

Abstract
Previous studies have shown that IRP1+/− IRP2−/− knockout mice develop progressive neurodegenerative symptoms similar to those observed in human movement disorders such as Parkinson's disease. Histological investigations using optical microscopy show that these IRP knockout mice display accumulation of ferritin in axonal tracts in the brain, suggesting a possible role for excess ferritin in mediating axonal degeneration. Direct observation of the 3D distribution of ferritin by electron tomography indicates that ferritin amounts are increased by 3- to 4-fold in selected regions of the brain, and structural damage is observed within the axon as evidenced by the loss of the internal network of filaments, and the invaginations of neighboring oligodendrocyte membranes into the axonal medium. While optical microscopic investigations suggest that there is a large increase in ferritin in the presumptive axonal regions of the IRP knockout mice, electron tomographic studies reveal that most of the excess ferritin is localized to double-walled vesicular compartments which are present in the interior of the axon and appear to represent invaginations of the oligodendrocyte cells into the axon. The amount of ferritin observed in the axonal space of the knockout mice is at least 10-fold less than the amount of ferritin observed in wild-type mouse axons. The surprising conclusion from our analysis, therefore, is that despite the overall increase in ferritin levels in the knockout mouse brain, ferritin is absent from axons of degenerating neurons, suggesting that trafficking is compromised in early stages of this type of neuronal degeneration.
Keywords: Iron regulatory protein, Neuronal degeneration, Electron tomography, Electron energy loss spectroscopy, Ferritin, Double-walled vesicle
1. Introduction
Iron is an important element for the central nervous system. Many processes in normal brain development, including oxidative respiration, myelination of axons, and neurotransmitter synthesis require iron (Hentze et al., 2004; van Gelder et al., 1998). Excessive brain iron, however, is neurotoxic, and produces free radicals that can cause oxidative damage of different cellular components. Elevated levels of brain iron are observed frequently in patients with Alzheimer's disease (Connor et al., 1992; Smith et al., 1997), Parkinson's disease (Berg et al., 2001; Hirsch and Faucheux, 1998), multiple sclerosis and other forms of neurodegenerative disease (Bourdon et al., 2003), highlighting the importance of regulation of iron metabolism in the maintenance of healthy neurons.
Iron regulatory proteins 1 and 2 (IRP1 and IRP2) are cytoplasmic mRNA binding proteins that regulate expression of ferritin and transferrin receptor (Hu and Connor, 1996; Rouault and Klausner, 1997; Schneider and Leibold, 2000), the two major proteins involved in maintaining intracellular iron homeostasis. By controlling the level of expression of these two proteins, the cell can stringently regulate the iron concentration and availability through uptake and sequestering mechanisms (Hentze et al., 2004; Rouault and Klausner, 1997; Schneider and Leibold, 2000). Ferritin, the principal intracellular iron storage protein, is particularly important in this regard because of its high iron binding capacity, serving the dual functions of sequestering iron for storage purposes, and protecting the cell from excess iron (Theil, 1987; Torti and Torti, 2002). Transgenic mice carrying knockouts of one copy of IRP1 and both copies of the IRP2 (IRP1+/− IRP2−/−; referred to in the rest of the text as IRP knockout mice) display an increased level of iron in the brain, and histological studies with optical microscopy have established the presence of high levels of iron and ferritin in white matter tracts (Smith et al., 2004). These IRP knockout mice are even more severely affected than the previously analyzed IRP2 knockout mice in which both copies of IRP1 are still present (LaVaute et al., 2001). These mice develop a progressive neurodegenerative disease whose features resemble those found in several human diseases including Parkinson's disease. Understanding the structural and molecular basis of neuronal degeneration in IRP knockout mice is therefore likely to be of fundamental and broad significance to human brain disorders.
Electron tomography is an emerging imaging method for 3D localization and visualization of cellular and molecular components at resolutions about an order of magnitude higher than that obtained with conventional optical microscopic methods (Baumeister, 2002). Electron tomography thus offers the possibility of direct 3D visualization of axon structure in degenerating neurons. Further, electron microscopy allows direct detection of ferritin in the axon due to the contrast arising from the cluster of iron atoms at the core. Here, we have combined both modes of electron microscopic imaging to analyze neuronal structure and ferritin distribution in fixed brain tissue sections of normal and diseased mouse brains. Our findings, which show that there are dramatic differences in the cellular location of ferritin molecules in the IRP knockout as compared to wild-type mice, begin to provide a structural context to understand the mechanism of degeneration.
2. Materials and methods
2.1. Specimen preparation
Age-matched mice of wild-type and IRP1+/− IRP2−/− knockout were perfused with 2% paraformaldehyde and 1% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). After perfusion, brain tissues were further fixed in the same buffer for 24 h. The fixed mouse brain cerebellar tissue was then cut into 1 mm cubes, and washed six times with buffer containing 0.1 M sodium cacodylate to remove the aldehyde fixatives. The tissue cubes were additionally fixed for 1 h in sodium cacodylate buffer containing OsO4 (at concentrations ranging from 0.04 to 2%) and then for 1 h in sodium cacodylate buffer containing 1% uranyl acetate. The tissue blocks were dehydrated using acetone and embedded in epon resin. Thick sections were cut with a Leica Ultracut T Microtome (Leica Microsystems, Vienna, Austria) and stained with Prussian blue for locating white matter regions using a light microscope. Sections with thicknesses in the range of 70–200 nm were prepared for investigation of ultrastructure by 2D and 3D electron microscopy.
Tissue samples were also used for histology (7 μm thick sagittal sections prepared by American Histo Labs, Gaithersburg, MD). For iron detection, enhanced Perls' DAB ferric iron staining was performed. Slides were dewaxed in xylene, rehydrated in decreasing concentrations of ethanol, washed in TBS buffer, and treated in a 1:1 solution of 2% HCl and 2% KFeCN for 30 min. The tissues were again washed in the same buffer and treated for 35 min with DAB–Tris–H2O2. Slides were washed multiple times in distilled water and then dehydrated in increasing concentrations of ethanol, cleared in xylene, and mounted with coverslips using permount. For ferritin immunohistochemistry, a prediluted ferritin antibody was used (Biogenesis, Kingston, NH). Goat anti-rabbit IgG conjugated to biotin was used as the secondary antibody (Vector Laboratories, Burlingame, CA), followed by avidin–biotin complex la- beled with HRP (horseradish peroxidase; ABC Elite Kit, Vector Laboratories). HRP was visualized using the substrate diaminobenzidine (DAB, Vector Laboratories) with incubation at room temperature for 1 h and counterstained with methyl green.
2.2. Ferritin detection by EELS
Electron energy loss spectroscopy (EELS) and elemental mapping were performed using a VG Microscope HB501 STEM equipped with a cold-field emission electron source operating at 100 kV with a nanometer-sized probe. Hyperspectral images were recorded using a Gatan UHV Enfina electron spectrometer, incorporating a cooled 1340 × 100 channel CCD detector. For each pixel in the scanned image, spectral channel intensities were summed in the direction perpendicular to the energy dispersion and were corrected for dark-current and channel gain variations. Specimens were cooled to −160 °C to minimize mass loss of the tissue sections under exposure of the electron probe and also to avoid polymerization of diffusible contaminants on the surfaces of the specimen during irradiation. Specimen drift was compensated by cross correlating dark-field STEM images acquired at the end of each scan line.
For testing the presence of iron in the specimen, a dark field image was first obtained. Electron energy loss spectra (EELS) of small regions containing electron dense particles showed the presence of the iron L2,3 edge absorption peak at 710 eV, whereas this peak was absent in control regions not containing the electron dense particles. Iron maps were then obtained by integrating the spectral intensity over a 10 eV energy window that included the L2,3 peak intensity and subtracting from this, a corresponding background intensity obtained by extrapolating the pre-edge spectrum into the region of the iron L2,3 edge. The bright spots in the iron map corresponded exactly to the electron dense particles in the dark field STEM image, confirming the identification of these electron dense particles as iron-containing ferritin molecules (Fig. 3). To estimate the number of iron atoms in each particle, the core edge signals of iron and carbon were integrated over boxed areas. The fraction of Fe relative to C atoms is given by
where, Nx is the number of x atoms, Ix is the core edge signal integrated over scattering angle β and energy window Δ, and σx is the partial ionization cross section. The number of carbon atoms in each boxed area is ∼2.25 × 105 as estimated from measurement of the specimen thickness, which was determined from the low-loss spectrum to be 0.29 ± 0.02 inelastic mean free paths. Therefore, the estimated number of Fe atoms in each particle ranges from 1200 to 3600, consistent with the known composition of the ferritin core, which can hold up to a maximum of 4500 iron atoms (Harrison and Arosio, 1996).
Fig. 3.
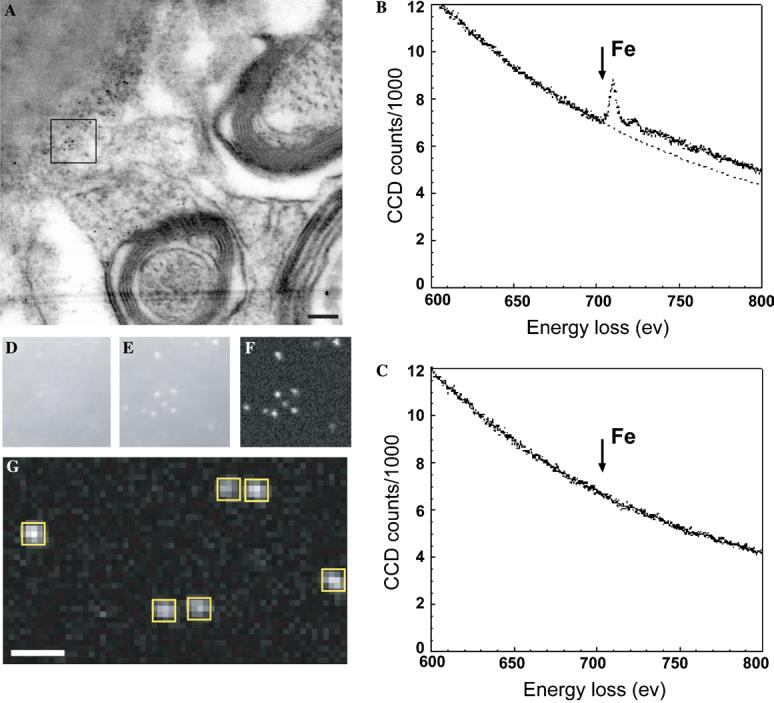
Identification of ferritin molecules with electron energy loss spectroscopy (EELS). (A) A dark field STEM image (contrast reversed) of a thin tissue section from the IRP knockout mouse brain. (B and C) EELS from regions 6 nm in diameter containing an electron dense particle (B) and a control region immediately adjacent to the particle (C). Arrows in B and C indicate the position of the energy loss peak for Fe L2,3 edge excitation. (D–F) Fe L2,3 edge elemental mapping of the region marked with a box in A. The maps were obtained by integrating the spectral intensity over a 10 eV energy window just below the edge (D), and just above the Fe L2,3 edge (E). The iron elemental map (F) was obtained by subtracting the background intensity, extrapolated from the pre-edge regions by using an inverse power law, from the Fe L2,3 edge intensity. (G) An iron elemental map obtained from a different region for estimating the number of iron atoms in each particle. The iron signal from each particle was integrated over the boxed region. Scale bars are 100 nm.
2.3. Electron microscopy and tomographic reconstruction
Projection images from the sections were recorded with a Gatan 2K CCD camera mounted on a Tecnai 12 electron microscope (from FEI Corporation, Oregon USA) equipped with a LaB6 filament operating at 120 kV, at nominal magnifications of 26 000 or 30 000×. For tomography, a series of tilted images was recorded at nominal magnifications of ∼15 000–26 000× by tilting the specimen from −70° to +70° with increments of 1°. The images were processed using a fiducial-bead based alignment procedure and back-projection algorithm, as implemented in the IMOD reconstruction package (Kremer et al., 1996) to convert the information present in the series of tilted projection images into a 3D density map.
2.4. Segmentation and rendering
Tomograms were initially visualized using IMOD, and the locations of ferritin molecules could be easily identified based on their characteristic 7 nm core-diameter and high contrast. For obtaining estimates of the relative distribution of ferritin within the brain, three types of sub-volumes were defined within each tomogram: regions within intact axons, regions outside the myelin sheath, and regions between the axon membrane and in- ner myelin sheath—including areas in which the oligodendrocyte cytoplasm invaginates into the axonal space. For each of the sub-volumes in each tomogram, a boundary of each region was drawn manually in the tomogram and the overall volume of each region was determined. The number of ferritin molecules present in these regions was counted manually. These measurements thus provided measures of the total ferrritin present in each tomogram, as well as volume-normalized amounts (i.e., concentration) of ferritin in each of the three regions. For 3D visualization of the overall axon, tomograms were semiautomatically segmented in the environment of Amira (TGS, San Diego, CA) by identifying all the components inside an axon, including microtubules, neurofilaments, mitochondria, vesicles, bilayer membranes, and ferritin molecules.
3. Results
3.1. Visualization of iron accumulation in IRP knockout mice by optical microscopy
IRP knockout mice show severe neurodegeneration associated with misregulation of iron metabolism, as previously reported (LaVaute et al., 2001; Smith et al., 2004). Optical microscopic analysis of white matter sections stained with anti-ferritin antibodies shows that there is an accumulation of ferritin in axonal regions of the knockout mice, as compared with wild-type mice (Figs. 1A and B). Similarly, chemical staining of sections for ferric iron shows an apparent accumulation of iron in the axonal tracts of the IRP knockout mice (Figs. 1C and D). These findings suggest that neuronal degeneration in the IRP knockout mice could be associated with the presence of excess ferritin and iron in the axon, although the precise location of ferritin within the axonal space cannot be deduced at this resolution.
Fig. 1.
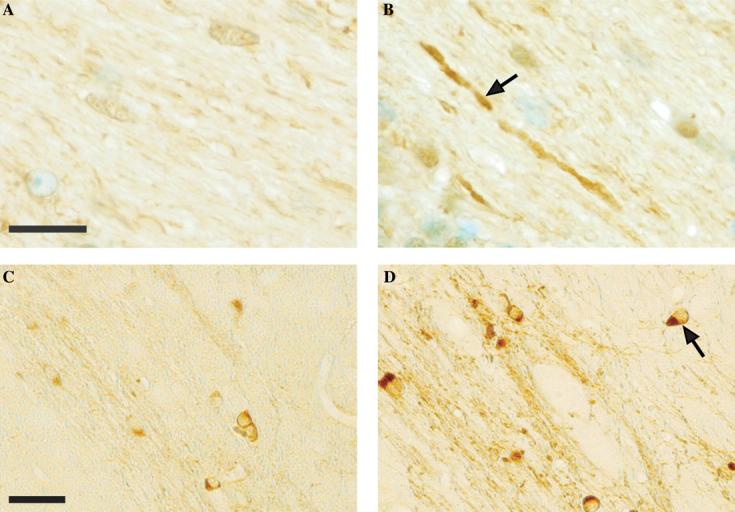
Visualization of ferritin and iron accumulation in axonal tracts of forebrain regions from the IRP knockout mice using optical microscopy. (A and B) Immunolabeling of sections (thickness ∼7 μm) from wild-type (A) and IRP knockout mice (B). The arrow in B points to a degenerating axon that appears to have a high level of ferritin. Methyl green counterstain identifies cell nuclei. (C and D) Sections as in A and B from wild-type (C) and IRP knockout (D) mice stained for ferric iron. The arrow in D points to oligodendrocyte cell bodies. Scale bars 10 μm in A and B, and 20 μm in C and D.
3.2. Identification of ferritin by 2D and 3D electron microscopy
To better visualize the 3D architecture of healthy and diseased axons, as well as detect individual ferritin molecules, we carried out 2D and 3D imaging of axonal sections using transmission electron microscopy. Conventional electron microscopic projection images of fixed, osmium stained axonal sections typically reveal architectural details of the myelin sheath, the axonal filament network, and several internal organelles (Bunge et al., 1961). Initial evaluation of specimens with conventional fixation protocols revealed that the use of osmium at concentrations high enough to provide good contrast and preservation interfered with reliable identification of ferritin because of the similarity of contrast between heavily stained tissue and ferritin molecules. We therefore tested a range of staining conditions (with osmium concentrations ranging from 0.04 to 2%) to identify osmium concentrations that were merely sufficient to detect the location of the myelin sheath, but low enough to allow unambiguous detection of ferritin without interference from heavily stained tissue. The use of the low osmium concentrations between 0.05 and 0.1% satisfied this requirement.
As shown in Figs. 2A and B, dark “spots” with diameters of ∼5–7 nm corresponding to ferritin particles could be detected in sections obtained from wild-type as well as knockout mice. In wild-type mice, ferritin particles were often detected within axonal regions, whereas in the knockout mice, they were detected most often outside the myelin sheath (see also results from Fig. 5 for a variation of this theme). To confirm that these electron dense particles were iron-containing ferritin mole-cules, thin sections (Fig. 3A) were analyzed with electron energy loss spectroscopy (EELS) (Leapman, 2003). EELS spectra of regions (diameter of 6 nm) boxed to include putative ferritin molecules show an unambiguous peak from the iron L2,3 edge excitation (Fig. 3B); this peak is absent in an equivalent area from a nearby control region without ferritin (Fig. 3C). The averaged intensity of the spots in the iron map suggests that they arise from an iron core containing between 1200 and 3600 atoms of iron (see Figs 3D-G and Section 2), in excellent agreement with the known values for the number of iron atoms in ferritin (Harrison and Arosio, 1996).
Fig. 2.

Electron microscopy of sections from wild-type and IRP knockout mice. (A and B) Transmission electron microscopic images of thin sections prepared using 0.05% osmium during fixation and tissue preservation from wild-type (A) and IRP knockout (B) mice. The arrows in A and B are pointing to ferritin molecules. A and B have the same magnification. Scale bar 150 nm.
Fig. 5.
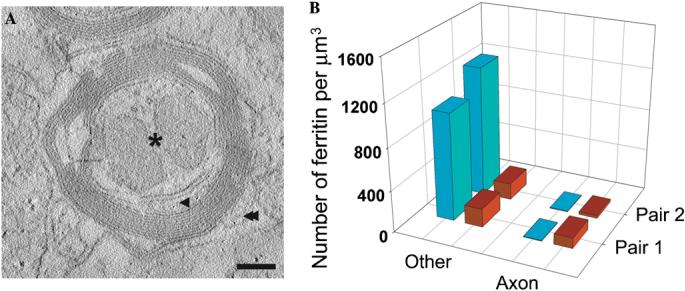
Quantitative analysis of ferritin distribution in IRP knockout and wild-type mice. (A) A slice from a reconstructed tomogram containing an axon from an IRP knockout mouse shows three different regions where ferritin molecules were counted manually: axon interior (asterisk), region between the axon and myelin sheath (single arrow head), and region outside axon (double arrowhead). The regions in the axonal interior represent cross-sections through mitochondria (seen as the two pear-shaped regions inside the axon), which were often found to be more enlarged in IRP knockout mice than in wild-type mice. Scale bar is 150 nm. (B) Averaged pair-wise comparison of volume-normalized ferritin distribution within the axons and other regions in wild-type mice (red) and IRP knockout mice (blue). The ferritin particles in regions marked by both single and double arrowheads (A) were merged to calculate ferritin amounts in regions outside the axonal regions. Data shown are from two pairs each (wild-type and knockout variety) of age-matched mice. Tomograms of sections from the two wild-type mice resulted in ferritin counts of 133 and 130 particles/μm3 from averaging over 15 and 17 tomograms, respectively. Tomograms of sections from the two knockout mice resulted in ferritin counts of 402 and 434 particles/μm3 from averaging over 26 and 19 tomograms, respectively.
3.3. Electron tomographic studies of axonal structure in 3D
To visualize the 3D distribution of ferritin in the sections, we recorded a series of images from the sections by tilting the specimen relative to the electron beam from −70° to +70°. The series of images was then computationally processed to generate a 3D volume (i.e., tomogram) of the region imaged (Fig. 4A). A slice from a typical tomogram obtained from wild-type axonal sections is shown in Fig. 4B. The outline of the myelin sheath is visible, and the dark spots with widths of ∼10–15 nm present throughout the interior represent the cross-sectional view of filaments that run along the length of the axon, as seen by the segmented rendering of the tomogram presented in Fig. 4C. The contrast in the image presented in Fig. 4B was computationally enhanced to visualize the myelin sheath and the axons. In the absence of contrast enhancement, the main features in the tomogram are small, rounded objects, with sizes in the range of 5–7 nm, which we had already established to be ferritin particles using EELS imaging (Fig. 3). Because of their high signal, and the low osmium background, the ferritin particles are easy to locate in the tomogram, as shown in the inset to Fig. 4B, which is an expanded view of the boxed region. Thus, by visualizing the tomogram with and without computational contrast enhancement, it was possible to determine both the overall 3D architecture of the axonal sections (Fig. 4C) as well as the 3D distribution of ferritin particles in the corresponding regions (inset Fig. 4B).
Fig. 4.

3D imaging of wild-type neuronal axons by electron tomography. (A) Cut-away view of a tomogram obtained from a 150 nm thick mouse brain section prepared using 0.05% osmium as a fixative. The tomogram is composed of a stack of slices representing the 3D structure of the section. A single slice from the tomogram is shown in (B). The contrast in the slice was computationally enhanced to show the myelin sheath and filaments inside the volume. The insets show that ferritin molecules can be easily detected in the raw tomographic slices when no contrast enhancement is applied. (C) Volume-rendered representation of a myelinated axon from a wild-type mouse. 3D tomographic volume is partially segmented within the axon region for neurofilaments (pink), axonal membrane (orange), and ferritin molecules (light blue). Myelin sheaths and other unsegmented regions are in blue. Scale bar 250 nm.
3.4. Quantitative analysis of ferritin concentration
To investigate differences in the spatial distribution of ferritin between wild-type and the IRP knockout mice, we obtained electron tomographic data from a number of regions from brain sections in two sets of age-matched wild-type and knockout mouse pairs. Inspection of these tomograms revealed that in wild-type mice, the ferritin distribution was relatively uniform in regions inside and outside the axon. In contrast, ferritin particles in the IRP knockout mice were predominantly clustered in the cytoplasmic regions of the oligodendrocytes bordering the inner and outer surfaces of the myelin sheath. Very few ferritin particles were detected inside the axonal region. To describe these distributions quantitatively, ferritin particles were classified as either being inside the axon itself, as defined by the recognizable axonal membrane, or in regions outside the axonal membrane. The latter category included the area between the axonal membrane and the inner myelin sheath where the inner tongue processes of oligodendrocyte that lie in between the myelin sheath and the axonal membrane. Absolute ferritin counts in these two regions, as well as volume-normalized counts; i.e., “concentrations” were obtained from a total of 32 tomograms (Fig. 5). A total of 4258 ferritin molecules were individually identified in these volumes. A clear result to emerge from these measurements is that the overall amount of ferritin is ∼3–4 times higher in the IRP knockout mice than in wild-type mice, consistent with previous ferritin immunostaining studies (LaVaute et al., 2001; Smith et al., 2004). However, despite this increase, only a negligible amount of ferritin in the IRP knockout mouse was present within the axon itself. Almost all of the additional ferritin was either in the oligodendrocyte cell bodies or in extensions of the oligodendrocyte cytoplasm that were found on the periphery of the axon (see for example the region indicated by the arrow in Fig. 2B). In some instances, ferritin particles were detected in regions expected to constitute the interior of the axon, but as described in the next section, the inside of these ferritin-containing compartments is topologically equivalent to the cytoplasm of the oligodendrocytes (Fig. 6).
Fig. 6.
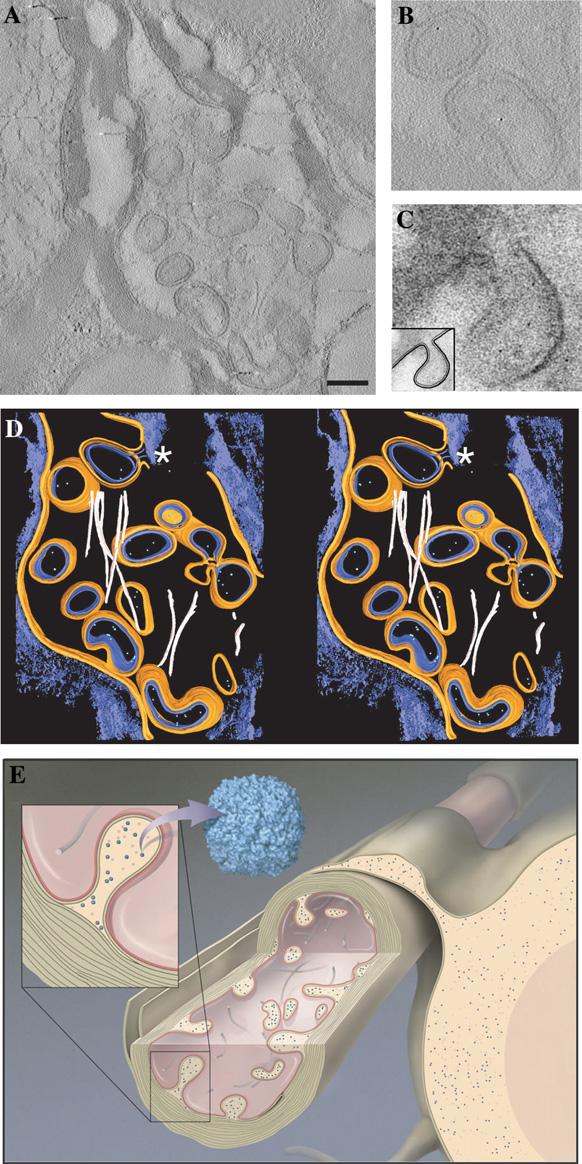
Electron tomography of degenerating neurons in an IRP knockout mouse brain. (A) A single tomographic slice showing invasion of the interior of the axon by what appear to be “double-walled vesicles.” The tomogram was recorded from a 150 nm thick slice from brain tissue. Scale bar 250 nm. (B) Enlarged views of a selected slice from the tomogram illustrating the presence of ferritin within these double-walled regions. (C) Enlarged view of a small region of a projection image recorded from a 70 nm slice showing the emergence of a double-walled vesicle from the junction of the axonal membrane and oligodendrocyte membrane at the innermost layer of the myelin sheath. The inset shows a schematic representation of this double-walled invagination. (D) Volume-rendered representation of the axonal section in stereo from the IRP knockout mouse axon, displaying the axonal membrane in orange, the oligodendrocyte membrane in blue, neurofilaments in pink, invaginated double-walled vesicles containing the oligodendrocytic cytoplasm and ferritin molecules in green. The myelin sheath and other unsegmented regions are in blue. The asterisk indicates a double-walled vesicle emerging from the junction of the two membranes. (E) Cartoon of axon and oligodendrocyte cell showing the anatomy of degeneration based on our electron tomographic studies of the IRP knockout mice. The blue dots represent ferritin particles.
3.5. Structural description of neuronal degeneration
Some of the neurons in brain sections from the IRP knockout mice showed a high level of structural damage (Fig. 6A), in contrast to sections from wild-type neurons (Figs. 4B and C) where the axonal interior is generally filled with filaments and occasional ferritin particles. The interior of these axons appears to be filled with structures that appear as double-walled compartments in projection images of the sections. Double-walled vesicles of this type have been previously reported in wild-type axons (Estable-Puig et al., 1971; Novotny, 1984), although in the present study we found them mainly in neurons obtained from knockout mice. Tomographic analysis of the sections reveals that these double-walled compartments arise by invagination of the oligodendro-cyte into the axonal space as shown in Fig. 6B. The outer layer of the compartments therefore represents the axonal membrane and the inner layer represents the cell membrane of the oligodendrocyte, consistent with the thickness and appearance of these two types of cell membranes. The appearance of these invaginations as double-walled vesicles is due to the fact that the images are from sections that slice through the extended protrusions. Although it appears probable that all of the double-walled compartments arise by invagination, we could not confirm that each one of the double-walled compartments in sections such as the one shown in Fig. 6A was physically connected to the oligodendro-cyte, given that the sections that we analyzed were only ∼ 200 nm thick, and some of the double-walled compartments may represent free-standing vesicles that have been pinched off from the axonal membrane. We also note that in our studies, such protrusions were rarely found in healthy myelinated axons.
Inspection of the computational slices (Figs. 6A and C), and the segmented 3D density map (Fig. 6D) reveals that all of the ferritin molecules detected in these axons are present in the interior of the double-walled structures, and not anywhere else in the surrounding medium. The interior of these structures is topologically equivalent to the cytoplasm of the oligodendrocytes. Thus, what might appear at first glance to be axonal localization of ferritin (Fig. 1B arrow), in fact arises from the cytoplasm of oligodendrocyte cells that have extended into the physical space normally occupied by axonal components. A further feature of neurons such as the one shown in Fig. 6A is the extensive damage of the internal filament structure of the axon, as highlighted in the segmented 3D representation of the axonal sections (Fig. 6D; compare with Fig. 4C, which shows the filament structure in wild-type axons). A schematic representation of the degenerating axons is presented in Fig. 6E. The cartoon represents our present understanding of the architecture of degenerating neurons such as the one shown in Fig. 6A, highlighting the observations of both the protrusions into the axonal space as well as the damaged internal filament network within the axon.
4. Discussion
In the present analysis, we have used electron tomography to establish the 3D architecture of neuronal axons and to determine the 3D localization of individual ferritin particles in these axons. The direct detection of individual molecules of various types in tissue by their structural signatures is a central goal in the emerging field of high resolution 3D electron microscopy. Ferritin presents a special case, because the thousands of atoms of iron in the central core provide a high absorption contrast for detection against the background signal of other cellular proteins. The use of EELS to provide local quantitation of the number of iron atoms also provides an independent and unambiguous approach to confirm the presence of ferritin in brain tissue. The success in detecting ferritin by electron tomography of whole tissue suggests that the use of metal cluster tags could be a valuable strategy worth exploring for visualization of the 3D localization of proteins and nucleic acids in a cellular context.
Our analyses show that in wild-type mouse brain sections, ferritin is present in the axon. None of the ferritin molecules in wild-type axonal sections were found to be compartmentalized or encapsulated in vesicles, implying that ferritin is very likely to be freely transported within the axon. Given that ferritin particles have a molecular weight of about 500 000 kD, ferritin would be unlikely to move into the axon by a process of simple diffusion, and our results thus imply that ferritin is likely to be actively transported into axons.
The increased amount of ferritin in the IRP knockout mice (∼3–4 times higher than in the wild-type) is in good agreement with observations from earlier light microscopic studies using immunolabeling and quantitation by Western blots of brain lysates (LaVaute et al., 2001; Smith et al., 2004). Despite this overall increase in ferritin in the IRP knockout, our analyses also show that the amount of ferritin within the axon is actually decreased by >10-fold at the expense of a ∼7-fold increase in ferritin amount in neighboring oligodendrocyte cells. This is an interesting finding because it provides a new insight into interpretation of the previous light microscopic studies of ferritin accumulation in the axonal tracts. Since the oligodendrocyte cytoplasm is very close to the myelin sheath, axonal vs. oligodendrocyte location of ferritin would not be distinguished at the lower resolutions used for optical imaging. Further, in neurons displaying extensive degeneration, ferritin can indeed appear to be present in the physical space occupied by the axon (Fig. 6) even though it is still in regions topologically equivalent to the oligodendrocyte cytoplasm. Electron tomographic studies of the type we report here may also be useful in the analysis of iron and ferritin distribution in patients with Parkinson's disease, where a ∼2.2-fold increase in brain ferritin level has been reported (Griffiths et al., 1999).
The electron tomographic studies we have carried out also permit a first glimpse into the cellular anatomy of degenerating neurons in the IRP knockout mouse. The axons containing these double-walled vesicles show classical signs of degeneration, including beading and fragmentation of axons and the elimination of most of the contents found in healthy axons (Novotny, 1980). Our data imply that the oligodendrocyte cells penetrating the interior of the axon and sections through these tubular intrusions would have the appearance of double-walled vesicles, with the inner layer arising from the oligodendrocytes and the outer layer arising from the axonal membrane. Ferritin molecules were found only in the interior of these vesicular regions, corresponding to the cytoplasm of oligodendrocytes. Double-walled vesicular entities such as those shown in Fig. 6 have been observed previously and interpreted either as freestanding vesicles (Estable-Puig et al., 1971) or as coated invaginations (Novotny, 1984). The present tomographic analysis shows that the double-walled compartments we have observed are, at least in some instances, clearly sections through the tubular protrusions into the axonal space.
What is the significance of the altered localization of ferritin? Abnormal iron accumulation in brain tissue is reportedly associated with a number of common neurological diseases including Alzheimer's, Parkinson's and multiple sclerosis. There is evidence that increased iron leads to oxidative damage in the brain. Further, mutations in the L-chain of ferritin lead to neuroferritinopathy, a dominantly inherited movement disorder that resembles Huntington disease (Crompton et al., 2002; Curtis et al., 2001). Patients with neuroferritinopathy have abnormal aggregates of ferritin and iron in brain. From our findings, we conclude that the mere presence of excess ferritin in a cell need not lead to damage, since large amounts of ferritin molecules are present in the oligodendrocyte cells in the IRP knockout mice, and these cells appear structurally normal. Previous immunohistochemical studies of these mice indicated that the ferritin content of neuronal cells increases markedly in regions of the brain that degenerate. However, here we show that the ferritin content of axons decreases markedly, and this is associated with significant loss of axonal integrity. One possible mechanistic origin of the observed degeneration is that the IRP knockout mice have a severe defect in transport of ferritin into the axon, which compromises the structural integrity of the axon. Conversely, it is also possible that the absence of normal levels of IRP1 and IRP2 result in damage to the structural elements of the axon, resulting in a defect in the trafficking of ferritin into the axon. At present, we cannot distinguish between these possibilities. Irrespective of whether ferritin transport is the cause or effect of degeneration, it is very likely that functional deficiency of iron associated with a lower level of axonal ferritin (Wade et al., 1991) would result in the impaired function of mitochondrial iron-containing proteins, which, in turn, could result in an inability to generate sufficient ATP to maintain axonal integrity. Although we detected no obvious differences in mitochondrial morphology at the present resolution of our studies, an intriguing finding from our tomographic analyses is that mitochondrial sizes in the IRP knockout mice are slightly larger than in wild-type mice (for example in Fig. 5A), consistent with possible mitochondrial stress in the IRP knockout mice. Further structural and functional studies of mice carrying defects in genes related to IRP1 and IRP2 will undoubtedly shed more light on the molecular mechanism of degeneration associated with defective iron metabolism.
References
- Baumeister W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Curr. Opin. Struct. Biol. 2002;12:679–684. doi: 10.1016/s0959-440x(02)00378-0. [DOI] [PubMed] [Google Scholar]
- Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P, Becker G. Brain iron pathways and their relevance to Parkinson's disease. J. Neurochem. 2001;79:225–236. doi: 10.1046/j.1471-4159.2001.00608.x. [DOI] [PubMed] [Google Scholar]
- Bourdon E, Kang DK, Ghosh MC, Drake SK, Wey J, Levine RL, Rouault TA. The role of endogenous heme synthesis and degradation domain cysteines in cellular iron-dependent degradation of IRP2. Blood Cells Mol. Dis. 2003;31:247–255. doi: 10.1016/s1079-9796(03)00161-x. [DOI] [PubMed] [Google Scholar]
- Bunge MB, Bunge RP, Ris H. Ultrastructural study of remyelination in an experimental lesion in adult cat spinal cord. J. Biophys. Biochem. Cytol. 1961;10:67–94. doi: 10.1083/jcb.10.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL, Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer's diseased brains. J. Neurosci. Res. 1992;31:75–83. doi: 10.1002/jnr.490310111. [DOI] [PubMed] [Google Scholar]
- Crompton DE, Chinnery PF, Fey C, Curtis AR, Morris CM, Kierstan J, Burt A, Young F, Coulthard A, Curtis A, Ince PG, Bates D, Jackson MJ, Burn J. Neuroferritinopathy: a window on the role of iron in neurodegeneration. Blood Cells Mol. Dis. 2002;29:522–531. doi: 10.1006/bcmd.2002.0589. [DOI] [PubMed] [Google Scholar]
- Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- Estable-Puig JF, de Estable-Puig RF, Romero C. Double-walled exo-endocytosis between synaptic terminals. Brain Res. 1971;28:172–175. doi: 10.1016/0006-8993(71)90534-8. [DOI] [PubMed] [Google Scholar]
- Griffiths PD, Dobson BR, Jones GR, Clarke DT. Iron in the basal ganglia in Parkinson's disease. An in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain. 1999;122(Pt 4):667–673. doi: 10.1093/brain/122.4.667. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Faucheux BA. Iron metabolism and Parkinson's disease. Mov. Disord. 1998;13(Suppl 1):39–45. [PubMed] [Google Scholar]
- Hu J, Connor JR. Demonstration and characterization of the iron regulatory protein in human brain. J. Neurochem. 1996;67:838–844. doi: 10.1046/j.1471-4159.1996.67020838.x. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, 3rd, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- Leapman RD. Detecting single atoms of calcium and iron in biological structures by electron energy-loss spectrum-imaging. J. Microsc. 2003;210:5–15. doi: 10.1046/j.1365-2818.2003.01173.x. [DOI] [PubMed] [Google Scholar]
- Novotny GE. Formation of cytoplasm-containing vesicles from double-walled coated invaginations containing oligodendrocytic cytoplasm at the axon–myelin sheath interface in adult mammalian central nervous system. Acta Anat. (Basel) 1984;119:106–112. doi: 10.1159/000145869. [DOI] [PubMed] [Google Scholar]
- Novotny GE. Observations on the lateral geniculate nucleus of the monkey (Macaca fasicularis) after eye removal: a light and electron microscopic study II. Axonal and dendritic degeneration. J. Hirnforsch. 1980;21:227–242. [PubMed] [Google Scholar]
- Rouault T, Klausner R. Regulation of iron metabolism in eukaryotes. Curr. Top Cell Regul. 1997;35:1–19. doi: 10.1016/s0070-2137(97)80001-5. [DOI] [PubMed] [Google Scholar]
- Schneider BD, Leibold EA. Regulation of mammalian iron homeostasis. Curr. Opin. Clin. Nutr. Metab. Care. 2000;3:267–273. doi: 10.1097/00075197-200007000-00005. [DOI] [PubMed] [Google Scholar]
- Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer's disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SR, Cooperman S, Lavaute T, Tresser N, Ghosh M, Meyron-Holtz E, Land W, Ollivierre H, Jortner B, Switzer R, III, Messing A, Rouault TA. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann. NY Acad. Sci. 2004;1012:65–83. doi: 10.1196/annals.1306.006. [DOI] [PubMed] [Google Scholar]
- Theil EC. Ferritin: structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu. Rev. Biochem. 1987;56:289–315. doi: 10.1146/annurev.bi.56.070187.001445. [DOI] [PubMed] [Google Scholar]
- Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- van Gelder W, Huijskes-Heins MI, Cleton-Soeteman MI, van Dijk JP, van Eijk HG. Iron uptake in blood–brain barrier endothelial cells cultured in iron-depleted and iron-enriched media. J. Neurochem. 1998;71:1134–1140. doi: 10.1046/j.1471-4159.1998.71031134.x. [DOI] [PubMed] [Google Scholar]
- Wade VJ, Levi S, Arosio P, Treffry A, Harrison PM, Mann S. Influence of site-directed modifications on the formation of iron cores in ferritin. J. Mol. Biol. 1991;221:1443–1452.. doi: 10.1016/0022-2836(91)90944-2. [DOI] [PubMed] [Google Scholar]