosphorylation, we examined UV-induced phosphorylation of ATM and H2AX in the presence of the specific ATM kinase inhibitor, KU55933 (ATMi) (Hickson et al, 2004). Neither phosphorylation was perturbed by this treatment, substantiating that both events are mediated by ATR rather than ATM (Figure 1A and Supplementary Figure S1A). In contrast, treatment of IR-irradiated control cells with ATMi abolished ATM-S1981 phosphorylation but had no impact on γ-H2AX formation, consistent with the former event representing autophosphorylation and the latter being redundantly DNA-PK/ATM dependent (Figure 1B and Supplementary Figure S1B). It is noteworthy that the residual phosphorylation of ATR-dependent substrates routinely observed in ATR-Seckel cells, as demonstrated here using α-p-S1981-ATM and α-γ-H2AX antibodies, is unperturbed by treatment with the ATM inhibitor verifying that it represents ‘leaky' ATR-dependent phosphorylation rather than ATM-dependent phosphorylation (Figure 1A and Supplementary Figure S1A). Such ‘leakiness' is likely due to the hypomorphic mutation in the ATR-Seckel cell line. Treatment of lymphoblastoid cell lines (LBLs) with HU (1 mM, 2 h), an agent that causes replication fork arrest due to nucleotide depletion, similarly induced ATM-S1981 and H2AX phosphorylation in control but not ATR-Seckel cells, and transfection with ATR cDNA concomitantly corrected both defects in ATR-Seckel cells (Figure 1C and D). LBLs were used to obtain sufficient numbers of replicating cells. Control and ATR-Seckel LBLs had similar numbers of S-phase cells (∼35%), consistent with the percentage of cells displaying a phosphorylation signal (Supplementary Figure S2). To verify the specificity of the α-p-S1981-ATM antibodies, we also examined an A-T LBL and observed no signal with α-p-S1981-ATM antibodies, but efficient γ-H2AX formation following HU (Figure 1C). We verified these results by immunoblotting using α-p-S1981-ATM antibodies and observed ATM phosphorylation in HU-treated control cells in the presence and absence of the ATM inhibitor, but not in HU-treated ATR-Seckel or A-T cells (Figure 1E). Immunoblotting also confirmed that the level of ATM-S1981 phosphorylation in NBS LBLs was reduced compared to control cells (Figure 1E). Similar results were found after treatment with UV irradiation (5 J m−2, 1 h post-treatment) (Supplementary Figure S1C). To consolidate these findings obtained using ATR-Seckel cells, we also carried out ATR siRNA in LBLs and observed decreased UV and HU-induced but normal IR-induced ATM-S1981 phosphorylation (Figure 1F). Finally, we compared the level of phosphorylated ATM after UV, HU or IR and observed by immunofluorescence that 0.5 Gy IR gave a similar signal to that produced by 1 J m−2 UV or 1 mM HU for 2 h (Supplementary Figure S3).
Figure 1.
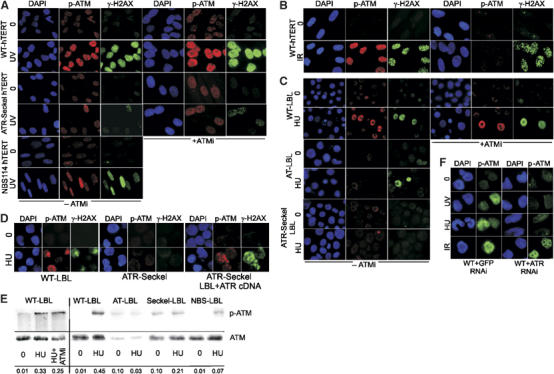
ATM S1981 phosphorylation after replication fork stalling or UV is ATR dependent and does not require ATM kinase activity. (A) Control (WT-hTERT), ATR-Seckel (ATR-Seckel hTERT) and NBS (NBS114 hTERT) fibroblasts were treated with UV (5 J m−2) and processed 1 h later for immunofluorescence using α-p-S1981 ATM or α-γ-H2AX antibodies either in the absence or presence of the ATM inhibitor, KU55933 (ATMi). We also added a DNA-PK inhibitor (NU7441) and observed the same pattern of phosphorylation (data not shown). Phosphorylated ATM and H2AX is ATR dependent and is not compromised by the presence of ATMi. Phosphorylated ATM and H2AX is diminished in the NBS114 cell line. Quantification of the results is shown in Supplementary Figure S1. All hTERT cell lines showed a similar percentage of S-phase cells (Supplementary Figure S2). (B) Control (WT-hTERT) fibroblasts were exposed to 3 Gy IR and examined 15 min later for immunofluorescence using α-p-ATM or α-γ-H2AX antibodies. Phosphorylation of ATM on S1981 is inhibited by ATMi, consistent with the anticipated autophosphorylation. γ-H2AX phosphorylation is not impaired consistently with the reported redundancy of this event with DNA-PK. (C) Control (WT-LBL), ATR-Seckel (ATR-Seckel LBL) and A-T (AT-LBL) LBLs were treated with HU (1 mM) and processed 2 h later for immunofluorescence as in (A). Approximately 30% of the control cells, most likely representing those that have progressed into S phase, show phosphorylation of ATM and H2AX either in the presence or absence of ATMi. This phosphorylation is ATR dependent and ATM independent. No signal is detected in A-T cells using α-p-ATM antibodies. As most phosphorylation after HU is ATR dependent and ATM is lacking, the loss of the signal confirms the specificity of these antibodies for ATM. All LBLs showed a similar percentage of S-phase cells, which is similar to the percentage of γ-H2AX responding cells (Supplementary Figure S2). (D) ATR-Seckel LBLs were transfected with empty vector or ATR cDNA and 48 h later subjected to HU and processed as described in (C) above. Approximately 10–20% of the transfected cells showed normal γ-H2AX phosphorylation reflecting the low transfection efficiency. However, close to 100% of the γ-H2AX-positive cells also recovered a signal with α-p-ATM antibodies. A positive and negative cell is shown. Cells transfected with empty vector looked identical to untransfected cells (data not shown). (E) Cells were treated as in (C), and samples prepared for Western blotting using α-p-ATM antibodies. The results confirm the findings shown in (C). The p-ATM signal intensity is quantified at the bottom of the panel, normalised to the total ATM signal. (F) LBLs were subjected to two rounds of siRNA with GFP control or ATR-specific oligonucleotides. After 48 h, cells were subjected to UV, HU or IR treatment and examined using α-p-ATM antibodies. Reduced ATM phosphorylation was observed after HU and UV, but not after IR, treatment only in cells treated with ATR siRNA.
We have shown previously that NBS cells display reduced ATR-dependent phosphorylation events after HU or UV treatment arguing that Nbs1 is required for ATR as well as ATM signalling (Stiff et al, 2005). Thus, an NBS-transformed cell line (NBS-ILB1) shows impaired UV-induced phosphorylation events demonstrated by reduced phosphorylation of Chk1 and SQTQ sites, the consensus PIKK phosphorylation motif (Figure 2A; Stiff et al, 2005). As described previously, HU- and UV-induced γ-H2AX phosphorylation is also reduced in NBS-ILB1 cells, although the defect is less marked compared to that observed for other substrates (Figure 2B; Stiff et al, 2005). These defects are fully complemented in cells expressing full-length Nbs1 (NBS-ILB1+Nbs1) (Figure 2A and B). Interestingly, we observed that C-terminally truncated Nbs652 cDNA, which lacks the ATM–Nbs1 interaction site (Desai-Mehta et al, 2001) can also complement these phosphorylation defects of NBS-ILB1 cells (Figure 2A and B). In contrast, Nbs652 was previously reported to be deficient for ATM signalling (Cerosaletti and Concannon, 2004). Thus, the C-terminal 100 amino acids of Nbs1 are dispensable for Nbs1 function in ATR signalling but are essential for ATM signalling. This allowed us to address whether the role of Nbs1 in ATM-S1981 phosphorylation after UV was due to its ability to facilitate ATM- or ATR-dependent signalling. NBS-ILB1 cells also show defective ATM-S1981 phosphorylation after UV or HU treatment, consistent with the findings in Figure 1A using NBShTERT cells (Figure 2B). Importantly, ATM-S1981 phosphorylation in response to HU or UV was restored in NBS-ILB1 cells expressing Nbs652 (Figure 2B). In contrast, ATM-S1981 phosphorylation was reduced in NBS-ILB1 cells and NBS-ILB1 cells expressing Nbs1-652 protein following IR, consistent with the previous findings that this represents ATM autophosphorylation requiring the C-terminus of Nbs1 (Cerosaletti and Concannon, 2004; Falck et al, 2005) (Figure 2C). γ-H2AX formation after IR was normal as DNA-PK redundantly phosphorylates H2AX (Stiff et al, 2004).
Figure 2.
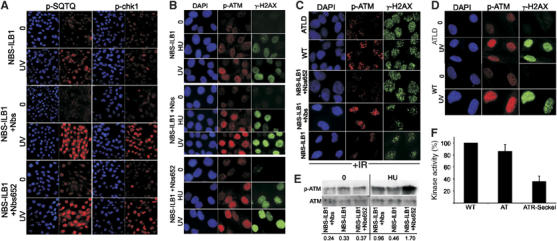
ATM phosphorylation on Ser1981 does not require the Nbs1–ATM interaction domain on Nbs1. (A) NBS-ILB1, a transformed NBS cell line either uncomplemented, complemented with full-length Nbs1 (NBS-ILB1+Nbs1) or expressing a C-terminally truncated Nbs1 cDNA (NBS-ILB1+nbs652) were treated with UV (5 J m−2) and processed 1 h later for immunofluorescence using α-p-Chk1 or α-SQTQ antibodies. NBS-ILB1 cells showed reduced phosphorylation of both substrates that is complemented by both full-length Nbs1 and nbs562 cDNA. (B) NBS-ILB1 and stably transfected derivatives described in (A) were treated with HU (1 mM) or UV (5 J m−2) and processed 2 or 1 h later, respectively, for immunofluorescence using α-p-ATM or α-γ-H2AX antibodies. These NBS cells show diminished ATM and H2AX phosphorylation that is fully restored following expression of either full-length Nbs1 or the C-terminally truncated protein. This strongly suggests that interaction between ATM and Nbs1 is dispensable for ATM phosphorylation after HU or UV treatment. (C) NBS-ILB1 and transfected derivatives described above, control and ATLD fibroblasts were exposed to 3 Gy IR and examined for γ-H2AX and ATM-S1981 phosphorylation 15 min later. Similar levels of γ-H2AX phosphorylation were observed in all lines as this can be carried out by DNA-PK. The reduced ATM-S1981 phosphorylation observed in NBS-ILB1 cells was corrected by full-length Nbs1 but not by Nbs652. ATLD cells showed reduced ATM-S1981 phosphorylation after IR. (D) In contrast to the results in (C) after IR, ATLD cells show normal ATM-S1981 phosphorylation after UV treatment. (E) Cells were treated with HU as in (B), and samples prepared for Western blotting using α-p-ATM and α-ATM antibodies. Although the NBS-ILB1 and derivative cells have a high endogenous level of phosphorylated ATM detectable by immunoblotting, an increased signal is detected after HU treatment in cells transfected with full-length or Nbs652 cDNA but not in untransfected cells. The results confirm the findings shown in (B). It is notable that NBSILB1 cells show low endogenous levels of p-ATM that is readily detectable by immunoblotting but less evident by immunofluorescence. The p-ATM signal intensity is quantified at the bottom of the panel, normalised to the total ATM signal. (F) ATR was immunoprecipitated using α-ATR antibodies from control (GMO2188), A-T (GMO3189D) and ATR-Seckel whole-cell extracts and used in a kinase assay using a peptide encompassing ATM-S1981. Reduced phosphorylation was observed in extracts from ATR-Seckel cells providing evidence that ATR can phosphorylate ATM-S1981 in vitro. The results represent the mean and standard error of two experiments normalising to the activity present in control cells.
The results also serve to demonstrate that neither the HU nor UV treatment that we have utilised activates ATM via DSB induction. These findings were reproduced by immunoblotting following HU treatment (Figure 2E). As Mre11-deficient ATLD1/2 cells have previously been shown to be defective in ATM-dependent but proficient in ATR-dependent events (Uziel et al, 2003; Stiff et al, 2005), we additionally examined an ATLD cell line. Consistent with this, ATLD1/2 cells showed proficient ATM-S1981 phosphorylation after UV but not after IR (Figure 2C and D).
Together, these findings demonstrate that the genetic requirements for Nbs1 in ATM compared to ATR signalling are distinct; the Nbs1 C-terminal region is essential for ATM signalling, while being dispensable for ATR signalling. The Nbs1 C-terminus is also dispensable for ATM-S1981 phosphorylation after HU and UV consolidating our notion that this represents an ATR-dependent phosphorylation event rather than ATM autophosphorylation.
Finally, we examined whether ATR is able to phosphorylate ATM-S1981 in vitro. As ATM can undergo autophosphorylation, we used a peptide encompassing S1981 as a substrate. ATR was immunoprecipitated using anti-ATR antibodies from control, A-T and ATR-Seckel LBLs and used in a kinase assay with the S1981 peptide as a substrate. Similar phosphorylation of the peptide was observed using control and A-T cells, which was significantly greater than that obtained when immunoprecipitation was carried out using ATR-Seckel cells providing strong evidence that ATR can phosphorylate ATM-S1981 (Figure 2F). The residual phosphorylation observed in ATR-Seckel cells is likely the result of residual ATR activity in this cell line. Immunoprecipitated ATM was also able to phosphorylate the S1981 peptide (data not shown).
ATM phosphorylation on Ser1981 after HU or UV does not require H2AX or 53BP1
Our findings strongly suggest that ATM is a downstream substrate in ATR-dependent damage response signalling. Previous studies have shown that the mediator proteins, H2AX and 53BP1, function to facilitate ATM and ATR signalling (Paull et al, 2000; Celeste et al, 2002; Ward et al, 2003). We, therefore, used mouse embryo fibroblasts (MEFs) defective for either H2AX or 53BP1 to examine ATM-S1981 phosphorylation after HU and UV treatment. We observed efficient ATM-S1981 phosphorylation in H2AX−/− and 53BP1−/− MEFs after HU or UV treatment by IF and immunoblotting (Figure 3A and B), suggesting that ATM is an upstream ATR substrate that does not depend upon γ-H2AX foci formation or the mediator protein 53BP1.
Figure 3.
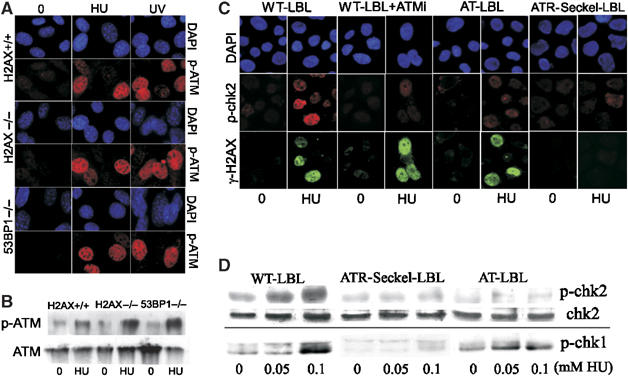
Phosphorylation of ATM on Ser1981 is not dependent upon H2AX or 53BP1; Chk2 phosphorylation is ATM and ATR dependent after HU treatment. (A, B) Wild type (H2AX+/+), H2AX −/− or 53BP−/− MEFs were treated with HU or UV as described in Figure 1 and examined for (A) immunofluorescence or (B) immunoblotting (after HU treatment) using α-p-S1981-ATM antibodies. Efficient ATM phosphorylation was observed in all cases. (C) Control (WT-LBL), control treated with the ATM inhibitor (WT-LBL+ATMi), ATR-Seckel (ATR-Seckel LBL) or A-T (AT-LBL) LBLs were treated with HU (1 mM) and 2 h later examined by immunofluorescence using α-p-Chk2 and γ-H2AX antibodies. Control cells efficiently phosphorylate both substrates after this treatment in contrast to ATR cells which show defective phosphorylation of both substrates. Loss of ATM results only in impaired Chk2 phosphorylation. (D) Cells were treated as in (C) and samples prepared for Western blotting. The results confirm the findings shown in (C) and show that, in contrast to Chk2, phosphorylation of Chk1 after HU is ATR dependent and ATM independent.
ATR-dependent ATM-S1981 phosphorylation activates ATM kinase activity in vivo
Although ATM and ATR have similar consensus phosphorylation motifs and target common substrates, evidence suggests that Chk1-S317 and Chk2-T68 are specific ATR and ATM substrates, respectively (Jazayeri et al, 2006). We next examined the PIKK dependency for Chk2 phosphorylation after HU and UV treatment using control, control treated with ATM inhibitor, A-T or ATR-Seckel LBLs. Using immunofluorescence, we observed that Chk2 phosphorylation following HU treatment was ATM and ATR dependent in contrast to γ-H2AX formation which is solely ATR dependent (Figure 3C) consistent with the notion that ATM is a downstream component in the ATR pathway and that ATM-S1981 phosphorylation by ATR activates ATM kinase activity, which specifically targets Chk2. To consolidate these findings, we examined Chk2-T68 phosphorylation using immunoblotting with phosphospecific antibodies (Figure 3D). We observed reduced Chk2-T68 phosphorylation in both ATR-Seckel and A-T LBLs in contrast to wild-type LBLs. In contrast, Chk1 phosphorylation is ATR dependent but ATM independent after HU treatment (Figure 3D) as reported previously (Stiff et al, 2005).
ATM-dependent Chk2 phosphorylation in response to UV functions redundantly with Chk1 to effect G2/M checkpoint arrest
Our findings demonstrate that both Chk1 and Chk2 are activated after UV with Chk1 being solely ATR dependent and Chk2 being ATR and ATM dependent. To determine whether ATM-dependent Chk2 activation after UV treatment might impact functionally, we investigated whether ATM and/or Chk2 might contribute to UV-induced G2/M arrest. We examined UV-induced G2/M checkpoint arrest after various treatments causing inhibition of ATM, Chk1, Chk2 or both Chk1 and Chk2. Previously, we found that analysis of mitotic index (MI) after UV treatment is an efficient monitor of G2/M arrest (Alderton et al, 2004; Stiff et al, 2005). Our previous time course analysis of G2/M arrest after UV treatment in the presence of nocodazole has shown it to be ATR-dependent and readily detectable 5 h post-treatment. First, we utilised wild-type and Chk2 knockout MEFs to confirm that UV-induced G2/M checkpoint arrest is ATM and Chk2 independent (Figure 4A). Consistent with previous findings using human A-T cell lines, we found that wild-type MEFs treated with the ATMi, Chk2-deficient MEFs and Chk2-deficient MEFs treated with ATMi effected UV-induced G2/M checkpoint arrest efficiently. Note that although Chk2−/− MEFs have a lower MI compared to wild-type MEFs, there was clear checkpoint arrest after UV treatment. To examine the contribution of Chk1 to UV-induced G2/M arrest, we utilised UCN-01, a drug which at low concentrations (100 nM) specifically inhibits Chk1 but not Chk2 (Feijoo et al, 2001). The inhibition of Chk1 is demonstrated by the fact that 53BP1foci formation after HU, a Chk1-dependent event, is inhibited by 100 nM UCN-01 (Supplementary Figure S4). UCN-01 (100 nM) did not impact upon G2/M arrest in control MEFs, but completely inhibited the arrest in Chk2-deficient MEFs. UCN-01 (600 nM) has been shown to inhibit both Chk1 and Chk2 (Yu et al, 2002; Adamson et al, 2005), and 600 nM UCN-01 inhibited arrest in control and Chk2−/− MEFs. Importantly, 100 nM UCN-01 also inhibited G2/M arrest in MEFs treated with ATMi, consistent with the notion that abolition of G2/M arrest requires loss of Chk1 and Chk2 function. Moreover, addition of debromohymenialdisine (DBH), another drug that jointly inhibits Chk1 and Chk2, resulted in loss of G2/M arrest after HU treatment (Figure 3A) (Curman et al, 2001). We also exploited siRNA to examine the impact of loss of Chk1 function. For this we utilised human cells, in which efficient Chk1 siRNA can be achieved. Chk1 siRNA resulted in significant knock down of Chk1 and impaired 53BP1 foci formation (Supplementary Figure S5). Diminished Chk1 activity alone, either via 100 nM UCN-01 or Chk1 siRNA, was without impact on UV-induced G2/M arrest in 1BR3neo cells (Figure 4B). Importantly, both 100 nM UCN-01 or Chk1 siRNA plus the presence of ATMi, to prevent Chk2 activation, now resulted in abrogated G2/M arrest. As anticipated, 600 nM UCN-01 also caused abrogated arrest. We have previously shown that ATR-Seckel cells fail to arrest at the G2/M checkpoint after UV. We show here that loss of ATM activity, Chk1 function or Chk2 function alone does not impact upon G2/M arrest, while the combined loss of Chk1 and Chk2, either directly or via inhibition of ATM, results in abolished arrest. Thus, our findings strongly suggest that Chk1 and Chk2 have overlapping functions in regulating mitotic entry. Hence, ATM can impact upon G2/M arrest after UV via its regulation of Chk2 but this role is downstream of ATR and functions in a cooperative manner with ATR-dependent Chk1 activation. Therefore, our findings suggest that there are two parallel pathways leading to G2/M arrest after UV irradiation, both ATR dependent and one ATM dependent.
Figure 4.
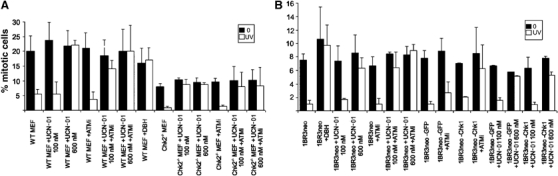
ATM-dependent phosphorylation of Chk2 and ATR-dependent phosphorylation of Chk1 redundantly overlap to effect UV-induced G2/M arrest. (A) Wild-type or Chk2−/− MEFs were treated with UV (2.5 J m−2) and the mitotic index was scored 5 h later. Cells were pretreated as indicated; +ATMi, addition of 10 μM KU55933; +UCN101 (100 or 600 nM); +DBH (30 μM) all for 30 min before UV treatment. Results represent the mean and s.d. of three experiments. (B) 1BR3neo cells, treated as indicated, were treated with UV and the mitotic index was scored as in (A). Treatment with ATMi, DBH and UCN101 was as in (A); +GFP, siRNA using GFP-specific oligonucleotides;+Chk1, siRNA using Chk1-specific oligonucleotides. The efficiency of knock down of Chk1 by Western blotting is shown in Supplementary Figure 5. This figure also shows that 53BP1 foci formation, which is Chk1 dependent, is abrogated after Chk1 siRNA but not after siRNA using GFP oligonucleotides. Neither knockdown of Chk1 nor loss of Chk2 has any impact on UV-induced G2/M arrest. However, loss of Chk1 and Chk2 (or ATM) function by various means abolishes G2/M arrest.
Chk2 phosphorylation after HU or UV treatment has also been reported in other studies (Matsuoka et al, 1998; Brown et al, 1999; Feijoo et al, 2001; Dodson and Tibbetts, 2006; Jazayeri et al, 2006). Although a dependency upon ATM has been observed (Dodson and Tibbetts, 2006; Jazayeri et al, 2006), this was not a universal finding (Brown et al, 1999; Feijoo et al, 2001). To address this, we examined the ATR/ATM dependency after prolonged HU treatment (5 mM, 24 h) and observed some ATM-independent activation and Chk2 phosphorylation (data not shown). Here, we utilise relatively mild treatment regimes (1 h after 5 J m−2 UV irradiation or 2 h post-treatment with 1 mM HU) to enable us to see a defect in the hypomorphic ATR-Seckel cell line. It is possible, therefore, that Chk2 is an ATR substrate at later times post-treatment, the conditions used in the two studies reporting ATM-independent Chk2 activation. It is also possible that DSBs may arise after high UV doses leading to ATM-dependent, ATR-independent activation. One study also reported ATM-S1981 phosphorylation in response to UV (Dodson and Tibbetts, 2006). Using siRNA of ATR to abrogate function, a partial ATR dependency for ATM-S1981 phosphorylation was reported. However, the ATR knockdown was partial with only modestly decreased Chk1-S317 phosphorylation. In contrast to our findings, the authors concluded that this did not represent direct phosphorylation of ATM by ATR, as a catalytically inactive ATM protein failed to undergo phosphorylation. It was notable that the ATM mutation that inactivated its kinase function also appeared to diminish ATM stability (Dodson and Tibbetts, 2006). It is, therefore, unclear whether the primary effect of this mutation is on the kinase activity of ATM or on the stability of the protein. Our findings demonstrate that ATR regulates ATM phosphorylation after UV and HU treatment but do not definitely show that this represents direct phosphorylation of ATM by ATR. However, we provide evidence that ATR is able to phosphorylate S1981-ATM in vitro. Moreover, given that the substrate specificities of ATM and ATR significantly overlap and that they share common targets in vivo, our findings are consistent with the notion that ATM itself may be a common substrate, and that ATR directly phosphorylates ATM. Interestingly, CREB was identified as a further substrate phosphorylated in an ATM-dependent manner after UV treatment providing evidence for additional ATM-specific functions (Dodson and Tibbetts, 2006).
In summary, we show that ATM is phosphorylated at Ser1981 and activated in an ATR-dependent manner following replication fork stalling and UV irradiation. Additionally, we show that both ATR and ATM can contribute to UV-induced G2/M checkpoint arrest via Chk1 and Chk2 in an overlapping manner (depicted in Figure 5).
Figure 5.
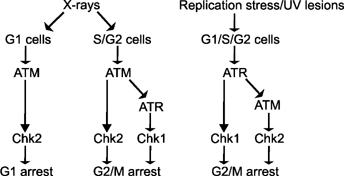
Model depicting the routes leading to ATM and ATR activation after different forms of DNA damage. X-rays induce DNA DSBs, which directly activate ATM via autophosphorylation in S and G2 phase. In S and G2 phase, ATM activates the MRN complex, which generates single-stranded DNA regions by end-resection leading to ATR activation. Replication fork stalling or UV irradiation activate ATR which phosphorylates and activates ATM. ATM has selectivity for Chk2 as a substrate and ATR shows Chk1 selectivity. These two kinases can contribute after replication fork stalling to G2/M arrest.
We provide strong evidence that, rather than representing autophosphorylation and activation, as occurs after exposure to IR, ATM becomes phosphorylated by ATR as (i) ATM-S1981 phosphorylation occurs even in the presence of an ATM inhibitor, (ii) cells lacking the C-terminus of Nbs1 and ATLD1/2 cells are proficient for UV-induced ATM-S1981 phosphorylation while being deficient for IR-induced ATM phosphorylation, (iii) Chk2 phosphorylation after UV is ATR and ATM dependent.
We also present evidence for a functional impact of ATM activation after UV by showing that ATM-specific phosphorylation of Chk2 has an overlapping function with Chk1 in contributing to UV-induced G2/M arrest. Both Chk1 and Chk2 phosphorylate and thereby inhibit the Cdc25A and C phosphatases, which dephosphorylate inhibitory Cdk1 phosphorylation and thereby regulate mitotic entry. However, the overlap between Chk1 and Chk2 and whether they have distinct impacts on Cdc25 inactivation is still unclear. It is likely, however, that the two kinases can function cooperatively although harsher treatments may preclude the ability to see a dual dependency as strong Chk1 activation alone may be sufficient to prevent Cdk1 dephosphorylation and activation.
In conclusion, we show that ATM is an ATR-dependent substrate under specific conditions potentially allowing its activation in the absence of DSB formation. Our findings are complementary to recent reports stating that following exposure of G2 cells to IR, ATR can be activated in a manner dependent upon ATM and CDKs (Jazayeri et al, 2006; Myers and Cortez, 2006). Thus, the two PIKKs do not function as independent signalling pathways to distinct forms of DNA damage, but rather act in concert. A full understanding of the substrate specificities of the two PIKKs is necessary to reveal their distinct roles.
Materials and methods
Cell culture
Fibroblasts utilised were 48BRhTERT (control), F02-98hTERT (ATR-Seckel) (O'Driscoll et al, 2003) and NBS114hTERT (Gennery et al, 2004). NBS114hTERT has a homozygous 1089C>A mutation in Nbs1 and was derived from a patient with clinical features that overlap with Fanconi anaemia. This cell line displays a marked defect in ATR signalling. LBLs were used for most of these studies to ensure sufficient numbers of replicating cells. GMO2188, GM3189D, LB195 and DK0064 were used as control, A-T, NBS and ATR-Seckel LBLs, respectively. NBS-ILB1 are transformed NBS fibroblasts from Dr M Zdzienicka. NBS-ILB1+Nbs1 and NBS-ILB1+Nbs652 are retrovirally complemented with full-length human Nbs1 or Nbs1 truncated at residue 652, respectively. ATLD1/2 has a truncating mutation in Mre11 (Stewart et al, 1999). ATLD1/2 LBL and primary fibroblast cells were kindly supplied by Dr M Taylor. 1BR3Neo is an SV40 immortalised cell line. H2AX−/− and 53BP1−/− MEFs were kindly supplied by Dr A Nussenzweig and Dr J Chen, respectively. LBLs were grown in RPMI medium supplemented with 15% foetal calf serum (FCS), penicillin and streptomycin. 1BR3Neo, MEFs and NBS-transformed cells were grown in MEM supplemented with 15% FCS, penicillin and streptomycin.
Treatment with DNA-damaging agents and ATM inhibitor
Irradiation was carried out using a UVC source (0.6 J m−2 s−1). HU was purchased from Sigma-Aldrich (Poole, UK). The KU55933 ATM inhibitor used was from Kudos Pharmaceuticals (Cambridge, UK) at a final concentration of 10 μM. Cells were pretreated for 30 min before irradiation. DBH was from Calbiochem (San Diego, CA) and used at a final concentration of 30 μM. UCN-01 was from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD.
Immunofluorescence and immunoblotting
α-chk1Ser317, α-p-SQTQ and α-chk2Thr68 antibodies were purchased from Cell Signaling Technology (Beverely, MA, USA). α-H2AXSer139 and α-Histone H3Ser10 antibodies were from Upstate Technology (Buckingham, UK). α-chk2, α-ATR and cjunSer63 antibodies were from Santa Cruz (Santa Cruz, CA). α-Ser1981 ATM antibodies were from Rockland Immunochemicals (Gilbertsville, PA). α-ATM antibodies were from Genetex (San Antonio, TX). α-53BP1 antibodies were from Bethyl Laboratories (Montgomery, TX). α-BrdU antibodies were from Abcam (Cambridge, UK). Anti-rabbit, anti-rat and anti-mouse secondary antibodies were purchased from Dako (Glostrup, Denmark).
Cells were fixed in 3% paraformaldehyde, 2% sucrose phosphate-buffered saline (PBS) for 10 min at room temperature and permeabilized in 20 mM HEPES pH 7.4, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton X-100 (Sigma-Aldrich, Poole, UK) for 2 min at 4°C. Thereafter, coverslips were washed in PBS before immunostaining. Primary antibody incubations were performed for 40 min at 37°C at 1:100 dilutions (1:800 for α-γ-H2AX) in PBS supplemented with 2% bovine serum fraction V albumin (BSA) (Sigma-Aldrich, Poole, UK) and followed by washing in PBS. Incubations with α-mouse TRITC and FITC or with α-rabbit FITC secondary antibodies (Sigma-Aldrich, Pool, UK) were performed at 37°C at 1:100 in 2% BSA for 20 min. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Poole, UK) for 10 min at 4°C. Coverslips were mounted in Vectashield (Vector Laboratories, Peterborough, UK). The error bars represent the standard deviation of the mean. A minimum of three experiments were carried out where error bars are shown.
Quantification of fluorescence intensity was performed on blind captured images using standardised capture settings, image processing and analysis performed on Simple PCI software. Variation in immunostaining between experiments was controlled by standardising to the background in untreated cells. At least 100 cells were quantified for each time point.
G2/M checkpoint arrest
Cells were exposed to 2.5 J m−2 UV and incubated for 5 h in complete medium containing 1.5 μM Nocodazole, followed by processing for immunofluorescence as detailed above. Mitotic cells were detected by α-Histone H3Ser10 antibodies and cells were counterstained with DAPI.
Small interfering RNA
Transfection of siRNA was carried out using siPORTTM NeoFXTM (Ambion, Austin, TX) following the manufacturer's instructions, with a final siRNA concentration of 10 nM. The siRNA duplexes were purchased from Invitrogen (Carlsbad, CA) and the sequence for CHK1 was as follows: AAUCGUGAGCGUUUGUUGAACTT. The negative control GFP siRNA duplexes were from Dharmacon (Lafayette, CO) and the target sequence was GGCTACGTCCAGGAGCGCACC.
ATR kinase assay
ATR or ATM was pulled down from 1 mg of whole-cell extract with 10 μl of anti-ATR/ATM antibody for 4 h at 4°C, then isolated using protein A sepharose beads (Amersham, UK) for 30 min. Beads were washed twice each with lysis buffer, lysis buffer with 500 mM NaCl, kinase buffer (20mM HEPES pH 7.5, 50 mM NaCl, 10 nM MgCl2, 1 mM DTT, 10 mM MnCl2) with 0.5 M LiCl, and kinase buffer. The kinase reaction was carried out in 30 μl kinase buffer with 0.5 μg ATM peptide (peptide sequence S-L-A-F-E-E-G-S-Q-S-T-T-I-S-S, Rockland Immunochemicals, Gilbertsville, PA) and 2 μM ATP for 30 min at 30°C. Reaction samples were spotted onto nitrocellulose paper and p-ATM signal was detected by immunoblotting with α-Ser1981 ATM antibodies. The blot was scanned and quantified using Simple PCI software normalised to background from protein A sepharose only pulldown.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Acknowledgments
The PAJ laboratory is supported by the Medical Research Council, the Human Frontiers Science Programme, the Leukaemia Research Fund, the International Agency for Cancer Research and EU grant (FIGH-CT-200200207). KC in the PC laboratory is supported by Grant CA57569 from the National Cancer Institute. EP is a member of the Keith Caldecott laboratory and collaborated on the experiments using UCN-01. AAG is supported by a fellowship from the Alberta Heritage Foundation for Medical Research.
References
- Abraham RT (2004) PI 3-kinase related kinases: ‘big' players in stress-induced signaling pathways. DNA Repair (Amst) 3: 883–887 [DOI] [PubMed] [Google Scholar]
- Adamson AW, Beardsley DI, Kim WJ, Gao Y, Baskaran R, Brown KD (2005) Methylator-induced, mismatch repair-dependent G2 arrest is activated through Chk1 and Chk2. Mol Biol Cell 16: 1513–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton GK, Joenje H, Varon R, Borglum AD, Jeggo PA, O'Driscoll M (2004) Seckel syndrome exhibits cellular features demonstrating defects in the ATR signalling pathway. Hum Mol Genet 13: 3127–3138 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Brown AL, Lee CH, Schwarz JK, Mitiku N, Piwnica-Worms H, Chung JH (1999) A human Cds1-related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc Natl Acad Sci USA 96: 3745–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A (2002) Genomic instability in mice lacking histone H2AX. Science 4: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerosaletti K, Concannon P (2004) Independent roles for nibrin and Mre11-Rad50 in the activation and function of Atm. J Biol Chem 279: 38813–38819 [DOI] [PubMed] [Google Scholar]
- Chun HH, Gatti RA (2004) Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 3: 1187–1196 [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213 [DOI] [PubMed] [Google Scholar]
- Curman D, Cinel B, Williams DE, Rundle N, Block WD, Goodarzi AA, Hutchins JR, Clarke PR, Zhou BB, Lees-Miller SP, Andersen RJ, Roberge M (2001) Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J Biol Chem 276: 17914–17919 [DOI] [PubMed] [Google Scholar]
- Desai-Mehta A, Cerosaletti KM, Concannon P (2001) Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol Cell Biol 21: 2184–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson GE, Tibbetts RS (2006) DNA replication stress-induced phosphorylation of cyclic AMP response element-binding protein mediated by ATM. J Biol Chem 281: 1692–1697 [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C (2001) Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol 154: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennery AR, Slatter MA, Bhattacharya A, Barge D, Haigh S, O'Driscoll M, Coleman R, Abinun M, Flood TJ, Cant AJ, Jeggo PA (2004) The clinical and biological overlap between nijmegen breakage syndrome and fanconi anaemia. Clin Immunol 113: 214–219 [DOI] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159 [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8: 37–45 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Myers JS, Cortez D (2006) Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem 281: 9346–9350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886–895 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99: 577–587 [DOI] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX following exposure to ioninsing radiation. Cancer Res 64: 2390–2396 [DOI] [PubMed] [Google Scholar]
- Stiff T, Reis C, Alderton GK, Woodbine L, O'Driscoll M, Jeggo PA (2005) Nbs1 is required for ATR-dependent phosphorylation events. EMBO J 24: 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22: 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Minn K, van Deursen J, Chen J (2003) p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 23: 2556–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, La Rose J, Zhang H, Takemura H, Kohn KW, Pommier Y (2002) UCN-01 inhibits p53 up-regulation and abrogates gamma-radiation-induced G(2)-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res 62: 5743–5748 [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
- Zou L, Liu D, Elledge SJ (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA 100: 13827–13832 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5