

Abstract
G-protein-coupled receptors (GPCRs) are key players in cell communication. Although long considered as monomeric, it now appears that these heptahelical proteins can form homo- or heterodimers. Here, we analyzed the conformational changes in each subunit of a receptor dimer resulting from agonist binding to either one or both subunits by measuring the fluorescent properties of a leukotriene B4 receptor dimer with a single 5-hydroxytryptophan-labeled protomer. We show that a receptor dimer with only a single agonist-occupied subunit can trigger G-protein activation. We also show that the two subunits of the receptor dimer in the G-protein-coupled state differ in their conformation, even when both are liganded by the agonist. No such asymmetric conformational changes are observed in the absence of G-protein, indicating that the interaction of the G-protein with the receptor dimer brings specific constraints that prevent a symmetric functioning of this dimer. These data open new options for the differential signaling properties of GPCR dimers.
Keywords: activation, asymmetry, dimer, GPCR, G-protein
Introduction
G-protein-coupled receptors (GPCRs) are versatile biological sensors that are responsible for the majority of cellular responses to hormones and neurotransmitters as well as for the senses of sight, smell and taste (Bockaert and Pin, 1999; Bockaert et al, 2002). Signal transduction is associated with a set of changes in the tertiary structure of the receptor that are recognized by the associated intracellular partners, in particular the G-proteins (Kobilka, 2002; Perez and Karnik, 2005). A growing body of evidence points to the fact that GPCRs exist as homo- or heterodimers (Bulenger et al, 2005), and the role of dimerization in receptor functioning is under extensive investigation (Hansen and Sheikh, 2004; Milligan, 2004; Terrillon and Bouvier, 2004). Receptor dimerization is, in some cases, required for a correct addressing of the receptor to the membrane. This is clearly demonstrated for class C GPCRs such as the GABAB receptor (Jones et al, 1998; Kaupmann et al, 1998; White et al, 1998; Robbins et al, 2001) and for some class A receptors (Grosse et al, 1997; Karpa et al, 2000; Lee et al, 2000). Dimerization is also likely required for an efficient interaction with intracellular partners including the G-protein (Banères and Parello, 2003; Jastrzebska et al, 2006) and certainly plays a role in receptor internalization (Perron et al, 2003; Stanasila et al, 2003).
There is evidence that receptor dimerization and activation are intricately associated. This has been clearly demonstrated for class C receptors (Pin et al, 2005). However, it is still unclear whether only one or both subunits in a receptor dimer need to be activated for function. For example, activation of both subunits in a δ–κ opioid receptor heterodimer is required for optimal activation of this complex (Jordan and Devi, 1999). Similarly, activation of both subunits in an M3 muscarinic receptor dimer appears to be required for arrestin recruitment (Novi et al, 2005), and activation of both D1 and D2 subunits in a D1–D2 dopamine heterodimer is required for phospholipase C activation (Lee et al, 2004). Consistent with both subunits in a receptor dimer being able to reach an active state, we recently reported, using the purified leukotriene B4 (LTB4) BLT1 receptor dimer in the absence of G-proteins, that both receptor protomers reach a similar conformational state upon binding the agonist (Mesnier and Banères, 2004).
Several reports, however, illustrate an asymmetric functioning of GPCR dimers. For example, it has been recently shown that a single heptahelical transmembrane domain within the homodimeric glutamate receptors reaches an active conformation at a time (Goudet et al, 2005; Hlavackova et al, 2005). Increasing number of data illustrating strong negative cooperativity in agonist binding and/or agonist action on GPCR dimers (El-Asmar et al, 2005; Urizar et al, 2005; Springael et al, 2006) also supports such an idea. An interesting model proposed on the basis of these results considers that intracellular proteins interacting with GPCR dimers, and in particular the heterotrimeric G-protein, are responsible for the receptor dimer asymmetric functioning. This would be in agreement with the proposed model for receptor: G-protein complex where a single heterotrimeric G-protein interacts with a receptor dimer (Banères and Parello, 2003; Filipek et al, 2004).
In this context, we have analyzed the conformational changes in each subunit of a purified 5-hydroxytryptophan-labeled BLT1 receptor dimer in the absence and in the presence of G-proteins. Whereas similar changes in fluorescence are observed in each subunit in the absence of G-protein, addition of the Gαβγ heterotrimer results in different conformational changes of the two subunits even when the LTB4 agonist occupies both. This strongly suggests that the G-protein restricts the possibility of receptor conformational changes in the receptor dimer and is therefore responsible for its asymmetric functioning.
Results
The R:R0 heterodimer
To distinguish the two protomers in the BLT1 dimer, we used here the previously described R.R0 heterodimer (Mesnier and Banères, 2004). R.R0 is a dimer composed of a wild-type protomer (R) and a mutant protomer (R0) where Cys97 in TM3 has been replaced by an alanine (Figure 1). The C97A mutation results in a ca. 100-fold decrease in the affinity of BLT1 for LTB4 without affecting the structure of the receptor in a way our methods can detect (Mesnier and Banères, 2004). We previously established that R.R0 is stable as a dimer under our experimental conditions (Mesnier and Banères, 2004).
Figure 1.
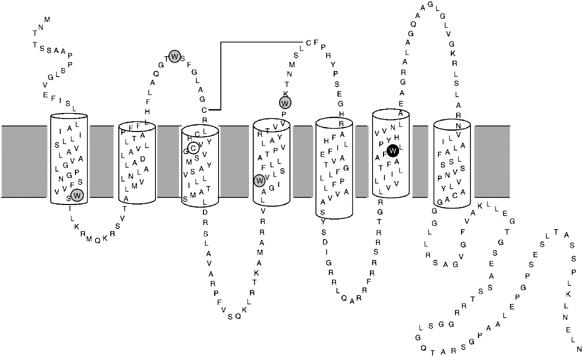
Secondary structure model of human BLT1 showing positions of the Trp residues. The Trp residues removed from the BLT1 receptor are circled in gray. The single Trp residue kept, Trp234, is circled in black. The position of Cys97 that is replaced by an alanine in R0 is also indicated.
We have used intrinsic fluorescence to monitor receptor activation. To simplify the analysis of the fluorescence profiles, all the tryptophan residues besides Trp234 were replaced by leucines in both R and R0 (Figure 1). Trp234 is located in TM6 and is highly conserved in class A GPCRs. It is the only Trp residue in BLT1 whose fluorescence properties are sensitive to the activation state of the receptor (Banères et al, 2003). Mutating all the Trp to Leu except Trp234 affects neither the ligand binding nor the structural properties of BLT1 (Banères et al, 2003).
We purified the R:R0 dimer as previously described (Mesnier and Banères, 2004). Briefly, R and R0 are labeled with two different purification tags, namely an S-tag (R) and a Strep-tag (R0). These two receptor molecules are mixed in equimolecular amounts before refolding and then refolded using the matrix-assisted refolding procedure described in Banères et al (2003). As stated above, the C97A mutation does not affect the structural features and dimerization properties of BLT1. We therefore obtain, after refolding, a mixture of the two R:R and R0:R0 homodimers and the R:R0 heterodimer. Since only R.R0 bears both the S-tag and the Strep-tag, it can be purified through two successive chromatographic steps involving each of these affinity tags.
Ligand binding to the BLT1 R:R0 heterodimer
We first analyzed the effects of the G-protein on the ligand-binding properties of R:R0. Ligand binding to the R:R0 heterodimer occurs in a stepwise manner whether Gαi2β1γ2 is present or not (Figure 2). In both cases, at low LTB4 concentration, the agonist first binds to the high-affinity site in the wild-type protomer R. Then, at higher LTB4 concentration, it binds to the low-affinity site in the mutant protomer R0. However, as clearly shown in Figure 2, Gαi2β1γ2 affects the affinity of the two protomers for LTB4 in a very different way. The affinity of R for LTB4 is increased by a ca. 10-fold when the G-protein is added (Kd=1.1±0.3 nM and 13.3±0.9 nM in the presence and absence of G-proteins, respectively; standard deviation from the mean value calculated from three independent experiments). This increase in affinity is in the same range than that measured when leukocyte membrane fractions containing BLT1 are reconstituted with exogenous G-proteins (Igarashi et al, 1999). In contrast, the affinity of R0 for LTB4 is not significantly affected by the addition of the G-protein trimer (Kd=227±9 and 215±8 nM in the presence and absence of G-proteins, respectively; standard deviation from the mean value calculated from three independent experiments). The coupling to the G-protein therefore affects the affinity of the R:R0 dimer for the agonist in a non-symmetric manner, with essentially only one of the protomers, R, being affected. No effect on the ligand-binding properties of the R.R0 dimer is observed when the Gαi subunit is replaced by GαS in the G-protein complex (see Supplementary data 1), clearly indicating that the increase in the affinity of R for LTB4 is a specific effect associated with the coupling to the Gαi subunit.
Figure 2.
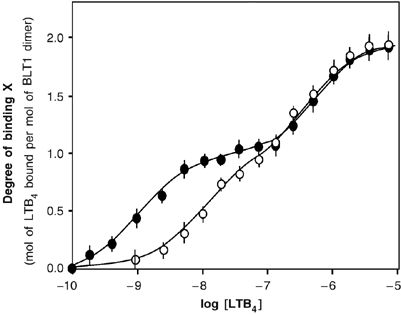
Agonist binding to R:R0 in the absence and presence of Gαi2β1γ2. Direct binding of LTB4 to the purified R:R0 dimer in the absence (open circles) or in the presence (closed circles) of purified Gαi2β1γ2. The binding data are presented as the degree of binding X (ratio of the moles of bound LTB4 per mole of receptor dimer) as a function of LTB4 concentration. The solid lines represent the theoretical profiles calculated from the experimental binding data. The error bars correspond to the standard deviation from the mean value calculated from three independent experiments.
G-protein activation
Functional coupling between the BLT1 receptor loaded with either one or two agonist molecules and the G-protein was then assessed by examining the ability of the purified receptor to stimulate GDP/[35S]GTPγS exchange on the α subunit.
To assess the biological relevance of our refolded BLT1 preparations, we first compared the [35S]GTPγS binding induced by the wild-type BLT1 receptor produced in Escherichia coli to that induced by membrane fractions of Chem-1 cells stably transfected with the human BLT1 sequence (the assays were carried out under the same experimental conditions; see Materials and methods). As shown in Figure 3, very similar [35S]GTPγS binding time course profiles are obtained in both cases. The time course profiles in Figure 3 are also similar to those reported for different Gαi-coupled receptors reconstituted in vitro with G-proteins (Kurose et al, 1991; Bae et al, 1999; Saidak et al, 2006). G-protein activation observed with the bacterially expressed BLT1 receptor is a specific effect as no noticeable GDP/GTP exchange occurs when Gαs is used instead of Gαi2 (not shown), in agreement with the fact that BLT1 does not activate Gαs in vivo (Masuda et al, 2003). All these results strongly suggest that the ability of our purified recombinant receptor preparation to activate G-proteins is biologically relevant.
Figure 3.
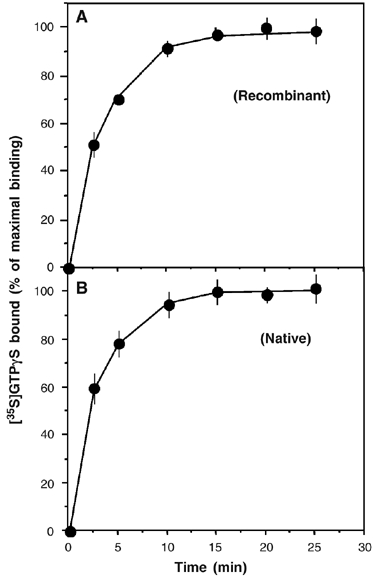
BLT1-catalyzed [35S]GTPγS binding to the G-protein. Time-dependent GDP/GTP exchange on Gαi catalyzed by the wild-type BLT1 receptor produced in E. coli (A) or in chem-1 cell membrane fractions (B). Data are expressed as the percent of maximal binding. Specific agonist-stimulated [35S]GTPγS was calculated by subtracting binding in the absence of agonist from binding in the presence of agonist at each point. In all cases, data represent the mean s.e. from three independent experiments.
To validate our GTPγS-binding assays, we also verified that, under the conditions used, the receptor rather than the G-protein concentration is rate limiting. For this, we carried out the GTPγS-binding experiments at two different receptor concentrations. As clearly shown in Figure 3 (inset in Figure 4A), doubling the amount of the receptor in the assay increases [35S]GTPγS binding by a ca. two-fold factor, establishing that under our conditions it is indeed the receptor concentration that is rate-limiting.
Figure 4.
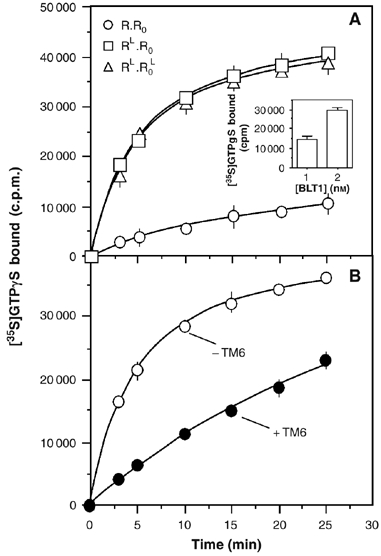
[35S]GTPγS binding to the G-protein catalyzed by the R.R0 dimer loaded with one or two agonist molecules or by the agonist-loaded BLT1 monomer. (A) Time-dependent GDP/GTP exchange on Gαi catalyzed by the R.R0 dimer in the absence of agonist (circles), after filling the high affinity sites (RL.R0 state; squares) or at saturating LTB4 concentration (RL.R0L state; triangles). Inset: [35S]GTPγS binding assay catalyzed by 1 or 2 nM of LTB4-saturated receptor after 3 min incubation. (B) Time-dependent GDP/GTP exchange on Gαi catalyzed by the agonist-loaded wild-type BLT1 receptor in the presence of saturating concentrations of LTB4 and in the absence (open circles) or the presence (closed circles) of the TM6 peptide. In all cases, data represent the mean s.e. from three independent experiments.
On the basis of the ligand-binding isotherm in Figure 2, three species can be distinguished. R:R0 is the ligand-free receptor. RL:R0 is the dimer where only the high affinity site in the R protomer is filled with LTB4 at low agonist concentration (nM range; see Materials and methods). RL:R0L is the BLT1 dimer fully loaded with LTB4 at high agonist concentration (μM range; see Materials and methods). We examined here the ability of R:R0, RL:R0, and RL:R0L to stimulate GDP/[35S]GTPγS exchange on Gαi. As shown in Figure 4A, both RL:R0 and RL:R0L induce a time-dependent increase in [35S]GTPγS binding. More important, the time course [35S]GTPγS-binding profile is very similar whether LTB4 is bound or not to the low-affinity protomer, suggesting that agonist binding to only one of the subunits in the BLT1 dimer is sufficient to trigger full G-protein activation. It should be noted that the effect observed with the dimer where only R is filled with LTB4 is not the result of a residual population of fully loaded dimer at the agonist concentration used in the assay. Indeed, a similar [35S]GTPγS-binding profile is obtained with a receptor dimer where one of the protomers does not bind LTB4 (in the concentration range used here) owing to an additional mutation in the ligand binding pocket besides the C97A one (see Supplementary data 2).
As binding of only a single agonist molecule to the dimeric receptor induces [35S]GTPγS binding, we then analyzed whether the two subunits were nevertheless required for activating the G-protein. We previously showed that adding a peptide corresponding to the sixth transmembrane domain of BLT1 fully dissociates the receptor dimer (Banères and Parello, 2003). Whether this is a direct competition effect or the indirect consequence of steric hindrance effects in regions close to the dimer interface is still an open question. We analyzed here the ability of the monomeric BLT1 receptor in the presence of the TM6 peptide to catalyze GDP/GTP exchange by the αi subunit. As shown in Figure 4B for the wild-type receptor R, some exchange is observed in the presence of the TM6 peptide. However, GTPγS binding occurs at much more slower rates in the presence of the TM6 peptide compared to that observed with the dimeric receptor. As expected, the same behavior is observed with the C97A mutant of BLT1 in the presence of the TM6 peptide (not shown). Although subtle effects of the TM6 peptide on the monomer conformation that would lead to a decreased receptor-G-protein coupling efficiency cannot be excluded, it is likely that, as previously reported for rhodopsin (Jastrzebska et al, 2006), full G-protein activation requires the dimeric complex, even if the BLT1 monomer can activate it to some extent.
Agonist-induced changes in R conformation
Next, we analyzed the influence of the G-protein on the ligand-induced changes in receptor conformation. To selectively follow the changes in the conformation of one protomer in the R.R0 dimer, we used the particular fluorescence properties of 5-hydroxytryptophan (5HW). 5HW can be introduced in a protein produced in E. coli through biosynthetic labeling with no change in the structural properties of the labeled protein (Ross et al, 1992, 1997). 5HW has a significant shoulder in its absorption spectrum at 315 nm that is absent from that of tryptophan so that excitation at 315 nm in a mixture of 5HW-labeled and unlabeled proteins produces a fluorescence signal centered at 337 nm that is exclusively from the 5HW label (Ross et al, 1992). In our case, labeling one of the two protomers in the R.R0 complex with 5HW allows a fluorescence-monitored analysis of the changes in the conformation of the labeled protomer.
We first analyzed the changes in the conformation of the R protomer using a R.R0 complex where only the wild-type subunit R was labeled with 5HW. The changes in 5HW fluorescence intensity as a function of LTB4 concentration are given in Figure 5. As clearly shown in Figure 5, LTB4 induces the same changes on R conformation whether the G-protein is present or not. In both cases, binding of LTB4 to the high-affinity site in R is associated with a concomitant change in the emission properties of this subunit. The fluorescence emission intensity value reached after filling the ligand-binding sites in R with LTB4 (RL:R0 state) is that of the fully activated BLT1 (Mesnier and Banères, 2004). No subsequent changes in the fluorescence properties of R are observed upon binding of LTB4 to the low-affinity site in R0 (Figure 5A), indicating that agonist binding to R0 in the R.R0 complex is not associated with a modification of the conformation of R.
Figure 5.
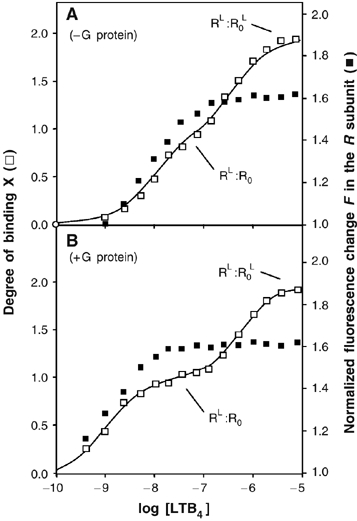
Activation of the R subunit in the R:R0 complex in the absence or presence of G-proteins. Binding of LTB4 to R.R0 (binding ratio X; open squares) and normalized change in 5HW fluorescence of the R subunit (F; closed squares) as a function of LTB4 concentration in the absence (A) or in the presence (B) of Gαi2β1γ2. The stage of the titration plot where the RL.R0 and RL.R0L states is reached is indicated.
Agonist-induced changes in R0 conformation
We then analyzed the changes in the conformation of R0 in the R:R0 dimer by using a complex where only R0 is labeled with 5HW. In contrast to what is obtained in the case of R, different effects are observed in the absence and presence of purified Gαi2β1γ2. In the absence of G-proteins, going from R:R0 to RL:R0L leads to two successive changes of the fluorescence emission intensity of R0 (Figure 6A). A first change is observed upon filling the high-affinity site in R (RL:R0 state). In this RL:R0 state, the emission intensity of R0 is intermediate between that of the inactive and that of the fully active states of the receptor. A second change in the fluorescence intensity of R0 then occurs upon binding of LTB4 to this protomer so that, in the RL:R0L state, the emission intensity of R0 is that of the fully active receptor.
Figure 6.
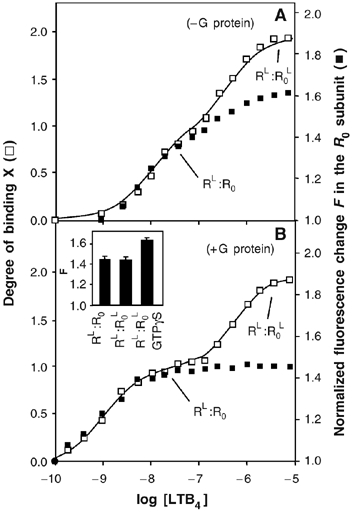
Activation of the R0 subunit in the R:R0 complex in the absence or presence of G-proteins. Binding of LTB4 to R.R0 (binding ratio X; open squares) and normalized change in 5HW fluorescence of the R0 subunit (F; closed squares) as a function of LTB4 concentration in the absence (A) or in the presence (B) of Gαi2β1γ2. The stage of the titration plot where the RL.R0 and RL.R0L states are reached is indicated. Inset in (B): F-values inferred from the titration plots for RL:R0, RL:R0L and RL:R0L in the presence of a 10-fold molar excess of GTPγS. The error bars in the insets correspond to the standard deviation from the mean value calculated from three independent experiments.
In the presence of purified Gαi2β1γ2, agonist binding to R induces the same change in the emission intensity of R0 than that measured in the absence of G-proteins (Figure 6B). The conformation of R0 in the RL:R0 state is therefore likely to be the same whether the G-protein is present or not. This observation indicates that coupling to the G-protein has no effect on the change in the conformation of R0 triggered by agonist-induced activation of R. However, in contrast to what is observed in the absence of G-proteins, subsequent binding of LTB4 to R0 induces no significant change in its fluorescence emission value (Figure 6B). This indicates that the agonist-induced change in R0 conformation that occurs in the absence of Gαi2β1γ2 no longer occurs in the presence of the G-protein. This is specific of the coupling of the receptor to the G-protein since it is not observed when GαSβ1γ2 is used instead of Gαi2β1γ2 (not shown). Finally, when GTPγS is added, an increase in the emission intensity is observed so that the emission intensity value measured under these conditions is similar to that measured in the absence of G-proteins, indicating that uncoupling the receptor from the G-protein allows the R0 protomer to reach its fully active conformation.
Conformational features of R and R0 in the agonist-loaded RL.R0L complex
We compared the fluorescence emission spectra of R and R0 in the LTB4 fully loaded state RL:RL0 (end of the titration plots in Figures 5 and 6). A strict comparison of the emission spectra of both protomers is possible under our present experimental conditions since, in R and R0, the same Trp, that is, Trp234, gives rise to the emission spectrum. In the absence of G-proteins, the emission spectra of the agonist-loaded R and R0 subunits of the R.R0 dimer are strictly superimposable and correspond to that of the fully activated receptor (Figure 7A), indicating that the two agonist-loaded subunits display similar active conformations. In contrast, in the presence of G-proteins, the emission spectra of the two agonist-loaded protomers in the RL:RL0 complex significantly differ in their emission intensity (Figure 7B). In this case, only the emission spectrum of R is that of the fully active conformation of BLT1. The data in Figure 7 therefore directly establish that the G-protein introduces an asymmetry in the conformational features of the receptor dimer: in the absence of G-protein, the two protomers display similar active conformations whereas the two agonist-loaded protomers in the BLT1 dimer display different conformations when associated to the G-protein, with only one, R, being in the fully active state.
Figure 7.

Conformation of R and R0 in the absence or presence of G-proteins. (A) Fluorescence emission spectra of 5HW-labeled R or R0 in the absence of G-proteins and in the absence or presence of saturating concentrations in LTB4. (B) Fluorescence emission spectra of the 5HW-labeled R or R0 in the presence of Gαi2β1γ2 and in the absence or presence of saturating concentrations in LTB4.
Receptor activation in the wild-type R.R dimer
We finally analyzed the conformational changes in the wild-type dimer using an R.R complex with a single protomer labeled with 5HW. As in R.R0, both subunits in the R.R dimer were devoid of all their Trp residues besides Trp234 To produce a homodimer with a single labeled subunit, we labeled the Strep-tagged R with 5HW, mixed it with unlabeled S-tagged receptor, and the homodimer with only one labeled protomer was purified as described for the R:R0 heterodimer (see Materials and methods). We then compared the fluorescence emission intensity of this labeled subunit in the agonist-free (R.R) and agonist-saturated (RL.RL) dimers.
As shown in Figure 8, in the absence of G-proteins, the emission value of the 5HW-labeled protomer in the RL.RL dimer is similar to that of either R0 or R measured under the same conditions (the difference in Figure 8 between these F-values is in the signal-to-noise ratio range and is therefore not statistically significant). This suggests that, in the absence of G-proteins, the two agonist-loaded protomers in the wild-type dimer are likely to be in the same active conformation. In the presence of Gαi2β1γ2 and saturating LTB4 concentrations, a rather different situation is encountered. As shown in Figure 8, the emission intensity value measured for the 5HW-labeled protomer in the agonist-loaded RL.RL dimer, that is, F=1.52, is the mean of the F-value measured for the fully activated receptor (F=1.62) and that obtained for the R0 protomer blocked in the intermediate conformation Owing to the interaction with the G-protein (F=1.46). The differences between these F-values are in this case totally significant from a statistical point of view as they are far above the signal-to-noise ratio range. This could mean that half of the R protomers in the agonist-loaded RL:RL complex reach a fully active conformation whereas the other half is blocked in the intermediate conformation owing to the interaction with the G-protein. In the case of the wild-type dimer, we therefore again probably have a non-symmetric dimer with only one subunit in the fully active conformation. As in the case of the R:R0 complex, adding GTPγS restores the subsequent change in emission, that is, the value obtained in this case is similar to that obtained in the absence of G-proteins (Figure 8).
Figure 8.
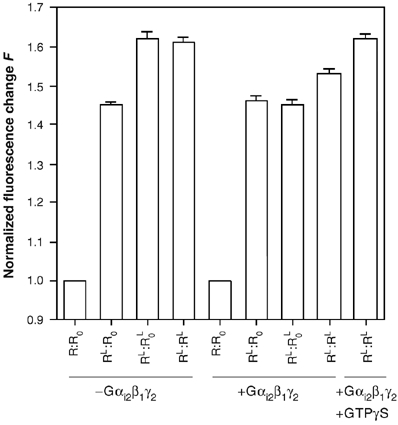
Receptor activation in the BLT1 dimer. Normalized change in 5HW fluorescence F in the R:R0 heterodimer and in the R:R homodimer in the absence of G-proteins, in the presence of Gαi2β1γ2 or in the presence of both Gαi2β1γ2 and a 10-fold molar excess of GTPγS, as a function of the LTB4 content of the receptor dimer. In all cases, only one of the protomers in the dimer is labeled with 5HW, that is, R0 in the R:R0 complex and one of the R protomers in the R:R homodimer. The error bar corresponds to the standard deviation from the mean value calculated from three independent experiments.
Discussion
We have used a purified receptor, the leukotriene B4 receptor BLT1, to analyze the activation mechanism in a GPCR dimer and the relationship between the conformational changes in each subunit of the dimer and the coupling to the G-protein. Our data show (i) that agonist-induced activation of a single protomer can trigger G-protein activation and (ii) that the G-protein restricts the conformational changes of the second receptor protomer so that, in the receptor dimer, only one subunit reaches the fully active state.
One of the main issues in elucidating the functional role of GPCR dimers is to know whether agonist binding to a single subunit is sufficient for G-protein activation or whether both subunits in a ligand-loaded state are required. Using the isolated BLT1 receptor dimer, we show here that agonist binding to a single subunit is sufficient for full activation of Gαi. Consistent with our observations, a single agonist per dimer is also sufficient for activation of the heterodimeric GABAB (Galvez et al, 2001; Kniazeff et al, 2002, 2004a) and T1R (Xu et al, 2004) receptors. In the case of an other homodimeric class C GPCR, the mGlu receptor, a single agonist per dimer is also sufficient for receptor activation, although in that case two agonists per dimer are required for full activity (Kniazeff et al, 2004b). That agonist occupation of a single subunit in a dimer is sufficient for G-protein activation is also consistent with a number of studies demonstrating trans-complementation between a receptor defective in ligand binding and a receptor defective in G-protein activation (Carrillo et al, 2003; Pascal and Milligan, 2005).
If activation of only one protomer is sufficient for an efficient activation of the G-protein, as clearly shown here, then why a receptor dimer? It has been recently shown that the oligomeric forms of rhodopsin couple more efficiently to transducin (Jastrzebska et al, 2006). In agreement with such a result, we show here that receptor-induced [35S]GTPγS binding is significantly less efficient when the BLT1 monomer is used instead of the dimer. As discussed by Jastrzebska et al (2006), a possibility would be that a single protomer is the dimer is responsible G-protein activation but that the dimer allows a more efficient coupling to occur through multiple interactions between the two receptor protomers and different regions of the trimeric G protein. Such a model would be in agreement with our previous observation of a reduced stability of the receptor:G-protein complex in the presence of the dimer dissociating TM6 peptide (Banères and Parello, 2003).
We recently proposed on the basis of functional studies with the glutamate receptor that only one subunit per receptor dimer can reach a fully active state at a time (Goudet et al, 2005; Hlavackova et al, 2005). This led us to propose that the G-protein is responsible for this asymmetric functioning of a receptor homodimer. We provide here an experimental demonstration of this model. Indeed, we show that, in contrast to what we observed in the absence of G-proteins (Mesnier and Banères, 2004), both protomers in the BLT1 dimer have different conformations when the G-protein is present. This directly demonstrates that in the reconstituted (BLT1)2.Gαβγ pentamer the active form of the receptor dimer is indeed a nonsymmetric one, with probably only one being in the fully active state. Such a restriction of the conformational landscape of one of the protomers in the dimer by the heterotrimeric G-protein is certainly due to a specific interaction of the G-protein with this protomer.
Our data with the reconstituted receptor:G-protein complex raise an important issue on the way the receptor dimer interacts with the G-protein trimer. We previously established that only one G-protein trimer interacts with the BLT1 dimer to make a stoichiometrically well-defined pentameric (BLT1)2.Gαβγ complex (Banères and Parello, 2003). We now show (i) that activation of one of the protomers triggers G-protein activation, indicating that the activated protomer interacts with Gα and (ii) that the conformational landscape of the second protomer in the dimer is restricted by the G-protein, indicating that specific contacts between this subunit and the G-protein occur. All these experimental observations therefore imply that the G-protein contacts both subunits of the receptor dimer.
According to our proposal of a nonsymmetric active form of a GPCR dimer, one is expecting a differential effect of the G-protein on the affinity of the two protomers for the agonist. In agreement with such a view, we observe that the affinity of essentially only one of the protomers in the R:R0 dimer is affected by the coupling to the G-protein. Such an effect could result in negative cooperativity in agonist binding. Consistent with this idea, several studies reported already in the 1970's, demonstrated negative cooperativity in agonist binding in native tissues (Limbird et al, 1975; de Meyts, 1976; Carayon et al, 1979). Such a negative cooperativity can result from receptor dimerization (Durroux, 2005), as shown for the glycoprotein hormone receptor (Urizar et al, 2005) or for the chemokine CCR5–CCR2 heterodimer (El-Asmar et al, 2005; Springael et al, 2006). Not only such a negative cooperativity was observed in heterologous expressing cells, but similar data were also obtained in native tissue, demonstrating that such a phenomena is not the simple consequence of receptor over-expression.
Other studies, however, appear not to be consistent with our observations, suggesting that the asymmetric functioning of GPCR dimers may not necessarily be true for all the receptors. For example, positive cooperativity has been reported for agonist binding to the μ-δ opioid receptor heterodimer, and both μ- and δ-agonists are required for efficient MAP kinase activation (Jordan and Devi, 1999). Similarly, activation of both subunits of a muscarinic M3 dimer is required for arrestin recruitment (Novi et al, 2005). However, because our present and previous data demonstrate that the G-protein is likely responsible for the asymmetric active conformation of the BLT1 dimer and that uncoupling from the G-protein through addition of GTPγS leads to a symmetric receptor dimer with two activated protomers, it is tempting to speculate that, whereas activation of the G-protein is associated with an asymmetry of the receptor dimer, a symmetric dimer may be associated to arrestin in the subsequent signaling steps. If confirmed, this would lead to the exciting idea that the signal transduction pathway of a receptor dimer may be controlled by the stoichiometry of subunit activation.
Taken together, our present observations on the BLT1 dimer are consistent with the idea that a single subunit reaches the active state at a time. Our data also provide a framework for the possible development of heterodimeric-preferring ligands and leave opened the possibility that depending on the stoichiometry of subunit activation, different signaling pathways may be activated by a GPCR dimer.
Materials and methods
Materials
LTB4 was purchased from BIOMOL laboratories. 5HW was from Sigma. Asolectin was purchased from Fluka. N-hexadecyl-β-D-maltoside (HDM) was from Anatrace. Chem-1 cells membrane fractions containing wtBLT1 were obtained from Chemicon. These are membrane preparations made from chem-1 cell line stably transfected with the coding sequence of the human BLT1 receptor. We systematically determined the receptor number in membrane preparations by saturation binding assays.
Buffers
Buffer A: 100 mM NaH2PO4, 10 mM Tris–HCl, 10% glycerol, 1 mM 2-mercaptoethanol, 0.5% SDS, pH 8. Buffer B: 12.5 mM Na-borate, 10 mM NaCl, 10% glycerol, pH 7.8 containing HDM/asolectin (1:2 detergent:protein w/w ratio). To prepare lipid/detergent mixed micelles, an appropriate volume of 1% HDM was added to a portion of powdered asolectin to yield a stock of 0.1% asolectin/0.05 mM HDM. The solution was passed multiple times through a fine needle to disperse the phospholipids, and was clarified by centrifugation (100 000 g, 20 min) before use.
R:R0 preparation
The R:R0 complex was produced as described in Mesnier and Banères (2004) with the exception that the receptor was refolded in HDM/asolectin mixed micelles (see buffer B). Lipids were added to increase the stability of the dimeric assembly. Briefly, BLT1 was expressed as fusion proteins with KSI as described for 5-HT4(a) (Banères et al, 2005), with an S-tag (wild-type receptor) or a Strep-tag (mutant receptor) sequence after the thrombin cleavage site. After removing KSI with thrombin (Banères et al, 2005) the wild-type and mutant receptors, solubilized in buffer A, were mixed in equimolecular amounts, loaded on a Ni-NTA superflow column and the protein refolded by exchanging buffer A by buffer B, as described in Banères et al (2003). Unfolded proteins were discarded and the functional receptor further purified first on a 5–20% isokinetic sucrose gradient and then by gel filtration chromatography on a Superdex S200 HR column (1 × 30 cm) using buffer B as the eluent. For R:R0 purification, the refolded proteins were first immobilized on S-protein agarose column previously equilibrated in buffer B. The flow-through fractions were discarded and the retained proteins were then eluted with buffer B containing 1 M MgCl2. The protein fraction recovered under these conditions was then loaded onto a Streptactin affinity column (5.0 × 0.6 cm), washed with buffer B and eluted with buffer B containing 2.5 mM dethiobiotin. The eluted protein was then extensively dialyzed in buffer B. The same method was used to prepare R.R homodimers with only one 5HW-labeled subunit. In this case, we simply mixed before refolding the unlabeled S-tagged receptor with the 5HW-labeled Strep-tagged receptor. In all cases, 5HW was introduced in the receptors and was carried out by biosynthetic labeling using the method described in Mesnier and Banères (2004).
The receptor dimer was also reconstituted in lipid vesicles by removing the detergent as described in Rigaud et al (1995). The proteoliposomes were purified on a 15, 20 and 45% (w/w) sucrose step density gradient. The BLT1 proteoliposome preparation was loaded at the top of the sucrose gradient and centrifuged overnight (200 000 g at 4°C). Fractions of 1 ml were collected from top to bottom of the centrifuge tube without disturbing the gradient. Sucrose in the BLT1 receptor proteoliposome fraction was removed by dilution with five volumes of Milli-Q water, and subsequent centrifugation for 30 min (80 000 g at 4°C) yielded a visible precipitate, which was subsequently stored as a pellet at −80°C.
Ligand-binding assays
LTB4 binding was assayed as previously described (Banères et al, 2003; Mesnier and Banères, 2004). All protein concentrations were calculated from UV-absorptivity values (Cary 400 spectrophotometer, Varian) using the extinction coefficient calculated by the method of Gill and von Hippel (1989). LTB4 concentration was calculated on the basis of an extinction coefficient of 5.0 × 104 l/mol/cm at 270.5 nm (Radmark et al, 1980). G-protein concentration in the 10 μM range were used to ensure an efficient coupling between the receptor and the G-protein, based on the Kd value measured for the receptor:G-protein interaction (unpublished Surface Plasmon Resonance measurements). The binding profiles are presented as the degree of binding X as a function of LTB4 concentration. The degree of binding X is defined as the moles of LTB4 bound per mole of BLT1 dimer (for a definition of the degree of binding, see Wyman and Gill, 1990). The titration data were analyzed using the PRISM software version 4.0 (Graphpad Inc.) by considering a set of usual models for describing the ligand:receptor interactions.
GTPγS-binding assays
GTPγS-binding assays were carried out as described by Glass and Northup (1999). The Gαi2β1γ2 was produced as described in Banères et al (2003). We used in these assays R:R0 either in the absence of agonist (R:R0), or with the high-affinity site loaded with LTB4 (RL:R0) or totally loaded with LTB4 (RL:R0L). The LTB4 concentrations required for reaching each of these states were defined from the titration profiles in Figure 2 (5 nM and 5 μM for the RL.R0 and RL.R0L states, respectively; receptor concentration 1 nM). We systematically checked that at these receptor and ligand concentrations one or two LTB4 molecules, respectively, were bound per receptor dimer. We also checked by FRET with labeled R and R0 receptors (Mesnier and Banères, 2004) that no dissociation of the receptor dimer occurs at this protein concentration. In the case of the dimer with a double C97A,E185A mutant protomer (see Supplementary data), the LTB4 concentration used was 50 nM. The assays were all carried out at 1 nM receptor concentration (unless otherwise stated), 50 nM Gα and 100 nM Gβγ. G-protein-binding activity was also systematically measured in the absence of LTB4. The assays were carried out at 30°C in a buffer 10 mM MOPS, pH 7.5, 2 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 0.5% (w/v) BSA, 4 μM GDP, and [35S]GTPγS (0.4–0.8 nM; 2–5 × 105 c.p.m.). [35S]GTPγS was added to the receptor preparations and the reaction incubated for the times indicated in Figures 3 and 4. Assays were stopped by the addition of 400 μl of a 0.5% cholate solution as described by Kurose et al (1991). For the experiment at different receptor concentrations, the assay was carried out as described above in the presence of 1 or 2 nM BLT1. In this case, a 3-min incubation time was used before stopping the reaction as described above. For the assays with the monomeric receptor, an excess (1:1000 receptor-to-peptide molar ratio) of the TM6 peptide obtained from the whole receptor as described in Banères and Parello (2003) was added after receptor refolding. We checked by ultracentrifugation that, under these conditions, the BLT1 receptor is essentially monomeric (data not shown). For the GTP binding assays with the receptor in the presence of TM6, 1 and 50 nM receptor and LTB4 concentrations were used in the case of the wild-type receptor and 1 nM and 5 μM receptor and LTB4 concentrations were used in the case of the C97A mutant.
Fluorescence measurements
Fluorescence emission spectra were recorded at 20°C on a Cary Eclipse spectrofluorimeter (Varian) with an excitation wavelength of 315 nm (bandwidth 2 nm). Emission was recorded 15 min after adding the ligand. Titrations were carried out by successive additions of LTB4 to a receptor stock solution so that dilution effects do not exceed 2% at the end of the titration profile. Receptor concentrations in the 10−8–10−9 M range were used. As in the ligand-binding measurements, a G-protein concentration in the micromolar range was used to ensure an efficient coupling between the receptor and the G-protein. Buffer contributions were subtracted under the same experimental conditions. The normalized fluorescence change F is defined as the ratio of the 5HW emission intensity at 337 nm at a given agonist concentration to that of the ligand-free receptor. As (i) the emission profiles of the ligand-free R and R0 are strictly identical whether the G-protein is present or not and (ii) R and R0 contain the same single Trp residue Trp234, using F allows a direct comparison of the changes in R and R0. In all the cases, a whole series of experiments was carried out at the same time and under the same experimental conditions (e.g. R and R0 fluorescence in the absence and presence of G-protein) to directly compare the emission intensities in the fluorescence spectra.
Supplementary Material
Supplementary Material 1
Acknowledgments
We thank J Bockaert and T Durroux for their constructive comments on this work. J Parello is also thanked for a critical reading of the manuscript. This manuscript also highly benefited from the constructive comments of the referees. This work was supported by the CNRS, the Ministère de la Recherche (Actions Concertées Incitatives ‘Molécules et Cibles Thérapeutiques' and ‘Biologie Cellulaire, Moléculaire and Structurale') and l'Association pour la Recherche sur le Cancer.
References
- Bae H, Cabrera-Vera TM, Depree KM, Graber SG, Hamm HE (1999) Two amino acids within the α4 helix of Gα1 mediate coupling with 5-hydroxytryptamine1B receptors. J Biol Chem 274: 14963–14971 [DOI] [PubMed] [Google Scholar]
- Banères JL, Martin A, Hullot P, Girard JP, Rossi JC, Parello J (2003) Structure-based analysis of GPCR function. Conformational adaptation of both agonist and receptor upon leukotriene B4 binding to recombinant BLT1. J Mol Biol 329: 801–814 [DOI] [PubMed] [Google Scholar]
- Banères J-L, Mesnier D, Martin A, Joubert L, Dumuis A, Bockaert J (2005) Molecular characterization of a purified 5-HT4 receptor: a structural basis for drug efficacy. J Biol Chem 280: 20253–20260 [DOI] [PubMed] [Google Scholar]
- Banères JL, Parello J (2003) Structure-based analysis of GPCR function. Evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J Mol Biol 329: 815–829 [DOI] [PubMed] [Google Scholar]
- Bockaert J, Claeysen S, Becamel C, Pinloche S, Dumuis A (2002) G-protein-coupled receptors: dominant players in cell–cell communication. Int Rev Cytol 212: 63–132 [DOI] [PubMed] [Google Scholar]
- Bockaert J, Pin JP (1999) Molecular tinkering of G-protein-coupled receptors: an evolutionary success. EMBO J 18: 1723–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulenger S, Marullo S, Bouvier M (2005) Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci 26: 131–137 [DOI] [PubMed] [Google Scholar]
- Carayon P, Guibout M, Lissitzky S (1979) The interaction of radioiodinated thyrotropin with human plasma membranes from normal and diseased thyroid glands. Relation of thyrotropin binding to adenylate cyclase activity. Ann Endocrinol (Paris) 40: 211–227 [PubMed] [Google Scholar]
- Carrillo JJ, Pediani J, Milligan G (2003) Dimers of class A G-protein-coupled receptors function via agonist-mediated trans-activation of associated G-proteins. J Biol Chem 278: 42578–42587 [DOI] [PubMed] [Google Scholar]
- de Meyts P (1976) Cooperative properties of hormone receptors in cell membranes. J Supramol Struct 4: 241–258 [DOI] [PubMed] [Google Scholar]
- Durroux T (2005) Principles: a model for the allosteric interactions between ligand binding sites within a dimeric GPCR. Trends Pharmacol Sci 26: 376–384 [DOI] [PubMed] [Google Scholar]
- El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M (2005) Evidence for negative binding cooperativity within CCR5–CCR2b heterodimers. Mol Pharmacol 67: 460–469 [DOI] [PubMed] [Google Scholar]
- Filipek S, Krzysko KA, Fotiadis D, Liang Y, Saperstein DA, Engel A, Palczewski K (2004) A concept for G-protein activation by G-protein-coupled receptor dimers: the transducin/rhodopsin interface. Photochem Photobiol Sci 3: 628–638 [DOI] [PubMed] [Google Scholar]
- Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prezeau L, Pin JP (2001) Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J 20: 2152–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182: 319–326 [DOI] [PubMed] [Google Scholar]
- Glass M, Northup JK (1999) Agonist selective regulation of G-proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol 56: 1362–1369 [DOI] [PubMed] [Google Scholar]
- Goudet C, Kniazeff J, Hlavackova V, Malhaire F, Maurel D, Acher F, Blahos J, Prezeau L, Pin JP (2005) Asymmetric functioning of dimeric metabotropic glutamate receptors disclosed by positive allosteric modulators. J Biol Chem 280: 24380–24385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse R, Schoneberg T, Schultz G, Gudermann T (1997) Inhibition of gonadotropin-releasing hormone receptor signaling by expression of a splice variant of the human receptor. Mol Endocrinol 11: 1305–1318 [DOI] [PubMed] [Google Scholar]
- Hansen JL, Sheikh SP (2004) Functional consequences of 7TM receptor dimerization. Eur J Pharm Sci 23: 301–317 [DOI] [PubMed] [Google Scholar]
- Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, Trojanova J, Prezeau L, Pin JP, Blahos J (2005) Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J 24: 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi T, Yokomizo T, Tsutsumi O, Taketani Y, Shimizu T, Izumi T (1999) Characterization of the leukotriene B4 receptor in porcine leukocytes. Separation and reconstitution with heterotrimeric GTP-binding proteins. Eur J Biochem 259: 419–425 [DOI] [PubMed] [Google Scholar]
- Jastrzebska B, Fotiadis D, Jang GF, Stenkamp RE, Engel A, Palczewski K (2006) Functional and structural characterization of rhodopsin oligomers. J Biol Chem 281: 11917–11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C (1998) GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 396: 674–679 [DOI] [PubMed] [Google Scholar]
- Jordan BA, Devi LA (1999) G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399: 697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpa KD, Lin R, Kabbani N, Levenson R (2000) The dopamine D3 receptor interacts with itself and the truncated D3 splice variant d3nf: D3–D3nf interaction causes mislocalization of D3 receptors. Mol Pharmacol 58: 677–683 [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B (1998) GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 396: 683–687 [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Galvez T, Labesse G, Pin JP (2002) No ligand binding in the GB2 subunit of the GABA(B) receptor is required for activation and allosteric interaction between the subunits. J Neurosci 22: 7352–7361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, Pin JP (2004b) Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol 11: 706–713 [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Saintot PP, Goudet C, Liu J, Charnet A, Guillon G, Pin JP (2004a) Locking the dimeric GABA(B) G-protein-coupled receptor in its active state. J Neurosci 24: 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK (2002) Agonist-induced conformational changes in the β2-adrenergic receptor. J Pept Res 60: 317–321 [DOI] [PubMed] [Google Scholar]
- Kurose H, Regan JW, Caron MG, Lefkowitz RJ (1991) Functional interactions of recombinant α2 adrenergic receptor subtypes and G-proteins in reconstituted phospholipid vesicles. Biochemistry 30: 3335–3341 [DOI] [PubMed] [Google Scholar]
- Lee SP, O'Dowd BF, Ng GY, Varghese G, Akil H, Mansour A, Nguyen T, George SR (2000) Inhibition of cell surface expression by mutant receptors demonstrates that D2 dopamine receptors exist as oligomers in the cell. Mol Pharmacol 58: 120–128 [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O'Dowd BF, George SR (2004) Dopamine D1 and D2 receptor co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem 279: 35671–35678 [DOI] [PubMed] [Google Scholar]
- Limbird L, de Meyts P, Lefkowitz RJ (1975) β-Adrenergic receptors: evidence for negative cooperativity. Biochem Biophys Res Comm 64: 1160–1168 [DOI] [PubMed] [Google Scholar]
- Masuda K, Itoh H, Sakihama TA, Kiyama C, Takahashi K, Fukudan R, Yokomizo T, Shimizu T, Kodama T, Hamakubo T (2003) A combinatorial G-protein-coupled receptor reconstitution system on budded baculovirus. Evidence for Gαi and Gαo coupling to a human leukotriene B4 receptor. J Biol Chem 278: 24552–24662 [DOI] [PubMed] [Google Scholar]
- Mesnier D, Banères J-L (2004) Cooperative conformational changes in a G-protein-coupled receptor dimer, the leukotriene B4 receptor BLT1. J Biol Chem 279: 49664–49670 [DOI] [PubMed] [Google Scholar]
- Milligan G (2004) G-protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol 66: 1–7 [DOI] [PubMed] [Google Scholar]
- Novi F, Stanasila L, Giorgi F, Corsini GU, Cotecchia S, Maggio R (2005) Paired activation of two components within muscarinic M3 receptor dimers is required for recruitment of β-arrestin-1 to the plasma membrane. J Biol Chem 280: 19768–19776 [DOI] [PubMed] [Google Scholar]
- Pascal G, Milligan G (2005) Functional complementation and the analysis of opioid receptor homodimerization. Mol Pharmacol 68: 905–915 [DOI] [PubMed] [Google Scholar]
- Perez DM, Karnik SS (2005) Multiple signaling states of G-protein-coupled receptors. Pharmacol Rev 57: 147–161 [DOI] [PubMed] [Google Scholar]
- Perron A, Chen ZG, Gingras D, Dupre DJ, Stankova J, Rola-Pleszczynski M (2003) Agonist-independent desensitization and internalization of the human platelet-activating factor receptor by coumermycin-gyrase B-induced dimerization. J Biol Chem 278: 27956–27965 [DOI] [PubMed] [Google Scholar]
- Pin JP, Kniazeff J, Liu J, Binet V, Goudet C, Rondard P, Prezeau L (2005) Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J 272: 2947–2955 [DOI] [PubMed] [Google Scholar]
- Radmark O, Samulesson B, Clark DA, Marfat A, Corey EJ (1980) Leukotriene A: stereochemistry and enzymatic conversion to leukotriene B. Biochem Biophys Res Commun 92: 954–961 [DOI] [PubMed] [Google Scholar]
- Rigaud J-L, Pitard B, Levy D (1995) Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim Biophys Acta 1231: 223–246 [DOI] [PubMed] [Google Scholar]
- Robbins MJ, Calver AR, Filippov AK, Hirst WD, Russell RB, Wood MD, Nasir S, Couve A, Brown DA, Moss SJ, Pangalos MN (2001) GABA(B2) is essential for G-protein coupling of the GABA(B) receptor heterodimer. J Neurosci 21: 8043–8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JBA, Senear DF, Waxman E, Kombo BB, Rusinova E, Huang YT, Laws WR, Hasselbacher CA (1992) Spectral enhancement of proteins: biological incorporation and fluorescence characterisation of 5-hydroxytryptophan in bacteriophage λ cI repressor. Proc Natl Acad Sci USA 89: 12023–12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JBA, Szabo AG, Hogue CWV (1997) Enhancement of protein spectra with tryptophan analogs: fluorescence spectroscopy of protein-protein and protein-nucleic acid interactions. Methods Enzymol 278: 151–190 [DOI] [PubMed] [Google Scholar]
- Saidak Z, Blake-Palmer K, Hay DL, Northup JK (2006) Differential activation of G-proteins by μ-opioid receptor agonists. Br J Pharmacol 147: 671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Nguyen PL, Urizar E, Costagliola S, Vassart G, Parmentier M (2006) Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol 69: 1652–1661 [DOI] [PubMed] [Google Scholar]
- Stanasila L, Perez JB, Vogel H, Cotecchia S (2003) Oligomerization of the α1a- and α1b-adrenergic receptor subtypes. Potential implications in receptor internalization. J Biol Chem 278: 40239–40251 [DOI] [PubMed] [Google Scholar]
- Terrillon S, Bouvier M (2004) Roles of G-protein-coupled receptor dimerization. EMBO Rep 5: 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urizar E, Montanelli L, Loy T, Bonomi M, Swillens S, Gales C, Bouvier M, Smits G, Vassart G, Costagliola S (2005) Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J 24: 1954–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396: 679–682 [DOI] [PubMed] [Google Scholar]
- Wyman J, Gill SJ (1990) Properties of binding curves In Binding and Linkage Functional chemistry of Biological Macromolecules, Kelly A (ed), pp 33–61. CA, USA: University Science Books [Google Scholar]
- Xu H, Staszewski L, Tang H, Adler E, Zoller M, Li X (2004) Different functional roles of T1R subunits in the heteromeric taste receptors. Proc Natl Acad Sci USA 101: 14258–14263 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1