

Abstract
The large T antigen (T-ag) protein binds to and activates DNA replication from the origin of DNA replication (ori) in simian virus 40 (SV40). Here, we determined the crystal structures of the T-ag origin-binding domain (OBD) in apo form, and bound to either a 17 bp palindrome (sites 1 and 3) or a 23 bp ori DNA palindrome comprising all four GAGGC binding sites for OBD. The T-ag OBDs were shown to interact with the DNA through a loop comprising Ser147–Thr155 (A1 loop), a combination of a DNA-binding helix and loop (His203–Asn210), and Asn227. The A1 loop traveled back-and-forth along the major groove and accounted for most of the sequence-determining contacts with the DNA. Unexpectedly, in both T-ag-DNA structures, the T-ag OBDs bound DNA independently and did not make direct protein–protein contacts. The T-ag OBD was also captured bound to a non-consensus site ATGGC even in the presence of its canonical site GAGGC. Our observations taken together with the known biochemical and structural features of the T-ag–origin interaction suggest a model for origin unwinding.
Keywords: DNA replication, origin binding protein, protein–DNA complex, replication origin, SV40 large T antigen
Introduction
The sequence of events that underlies the initiation of DNA replication has been derived from extensive biochemical and genetic experiments in both prokaryote and eukaryote systems (reviewed in DePamphilis, 1996; Bullock, 1997; Stenlund, 2003; Eichman and Fanning, 2004). In each of these systems, the initiation process begins with binding of specialized origin DNA-binding proteins (OBPs) to the origin (ori). The OBP serves two main roles: first, it binds to multiple recognition elements within the ori through higher order nucleoprotein interactions and induces local distortion or unwinding of the origin DNA; second, the OBP recruits to the ori the other components of the primosome, usually including an ATP-dependent DNA helicase. This helicase acts in concert with the OBP to unwind origin DNA. A mechanistic understanding of these events is limited owing to insufficient structural information about all the biochemically defined steps in early primosome assembly.
Functional origins of most eukaryotic genomes have not been characterized, and for many years, simian virus 40 (SV40) has been a favored laboratory model for studies of the initiation of DNA replication (Fanning and Knippers, 1992; Bullock, 1997; Butel and Lednicky, 1999). The viral OBP, named large T antigen (T-ag), is a 708-amino-acid (80.2 kDa) protein that also acts as the viral replicative helicase (Figure 1A). Insofar as is known, the T-ag binds to the origin first as a monomer to its pentanucleotide recognition element (Gidoni et al, 1982; Scheidtmann et al, 1984; Runzler et al, 1987). The monomers are then thought to assemble into hexamers and double hexamers, which constitute the form that is active in initiation of DNA replication. When bound to the ori, T-ag double hexamers encircle DNA (Mastrangelo et al, 1989; Bullock, 1997). Hexamer assembly nucleates at T-ag recognition pentanucleotides (Parsons et al, 1991). Preformed T-ag hexamers are incapable of forming double hexamers on DNA (Huang et al, 1998). A model has been proposed in which double hexamer assembly occurs by successive binding of 12 monomers (Huang et al, 1998).
Figure 1.
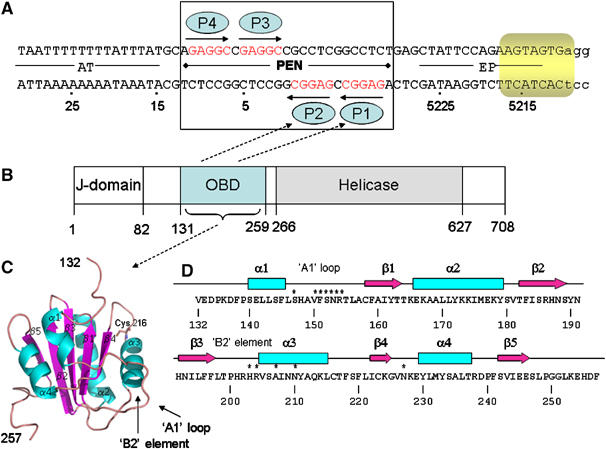
Structural and functional elements of SV40 DNA replication system. (A) Sequences present in the 64-bp core origin of SV40 replication. Locations of the AT-rich region, the pentanucleotide box (PEN) and EP sites, numbered as in the genome of reference strain 776. Arrows depict the four GAGGC pentanucleotides—the binding sites for T-ag OBDs (blue ellipses). Eight nucleotides within the EP that melt upon the T-ag double hexamer assembly are highlighted in yellow. The 23 bp PEN element structure reported in this work is boxed. (B) Functional domains of SV40 large T antigen (T-ag). ‘J Domain', region not required for in vitro DNA replication; ‘OBD' origin DNA-binding domain, minimal region required for binding to SV40 Ori DNA; ‘Helicase', minimal region required for the helicase activity; the numbers given are the amino-acid residues. (C) T-ag OBD structure (apo form) ribbon diagram showing the domain containing residues 134–259 (in figure it is 132–257). α-Helices are in cyan and β-strands in magenta. Position of Cys 216 and DNA-binding components ‘A1 loop' and ‘B2 element' are indicated. (D) Secondary structure elements of T-ag OBD. The β-strands are indicated by arrows and the α-helices by boxes. Amino acids contributing to DNA binding are labeled with ‘*'.
The functions of T-ag are mediated by several structural and functional domains. The N-terminal region (J-domain) is not required for in vitro DNA replication (Chen et al, 1997; Kim et al, 2001). The origin-binding domain (OBD) maps to amino acids 131–259 (Arthur et al, 1988; Joo et al, 1997). Mutagenesis studies of this domain in the context of the full-length protein also suggest a role for this domain in oligomerization and DNA structural perturbations. Helicase activity is associated with the helicase domain (aa 271–627; Figure 1D) (Li et al, 2003). Coordinated action of the OBD and helicase domains is required for melting of the SV4O origin (Chen et al, 1997).
The SV4O core origin of replication (ori) is 64 bp in length, and is required for both in vivo and in vitro replication (Bullock et al, 1989; Bullock, 1997). There are three functional regions of the ori: the early palindrome (EP), the 23 bp pentanucleotide palindrome (PEN), and a 17 bp A/T-rich domain (Figure 1B). The PEN contains a cluster of four GAGGC pentanucleotides (P1–4), depicted by arrows in Figure 1B, which are arranged in two pairs inverted towards each other. All four pentanucleotides are required for initiation of DNA synthesis, and the spacing between the GAGGC elements is critical; substitution of the single base pairs separating the pentanucleotides did not affect DNA replication, whereas duplicating the same base pairs drastically reduced replication (Bullock, 1997). T-ag binds to the SV40 ori through the PEN element; it binds to the GAGGC pentanucleotide with high affinity (57–150 nM) (Titolo et al, 2003b). The OBD is also capable of binding in a non-sequence-specific way to both dsDNA and ssDNA, albeit with lower affinity (about 10-fold lower affinity for random dsDNA) (Titolo et al, 2003b).
Origin unwinding by T-ag is dependent on the three functional regions of the origin, including four T-ag binding sites arranged as in PEN. T-ag binds cooperatively to the PEN element, in the presence of ATP oligomerizes into a double hexamer (SenGupta and Borowiec, 1994; San Martin et al, 1997; Gomez-Lorenzo et al, 2003) and induces melting of 8 nt within EP (Borowiec and Hurwitz, 1988) (Figure 1B). Electron microscopy studies revealed that the double hexamer spans about 240, or 120 Å per hexamer, along the DNA axis (Valle et al, 2000), which is consistent with the biochemical observation that T-ag protects 74 bp DNA (Fanning and Knippers, 1992).
Dissection of the ori functional regions revealed that a single pentanucleotide is sufficient to mediate an assembly of T-ag hexamer (Joo et al, 1998) and the presence of two sites, 1 and 3 (Figure 1B), is sufficient for the formation of two T-ag hexamers (T-ag double hexamer). However, neither one nor two pentanucleotides is sufficient for unwinding of the SV40 ori (Joo et al, 1998). Only in the presence of all four sites, appropriately spaced, can T-ag partially melt 8 bp of the SV40 ori through its helicase activity (Borowiec and Hurwitz, 1988).
The current state of structural knowledge about how OBPs from different species bind to their cognate DNA origins is limited. NMR and X-ray structures of the T-ag OBD in apo form are available (Luo et al, 1996; Meinke et al, 2006). In the crystal structure (Meinke et al, 2006), T-ag OBD adopts a left-handed spiral with six OBDs per turn and pitch of 35.8 Å per turn. Apo structures for OBD are also known for Epstein–Barr virus (EBV) OBP, EBNA1 (Bochkarev et al, 1995), bovine papilloma virus (BPV) E1 (Enemark et al, 2000), and the Rep proteins from adeno-associated virus 5 (AAV-5) (Hickman et al, 2002) and tomato yellow leaf curl virus (TYLCV) (Campos-Olivas et al, 2002). The crystal structures of EBNA1, BPV E1, and E2 (E1 binding partner on the origin) were also determined in complex with DNA (Hegde et al, 1992; Bochkarev et al, 1996; Enemark et al, 2002), but structural basis for T-ag binding to DNA remains unknown.
The mechanisms of origin unwinding are best described for the bovine papilloma viruses (BPV). Like for SV40, the BPV origin also contains four binding sites arranged in two pairs and is bound by an OBP (E1) that contains both a DNA-binding and a helicase domain. E1 is a monomer in solution and dimerizes upon binding the origin. Assembly of the double hexameric E1 helicase proceeds in an ordered manner via a double-monomer, double-dimer, double-trimer, and finally a double-hexamer. Assembly of the E1 protein along this pathway is associated with a helix–to–ring transition (Schuck and Stenlund, 2005).
Most studies on the SV40 system suggest general functional similarities with the BPV system, with minor differences. SV40 has a different arrangement of the binding sites in the origin and a higher affinity of its T-ag OBD to a single binding site (57–150 nM) than does E1 for its sequences (∼500 nM) (Titolo et al, 2003a, 2003b). In addition, the initial loading of E1 onto the ori is mediated through cooperative binding of E1 (the affinity for two properly spaced sites is ∼32 nM) but also through interactions with the viral transcription factor E2 (Sanders and Stenlund, 2001; Titolo et al, 2003a).
Here, we report the high-resolution structure of the T-ag OBD in its apo form (1.5 Å) and the structure of the functional PEN palindrome DNA bound by T-ag OBD (1.65 Å). We also present a medium-resolution (2.3 Å) structure in which T-ag OBD was captured bound to the DNA in a nonspecific binding mode. Taken together with the structures of the T-ag helicase domain, the T-ag OBD spiral hexamer, and with biochemical data available in literature, our data suggest a molecular model for the T-ag double hexamer and for SV40 origin unwinding.
Results
Structure solution
T-ag OBD was crystallized in two crystal forms belonging to the p21 and p212121 space groups and diffracted to 1.5 and 2.5 Å, respectively. The p21 structure was phased by a single isomorphous replacement with anomalous scattering (SIRAS) using a HgCl derivative and refined against 1.5 Å data to R=17.3% and R_free=19.5% (Figures 1C and D). The p212121 structure was solved by molecular replacement using the p21 structure as a model and refined against 2.5 Å data to R=21.6% and R_free=29.3%.
The T-Ag OBD was also crystallized in complex with two different origin fragments. The first complex was with a 23 bp PEN palindrome (PEN-4; with a single base overhang at the 5′-end) and belonged to space group c2 and diffracted to 1.65 Å. The structure was solved by molecular replacement using the apo form T-ag OBD structure as a search model. The structure was refined against 1.65 Å data to R=20.1% and R_free=25.1% (Figure 2).
Figure 2.
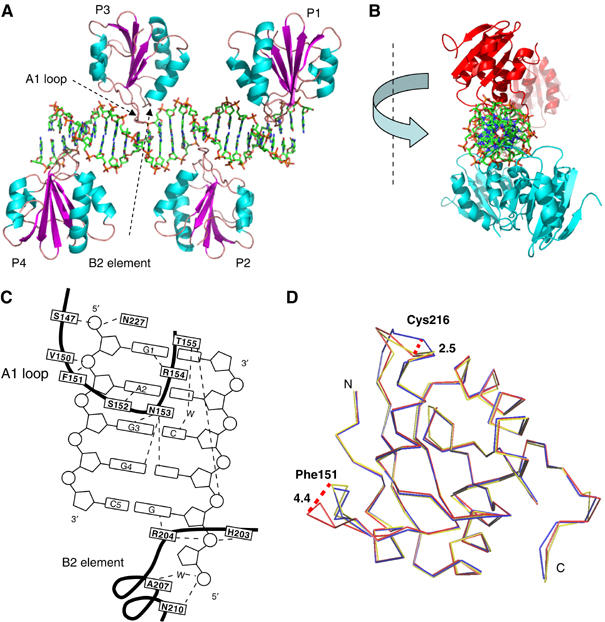
Structure of four T-ag OBDs bound to the 23 bp PEN box of ori DNA (PEN-4). (A) Four copies of OBD are represented as ribbon models and colored as in Figure 1C. DNA is shown as a stick model. (B) Same structure, PEN-4, viewed along the DNA axis. OBDs bound to sites 1 and 3 are colored in cyan and those bound to sites 2 and 4 are in red. (C) A schematic diagram showing the contacts of T-ag OBD with the GAGGC pentanucleotide. The bases are numbered 1–5 starting from the 5′ end of the recognition element. The amino acids are boxed and the trace of polypeptide backbone is indicated with a thick dark line. The broken lines indicate hydrogen bonds. ‘W' for water molecules. (D) Conformational changes in T-ag OBD induced by DNA binding. A superposition of free and bound OBD was generated as discussed in the text. Shown is the Cα trace of the free (blue and yellow for the P21 and P212121 crystal forms, respectively) and bound (red) domain. Important amino acids are labeled. Maximal shift of the loops are indicated with dashed lines, and the size of the shift is indicated in Å (large numbers).
The second OBD/DNA complex comprised a T-ag OBD mutant with a Cys216 to Ser substitution (C216S) and a palindromic DNA that contained sites 1 and 3 (PEN-2). Site 2 in this DNA, which would be oriented on the opposite face of the DNA to the Site 1/3 palindrome, was mutated to ATgAT in order to create a perfect palindrome (Figure 3A). T-ag OBD (C216S) bound to this mutated DNA, was crystallized in space group p43212 and diffracted to 2.3 Å. The structure was solved by molecular replacement using the apo form T-ag OBD structure as a search model and refined against 2.3 Å data to R=22.4% and R_free=29.8% (Figure 3B). Additional crystallographic statistics are given in Supplementary Table 1.
Figure 3.
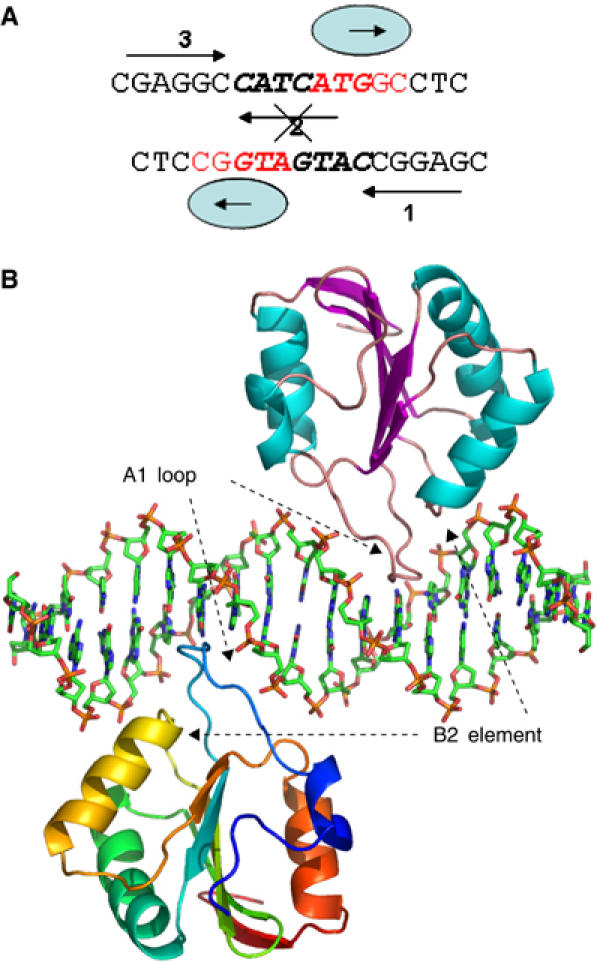
Non-sequence-specific binding of T-ag OBD to PEN-2. (A) DNA used for cocrystallization of the complex. Arrows depict two GAGGC pentanucleotides, sites 1 and 3. Mutated site 2 is crossed out. Non-canonical binding sites bound by T-ag OBD in the structure are shown in red. (B) T-ag OBD–PEN-2 complex structure. Two copies of OBD are represented as ribbon models. One monomer is colored according to the secondary structure; α-helices in cyan and β-strands in purple. The other monomer is rainbow colored from blue (N-terminus) to red (C-terminus).
Structure overview
T-ag OBD in the p21 apo crystal structure is similar to that in the NMR (PDB Id: 1TBD) (Luo et al, 1996) and crystal (PDB Id: 2FUF) (Meinke et al, 2006) structures reported previously. The RMS deviation between the apo structure and NMR structures is 1.2 Å with 121 Cα atoms and between the two X-ray structures of T-ag OBD is 0.9 Å with 122 Cα atoms. The structures of T-ag and BPV E1 OBDs are also similar (PDB Id: 1F08) (Enemark et al, 2000); the RMS deviation among 70 Cα was 1.8 Å. The two crystallographically independent molecules of T-ag OBD in the unit cell were connected by a disulfide bond between a cysteine on each molecule (Cys 216–Cys 216). Cys 216 is completely conserved across the polyoma virus family and the mutation C216G is defective in DNA unwinding (Wun-Kim et al, 1993). The p212121 and p21 structures were very similar (RMS deviation of 0.5 Å with 126 Cα atoms). The structures differed in that a Cys 216–Cys 216 disulfide bond was observed only in the p21 but not in the p212121 structure, and the loop containing Cys 216 had a slightly different conformation (discussed below).
In the T-ag OBD/PEN-4 complex, four OBDs bind individually to the four GAGGC elements (Figure 2A). Surprisingly, contrary to what was anticipated from biochemical studies and the structure of the BPV E1-DNA complex, the T-ag OBDs bound to PEN-4 elements were not dimers and indeed were not even in direct contact. Two pairs (sites 1, 3 and sites 2, 4) of T-ag OBDs were oriented face-to-face on approximately opposite (upper and lower) sides of the DNA (Figure 2B). As predicted from mutagenesis studies, the protein–DNA contacts are mediated by two DNA-binding elements: a DNA-binding loop (S147–T155), known in the literature as ‘A1 loop' (Simmons et al, 1990), and a DNA-binding helix with a portion of the loop (‘B2 element') N-terminal to it (H203–N210), with additional contribution from Asn 227. The A1 loop travels back-and-forth in the major groove and accounts for most of the sequence-determining contacts with the DNA (Figure 2C).
Protein–DNA interaction
There are two crystallographically independent T-ag monomers in the T-ag/PEN-4 co-crystal structure. Direct protein–base contacts are virtually identical in both. Five residues of T-ag OBD make a total of eight direct, and one water-mediated, hydrogen bonds with the five bases in the major groove of the pentameric DNA recognition element. Specific protein–base recognition is provided by two arginines, Arg 154 and Arg 204, and two flanking guanines, G1 (the first G in the GAGGC pentamer) and the G complementary to C5. Arg 154 forms two sequence-specific hydrogen bonds with G1 (Figure 4A). Arg 204 forms two similar contacts with the G complementary to C5 (Figure 4G). Ser 152 binds specifically to A2 (Figure 4C). Asn 153 contributes three hydrogen bonds to specific interaction with G3 and G4 (Figure 4E and F).
Figure 4.
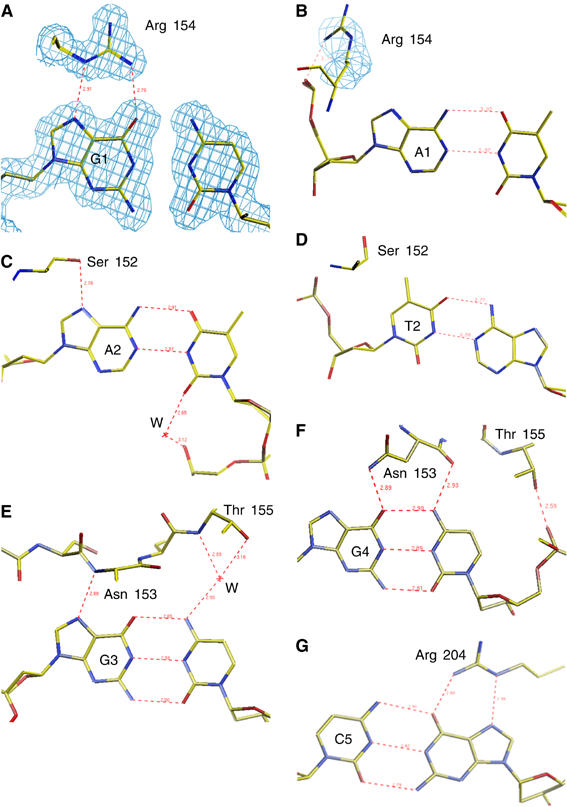
Structural details of T-ag interaction with DNA bases in the PEN-2 and PEN-4 structures. (A) Sequence-specific interaction of Arg 154 with G1 (the first G in the GAGGC pentamer) in the PEN-4 structure. A representative electron density map (shown in blue) is superimposed on the model. (B) Nonspecific interaction of Arg 154 with A1 (A in position of G1) in the PEN-2 structure. A representative electron density is superimposed on Arg 154. (C) Sequence-specific interaction of Ser 152 with A2. (D) Nonspecific interaction of Ser 152 with the T2 in the PEN-2 structure. Sequence-specific interactions, which involve (E) the G3 and Asn 153, (F) the G4 and Asn 153, and (G) the G (complementary to C5) with Arg 204. The protein and DNA are shown as stick models and colored by atom type; yellow for carbon, blue for nitrogen, red for oxygen, and purple for phosphorus. A representative electron density as captured from 2Fo−Fc map is shown with contours drawn at the 1.25σ level. Hydrogen bonds are indicated with red dashed lines, and the length of the bonds is indicated in Å.
T-ag OBD also makes extensive nonspecific contacts to DNA. There are 10 direct and water-mediated hydrogen bonds to phosphates (Figure 2C). Nonspecific contacts to phosphate groups are mediated by Ser 147, Val 151, Phe 150, Thr 155, His 203 (ND1), Asn 210, and Asn 227. Four direct H-bonds and one water-mediated contact are originated from the A1 loop (Ser 147, Val150, Phe151, and Thr 155). Three direct and one water-mediated contacts are originated from the B2 element, and one comes from Asn 227.
Our structural studies are consistent with the DNA-binding properties of T-ag OBD in solution as determined at near-physiological conditions (Titolo et al, 2003b). These studies showed that binding of multiple T-Ag OBDs to the origin was not cooperative. Similarly, in the structures, we did not observe any interaction between different OBDs in the protein–DNA complex. In contrast, the full-length T-ag does bind cooperatively to the origin as a double hexamer (Valle et al, 2006 and references cited there).
Comparing the structure of T-ag OBP in the presence and absence of origin DNA
Structural differences between the p21 apo and DNA-bound form of the T-ag OBD (aa 134–253) were identified by superimposing the two structures (Figure 2D, red and blue). The RMS deviation between the two forms was 0.59 Å with all (119) Cα atoms included. The biggest conformational change was detected in the DNA-binding loop (centered on alanine 151) and the loop containing Cys216 (C216 loop), which forms a disulfide bond in the apo structure. The conformational changes seen in the DNA-binding loop A1 were centered on phenylalanine 151 and involved residues 150 through 152. The maximal shift between the bound and free conformations for Phe 151 was 4.4 Å (Val150—3.7 Å; Ser152—1.7 Å).
C216 forms a disulfide bond in one crystal form of the T-ag OBD. This residue has been implicated in T-Ag function; a mutant T-ag with C216G substitution was not functional in origin DNA unwinding, although the mutated protein bound the ori with wild-type affinity, oligomerized into hexamer, and even unwound an ori-containing linear DNA (Wun-Kim et al, 1993). C216 is on the surface of the OBD important for cooperative interactions between hexamers in assembly on the origin (Weisshart et al, 1999). Our observation that the C216 was capable of forming a disulfide bond perhaps suggests a mechanism for its importance. However, we do not believe that its ability to form a disulfide bond is relevant for DNA binding for the following reasons. First, the C216 region is on the opposite side of the DNA binding face of the OBD more than 15 Å away. Second, although the orientation of the loop that contains the C216 residue is different in our various T-Ag structures, the orientation does not correlate with DNA-binding (Figure 2D). In one of the crystal forms of the protein not bound to DNA, the C216 loop is in the same position as in the DNA-bound form. Thus, we can conclude first that the C216 loop is quite flexible and second that its position in the crystal structure is more a function of the crystallization conditions than whether the protein is bound to DNA or not.
The DNA structure
The DNA bends slightly around each OBD. The extent of the bending around binding sites 3 and 4 and sites 1 and 2 was equal and opposite; the sum of the four bends was equal to zero. The binding of the T-ag OBD alone to its origin therefore appears insufficient to alter the DNA structure and enable origin unwinding. It is also known that unwinding cannot be initiated by the helicase domain alone, confirming that origin melting likely results from an interplay among the ori, OBD, and helicase (Li et al, 2003).
Nonspecific binding in the PEN-2 structure
In the T-ag/PEN-2 complex structure, site 2 was mutated to ATgAT to create a palindromic DNA sequence more suitable for crystallization (Figure 3A). T-ag OBD was expected to bind to sites 1 and 3 because of the known affinity for these binding sites, and because of their co-localization on the same face of the DNA. Although a two-site origin cannot be melted by the T-ag double hexamer, it is sufficient for double hexamer assembly. We expected that the structure of the T-ag OBD bound to this element would provide insight into the protein–protein interactions that preceded double hexamer assembly. Surprisingly, T-ag OBD was bound not to the canonical GAGGC sequences in sites 1 and 3, but to an abutting non-canonical sequence—ATGGC (Figure 3B). Although we cannot exclude that this binding may be an artifact of crystallization, the PEN-2 crystal structure does reveal aspects of the plasticity of the DNA binding, a plasticity that might be exploited during origin unwinding.
The general character of the non-canonical binding was similar to that observed in the binding to the consensus sequence. However, there was a significant difference in the interactions with the two ‘non-canonical' bases (AT). In binding to this non-consensus site, Arg 154 moved away from A1 and formed a hydrogen bond with the phosphate group of this nucleotide (Figure 4B). Ser152, which makes a hydrogen bond with canonical A2 (Figure 4C), is oriented away and does not form an H-bond with the non-canonical T2 base (Figure 4D). In short, the crystallographic experiments showed that the T-ag OBD exhibits some structural plasticity in its interactions with DNA, enabling it to bind to non-consensus sequences, albeit with lower affinity.
Model of SV40 ori bound by T-ag
With the structures of the individual domains of the functional SV40 double hexamer in hand, as well as with the information about origin/T-ag interaction, we attempted to build an atomic model of the functional T-ag hexamer (putative intermediate form) on DNA (Figure 5). Our starting point was the PEN-4 structure (black), as this comprises the specific T-ag binding portion of a functional origin. In the crystals, symmetry-related DNA molecules (23 bp) were arranged in a pseudo-continuous DNA helix, enabling us to model an extended piece of DNA by adding two symmetry-related DNA molecules to the left-hand and right-hand side of the parent DNA. The length of the modeled DNA is 71 bp (23+1/1 overhang+23+1/1 overhang+23) with one base overhang on each side. The OBDs were positioned as found in the PEN-4 complex, with their C-terminal domains oriented towards the ends of the DNA. Two T-ag helicase hexamers (blue) were threaded on these DNA extensions and moved as close to OBDs as possible, but not so close as to make sterical clashes (PDB Id: 1N25) (Li et al, 2003). The N-terminal part of the helicase domain (residue 266) is oriented towards the C-terminus (residue 253) of the nearest T-ag OBD. In this orientation, the DNA is predicted to run through the central tunnels of the hexameric helicase. In the model, the T-ag double hexamer is predicted to contact 70–73 bp of DNA, which is in a good agreement with biochemical observation that T-ag protects 74 bp. The predicted location of the EP fragment melted by T-ag hexamer is inside the helicase domain, in proximity of the β-Hairpin which moves along the central channel and pulls DNA into the helicase for unwinding (Gai et al, 2004).
Figure 5.
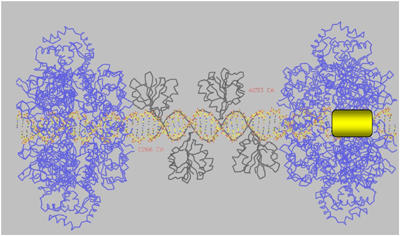
Molecular model of an initial step in the SV40 DNA replication. The PEN-4 structure is shown in black, two hexameric helicase domains are in blue (PDB Id: 1N25), modeled DNA is shown as a stick model and colored per atom type (carbon in yellow, oxygen in red, nitrogen in blue, and phosphorus in purple). Position of the initially melted 8 nt fragment of EP with respect to the PEN box is highlighted with a yellow rectangle. Relative positions of the C-terminus in the OBD (aa 253) and N-terminus in the helicase domain (aa 266) are indicated. See text for more detail.
Discussion
A combination of the available structural and biochemical data suggests a model for origin unwinding. In the first step, four OBD domains bind to the four consensus sites in the origin (Joo et al, 1997) (Figures 2A and 6A). These binding events are thought to bring the associated helicase domains to the DNA and induce the assembly of the helicase domains into two hexamers around the DNA (Figures 5 and 6B). However, there are only four DNA-binding sites; yet, a total of 12 T-ag molecules must be recruited to form the two predicted hexameric helicases. Accordingly, the DNA-bound T-ag must recruit other molecules from solution. The central channel of the T-ag helicase can be opened wide enough (at least about 22 Å) to accommodate dsDNA of about 20 Å in diameter (Gai et al, 2004). This putative passage of DNA is consistent with electron microscopy data (Mastrangelo et al, 1989; San Martin et al, 1997; Valle et al, 2000, 2006; Gomez-Lorenzo et al, 2003). Studies with individual DNA-binding elements reveal in part the mechanism. Biochemical data indicate that the presence of only one pentanucleotide is sufficient to induce hexamer assembly, and two pentanucleotides (binding sites 1 and 3) are sufficient to induce double hexamer assembly (Joo et al, 1998). Importantly though, double hexamer assembly on the two-site DNA is insufficient to induce origin melting. This must be mediated by the interaction between double hexamers on the four-site origin. The only obvious structural difference in the T-ag assembly mediated by two and four sites is the spacing between the helicases. In the case of two sites, the spacing would be predicted to be half a turn of DNA shorter (about 17 Å) as compared with the four-site origin. The assembled helicase would bring an additional eight T-ag OBDs in proximity of the PEN region. Although the eight additional OBDs may remain unbound, it is possible that they bind non-canonical DNA in the lower affinity mode revealed in the structure of OBD bound to two sites (Titolo et al, 2003b). If so, the higher order structure of the origin bound to 12 T-ag OBDs (four specifically and eight nonspecifically) may resemble the spiral arrangement of the T-ag OBDs seen in its apo structure (Figure 6C).
Figure 6.
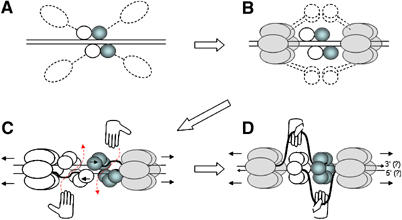
Model for origin melting and DNA unwinding by T-ag. (A) Four OBDs bind the origin and bring monomeric domains of helicase in proximity to the DNA. (B) Helicase domains are assembled in two hexamers encircling DNA; this brings an additional eight OBDs in proximity to the PEN box. (C) Using a nonspecific DNA-binding mode, the eight additional OBDs bind side by side with the four initial OBDs forming a spiral double hexamer. The moving helicase distorts and melts a fragment of EP, making it accessible for RPA (hands). RPA moves a melted strand of DNA out through the open gate (red dashed arrow). (D) The moving helicase pulls the most distant OBD (via the short linker comprising aa 253–266) and induces switching of an open (spiral) OBD hexamer to closed hexamer. OBDs are represented as circles. OBDs that mediate assembly of the right-hand hexamer are colored in blue and those for the left-hand hexamer are white. Monomers of the helicase domain are represented as ellipses and shadowed in gray. The direction of helicase movement is indicated by detached arrows in front of the helicase hexamers. A putative polarity of the DNA strands is shown by small arrows at the DNA ends.
With the helicase domains suitably positioned, the helicase would translocate and melt the dsDNA. In agreement with biochemical data and consistent with the structural model of SV40 ori bound by T-ag (discussed above), the predicted location of the EP fragment that is melted by T-ag hexamer is inside the helicase hexamer (Bullock, 1997).
Multiple EM experiments indicate that the T-ag forms a symmetrical double ring comprising 12 OBDs (San Martin et al, 1997; Valle et al, 2000, 2006; Gomez-Lorenzo et al, 2003). However, the initial binding of the four OBDs to PEN is asymmetric, and thus there must be some mechanism by which the OBDs undergo a conformtaional change. We postulate that the initially bound OBDs form a spiral hexamer, orienting the OBDs with respect to the helicase. Once the helicase translocates, perhaps the most distant OBD, which is predicted to be located in about one DNA turn (∼34 Å) away from the helicase, moves along in tow and causes a switch in conformation from the spiral hexamer of OBDs (open gate) to a closed hexamer (closed gate). This model parallels that of ‘helix–to–ring' switch in E1 unwinding (Schuck and Stenlund, 2005). After the origin is melted, but before the gate is closed, the ssDNA-binding protein RPA (hands; Figure 6C) binds the ssDNA and takes it out through the open gate. With the assistance of RPA, this DNA strand can be further pulled out of the central tunnel in the helicase domain (Enemark and Joshua-Tor, 2006). This intermediate structure could be consistent with the ‘rabbit ears' structure observed by EM (Figure 6D) (Wessel et al, 1992).
There are several lines of evidence in support of this hypothesis. First, T-ag OBDs form a ‘spiral hexamer' (Meinke et al, 2006), which is proposed to represent an important step along the path of assembly of the active dodecamer helicase. Second, the E1 BPV double-hexamer can assemble on the origin via either ‘productive' or ‘nonproductive' pathways (Schuck and Stenlund, 2005). The ‘productive' pathway is associated with a longer footprint and a double-trimer (DT) to double-hexamer (DH) ‘helix–to–ring' transition.
Although mechanistic by its nature, this model is in agreement with most of the data reported in literature. More detailed structural and functional analysis will be necessary to verify and refine the model.
Materials and methods
Constructs, proteins, and DNA
A plasmid encoding T-ag OBD (aa 131–159) was kindly provided by Dr Ellen Fanning (Vanderbilt University, TN) (Arthur et al, 1988). T-ag OBD (in its native form) was subcloned into a pET15b vector (Novagen), to generate the protein as a fusion with a His tag and a thrombin cleavage site. A mutant form of T-ag OBD (C216S) (pET15b-T-ag OBD (C216S)) was generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and the manufacturer's protocol. The OBD and OBD (C216S) were expressed in Escherichia coli and purified using a series of HiTrap affinity, Hi Trap Chelating, and Hi Trap Heparin columns (Amersham Biosciences), concentrated to 10 mg/ml, and stored frozen in a solution containing 5 mM HEPES (pH 7.5), 150 mM NaCl, and 10 mM DTT. The DNA for cocrystallization of the PEN-2 complex comprised 17 bp and a single nucleotide overhang at the 5′-end; 18-mer upper strand CGAGGCCATCATGGCCTC and 18-mer low strand CGAGGCCAT GATGGCCTC. That for PEN-4 had 23 bp and single nucleotide overhang at the 5′-end; 24-mer upper strand AGAGGCCGAGGCC GCCTCGGCCTC and 24-mer low strand AGAGGCCGAGGCGGC CTCGGCCTC (Sigma Genosys, The Woodlands, TX). The upper and low DNA strands were denatured by heating to 95°C for 2 min, and annealed by cooling to 35°C at a uniform rate of 0.2°C per minute. The complexes of T-ag OBD (C216S) with PEN-2 and T-ag OBD with PEN-4 were prepared by incubating protein solution with dry DNA at a 1.5 molar excess of DNA over protein.
Crystallization of T-ag OBD in p21 and p212121 apo forms
P21 crystals of the T-ag OBD were grown at 20°C by vapor diffusion as hanging drops prepared by mixing 1 μl of protein (10 mg/ml) with 1 μl of the crystallization buffer (20 mM NSCN, 10–14% PEG4K 0.1 Tris pH 8.3). The derivative (HgCl) was prepared by soaking the crystals in the crystallization buffer containing 0.05 mM HgCl2 for 100 min. P212121 crystals of the T-ag OBD were grown from freshly made protein at 20°C by vapor diffusion as hanging drops prepared by mixing 1 μl of protein (10 mg/ml) with 1 μl of the crystallization buffer (200 mM NaAcet, 20% PEG4K). Four percent p600 was used as an additive in the drops.
Crystallization of T-ag OBD bound to the 23 bp PEN-4 DNA. The PEN-4 complex crystals were grown at 4°C by vapor diffusion as sitting drops prepared by mixing 1 μl of protein (concentration of 10 mg/ml)/1 μl of the crystallization buffer: 200 mM di-ammonium hydrogen Citrate, 20% PEG3K, and 10 mM MgCl2. The crystals grew in 30 days at 4°C.
Crystallization of T-ag OBD (C216S) bound to the 17 bp DNA
The PEN-2 complex crystals were grown at 4°C by vapor diffusion as hanging drops prepared by mixing 1 μl of protein (concentration of 10 mg/ml)/1 μl of the crystallization buffer: 200 mM NaAcet, 20% PEG3K, and 4% PEG600. The crystals grew in 5–10 days at 4°C.
Structure solution and refinement
Data from flash-cooled T-ag OBD (p21 native and HgCl derivative; p212121 native), the PEN-2 complex, and the PEN-4 complex crystals at 100 °K were collected on Rigaku-H3R/Raxis-IV (Rigaku/MSC) using Cu radiation or at CHESS beamline A1 (for PEN-4) using 0.97 Å wavelength, integrated and scaled by using DENZO/SCALEPACK (Otwinoski and Minor, 1997). The mercury sites in HgCl derivative were manually located, and the experimental map was generated by using PHASES (Furey and Swaminathan, 1997). All other structures were solved by molecular replacement by using the T-ag OBD structure as a search model for MOLREP (Vagin and Teplyakov, 1997). All structures were initially traced by ARP/WARP (Perrakis et al, 1999) and then manually rebuilt in O (Jones et al, 1991). Final refinement was performed by using REFMAC (Murshudov et al, 1996). Additional crystallographic statistics are given in Supplementary Table 1. The drawings were generated with O and PYMOL (DeLano Scientific, South San Francisco, CA).
Coordinates
Coordinates of apo T-ag OBD (p21 and p212121 crystal forms), PEN-2 complex, and PEN-4 complex structures have been deposited to the PDB (PDB Id codes: 2IPR, 2ITJ, 2NL8, and 2ITL, respectively) and will be available from the PDB upon publication.
Supplementary Material
Supplementary Table1
Acknowledgments
We thank Aled Edwards, Lori Frappier, and Amy Wernimont for critical reading of the manuscript. This work was supported in part by a research grant from National Institute of General Medical Science R01-GM61192 awarded to AB.
References
- Arthur AK, Hoss A, Fanning E (1988) Expression of simian virus 40T antigen in Escherichia coli: localization of T-antigen origin DNA-binding domain to within 129 amino acids. J Virol 62: 1999–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, Edwards AM (1996) Crystal structure of the DNA-binding domain of the Epstein–Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84: 791–800 [DOI] [PubMed] [Google Scholar]
- Bochkarev A, Barwell JA, Pfuetzner RA, Furey W Jr, Edwards AM, Frappier L (1995) Crystal structure of the DNA-binding domain of the Epstein–Barr virus origin-binding protein EBNA 1. Cell 83: 39–46 [DOI] [PubMed] [Google Scholar]
- Borowiec JA, Hurwitz J (1988) Localized melting and structural changes in the SV40 origin of replication induced by T-antigen. EMBO J 7: 3149–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock PA (1997) The initiation of simian virus 40 DNA replication in vitro. Crit Rev Biochem Mol Biol 32: 503–568 [DOI] [PubMed] [Google Scholar]
- Bullock PA, Seo YS, Hurwitz J (1989) Initiation of simian virus 40 DNA replication in vitro: pulse-chase experiments identify the first labeled species as topologically unwound. Proc Natl Acad Sci USA 86: 3944–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butel JS, Lednicky JA (1999) Cell and molecular biology of simian virus 40: implications for human infections and disease. J Natl Cancer Inst 91: 119–134 [DOI] [PubMed] [Google Scholar]
- Campos-Olivas R, Louis JM, Clerot D, Gronenborn B, Gronenborn AM (2002) The structure of a replication initiator unites diverse aspects of nucleic acid metabolism. Proc Natl Acad Sci USA 99: 10310–10315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Joo WS, Bullock PA, Simmons DT (1997) The N-terminal side of the origin-binding domain of simian virus 40 large T antigen is involved in A/T untwisting. J Virol 71: 8743–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePamphilis M (1996) DNA replication in Eukaryotic Cells. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Eichman BF, Fanning E (2004) The power of pumping together; deconstructing the engine of a DNA replication machine. Cell 119: 3–4 [DOI] [PubMed] [Google Scholar]
- Enemark EJ, Chen G, Vaughn DE, Stenlund A, Joshua-Tor L (2000) Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Mol Cell 6: 149–158 [PubMed] [Google Scholar]
- Enemark EJ, Joshua-Tor L (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442: 270–275 [DOI] [PubMed] [Google Scholar]
- Enemark EJ, Stenlund A, Joshua-Tor L (2002) Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J 21: 1487–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning E, Knippers R (1992) Structure and function of simian virus 40 large tumor antigen. Annu Rev Biochem 61: 55–85 [DOI] [PubMed] [Google Scholar]
- Furey W, Swaminathan S (1997) PHASES-95: a program package for the processing and analysis of diffraction data from macromolecules. In Methods in Enzymology: Macromolecular Crystallography, Part B, Carter CWJ, Sweet RM (eds) pp 590–620. Orlando, FL: Academic Press [DOI] [PubMed] [Google Scholar]
- Gai D, Zhao R, Li D, Finkielstein CV, Chen XS (2004) Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119: 47–60 [DOI] [PubMed] [Google Scholar]
- Gidoni D, Scheller A, Barnet B, Hantzopoulos P, Oren M, Prives C (1982) Different forms of simian virus 40 large tumor antigen varying in their affinities for DNA. J Virol 42: 456–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Lorenzo MG, Valle M, Frank J, Gruss C, Sorzano CO, Chen XS, Donate LE, Carazo JM (2003) Large T antigen on the simian virus 40 origin of replication: a 3D snapshot prior to DNA replication. EMBO J 22: 6205–6213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde RS, Grossman SR, Laimins LA, Sigler PB (1992) Crystal structure at 1.7 Å of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature 359: 505–512 [DOI] [PubMed] [Google Scholar]
- Hickman AB, Ronning DR, Kotin RM, Dyda F (2002) Structural unity among viral origin binding proteins: crystal structure of the nuclease domain of adeno-associated virus Rep. Mol Cell 10: 327–337 [DOI] [PubMed] [Google Scholar]
- Huang SG, Weisshart K, Gilbert I, Fanning E (1998) Stoichiometry and mechanism of assembly of SV40 T antigen complexes with the viral origin of DNA replication and DNA polymerase alpha-primase. Biochemistry 37: 15345–15352 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Joo WS, Kim HY, Purviance JD, Sreekumar KR, Bullock PA (1998) Assembly of T-antigen double hexamers on the simian virus 40 core origin requires only a subset of the available binding sites. Mol Cell Biol 18: 2677–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo WS, Luo X, Denis D, Kim HY, Rainey GJ, Jones C, Sreekumar KR, Bullock PA (1997) Purification of the simian virus 40 (SV40) T antigen DNA-binding domain and characterization of its interactions with the SV40 origin. J Virol 71: 3972–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Ahn BY, Cho Y (2001) Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO J 20: 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhao R, Lilyestrom W, Gai D, Zhang R, DeCaprio JA, Fanning E, Jochimiak A, Szakonyi G, Chen XS (2003) Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature 423: 512–518 [DOI] [PubMed] [Google Scholar]
- Luo X, Sanford DG, Bullock PA, Bachovchin WW (1996) Solution structure of the origin DNA-binding domain of SV40 T-antigen. Nat Struct Biol 3: 1034–1039 [DOI] [PubMed] [Google Scholar]
- Mastrangelo IA, Hough PV, Wall JS, Dodson M, Dean FB, Hurwitz J (1989) ATP-dependent assembly of double hexamers of SV40 T antigen at the viral origin of DNA replication. Nature 338: 658–662 [DOI] [PubMed] [Google Scholar]
- Meinke G, Bullock PA, Bohm A (2006) Crystal structure of the simian virus 40 large T-antigen origin-binding domain. J Virol 80: 4304–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G, Vagin A, Dodson E (1996) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Otwinoski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, Part A, Carter Jr CW, Sweet RM (eds) pp 307–326. Orlando, FL: Academic Press [DOI] [PubMed] [Google Scholar]
- Parsons RE, Stenger JE, Ray S, Welker R, Anderson ME, Tegtmeyer P (1991) Cooperative assembly of simian virus 40 T-antigen hexamers on functional halves of the replication origin. J Virol 65: 2798–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Runzler R, Thompson S, Fanning E (1987) Oligomerization and origin DNA-binding activity of simian virus 40 large T antigen. J Virol 61: 2076–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martin MC, Gruss C, Carazo JM (1997) Six molecules of SV40 large T antigen assemble in a propeller-shaped particle around a channel. J Mol Biol 268: 15–20 [DOI] [PubMed] [Google Scholar]
- Sanders CM, Stenlund A (2001) Mechanism and requirements for bovine papillomavirus, type 1, E1 initiator complex assembly promoted by the E2 transcription factor bound to distal sites. J Biol Chem 276: 23689–23699 [DOI] [PubMed] [Google Scholar]
- Scheidtmann KH, Hardung M, Echle B, Walter G (1984) DNA-binding activity of simian virus 40 large T antigen correlates with a distinct phosphorylation state. J Virol 50: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Stenlund A (2005) Assembly of a double hexameric helicase. Mol Cell 20: 377–389 [DOI] [PubMed] [Google Scholar]
- SenGupta DJ, Borowiec JA (1994) Strand and face: the topography of interactions between the SV40 origin of replication and T-antigen during the initiation of replication. EMBO J 13: 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DT, Loeber G, Tegtmeyer P (1990) Four major sequence elements of simian virus 40 large T antigen coordinate its specific and nonspecific DNA binding. J Virol 64: 1973–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenlund A (2003) Initiation of DNA replication: lessons from viral initiator proteins. Nat Rev Mol Cell Biol 4: 777–785 [DOI] [PubMed] [Google Scholar]
- Titolo S, Brault K, Majewski J, White PW, Archambault J (2003a) Characterization of the minimal DNA binding domain of the human papillomavirus e1 helicase: fluorescence anisotropy studies and characterization of a dimerization-defective mutant protein. J Virol 77: 5178–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titolo S, Welchner E, White PW, Archambault J (2003b) Characterization of the DNA-binding properties of the origin-binding domain of simian virus 40 large T antigen by fluorescence anisotropy. J Virol 77: 5512–5518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30: 1022–1025 [Google Scholar]
- Valle M, Chen XS, Donate LE, Fanning E, Carazo JM (2006) Structural basis for the cooperative assembly of large T antigen on the origin of replication. J Mol Biol 357: 1295–1305 [DOI] [PubMed] [Google Scholar]
- Valle M, Gruss C, Halmer L, Carazo JM, Donate LE (2000) Large T-antigen double hexamers imaged at the simian virus 40 origin of replication. Mol Cell Biol 20: 34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshart K, Taneja P, Jenne A, Herbig U, Simmons DT, Fanning E (1999) Two regions of simian virus 40T antigen determine cooperativity of double-hexamer assembly on the viral origin of DNA replication and promote hexamer interactions during bidirectional origin DNA unwinding. J Virol 73: 2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel R, Schweizer J, Stahl H (1992) Simian virus 40 T-antigen DNA helicase is a hexamer which forms a binary complex during bidirectional unwinding from the viral origin of DNA replication. J Virol 66: 804–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wun-Kim K, Upson R, Young W, Melendy T, Stillman B, Simmons DT (1993) The DNA-binding domain of simian virus 40 tumor antigen has multiple functions. J Virol 67: 7608–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table1