

Abstract
The eukaryotic pre-replication complex is assembled at replication origins in a reaction called licensing. Licensing involves the interactions of a variety of proteins including the origin recognition complex (ORC), Cdc6 and the Mcm2-7 helicase, homologues of which are also found in archaea. The euryarchaeote Archaeoglobus fulgidus encodes two genes with homology to Orc/Cdc6 and a single Mcm homologue. The A.fulgidus Mcm protein and one Orc/Cdc6 homologue have been purified and investigated in vitro. The Mcm protein is an ATP-dependent, hexameric helicase that can unwind between 200 and 400 bp of duplex DNA. Deletion of 112 amino acids from the N-terminus of A.f Mcm produced a protein, which was still capable of forming a hexamer, was competent in DNA binding and was able to unwind at least 1 kb of duplex DNA. The purified Orc/Cdc6 homologue was also able to bind DNA. Both Mcm and Orc/Cdc6 show a preference for specific DNA structures, namely molecules containing a single stranded bubble that mimics early replication intermediates. Nuclease protection showed that the binding sites for Mcm and Orc/Cdc6 overlap. The Orc/Cdc6 protein bound more tightly to these substrates and was able to displace pre-bound Mcm hexamer.
INTRODUCTION
DNA replication in eukaryotes is a complex multi-step process. One of the key steps in ensuring replication occurs once and only once in each cell cycle is the licensing of origins (reviewed in 1). Origin licensing involves the co-operative interaction of many proteins to establish the pre-replication complex (Pre-RC), in which the MCM (Mini-Chromosome Maintenance) helicase is loaded onto origin DNA. Origins are bound specifically by the origin recognition complex (ORC), which comprises six different proteins, and in yeast the ORC complex remains bound to origins throughout the cell cycle (2–5). ORC associates with Cdc6 and Cdt1 to localise the Mcm protein in a cell cycle dependent manner (5–8). Cdc6 protein is a member of the AAA+ family of ATPases and as such is related to the Orc1, 4 and 5 subunits of the origin recognition complex, and all the subunits of the MCM complex (9).
The MCM helicase is a heterohexameric complex made up from Mcm 2–7 proteins, and is thought to be the eukaryotic replicative helicase (10), although it may not be the only helicase required for DNA replication initiation (11). The six Mcm subunits are highly related to each other, although each can be distinctly identified as belonging to a certain subfamily, and these sub-classes are themselves conserved across eukaryotes. Various complexes of the Mcm protein have been isolated from human cells, including a hexameric complex containing all the different Mcm 2–7 subunits. However, only complexes containing Mcm 4, 6 and 7 show activity in vitro (12–14), despite all the subunits being required at a replication fork in vivo (15). Similar results have been observed with the Mcm proteins from Schizosaccharomyces pombe (14).
Almost 20 archaeal genomes have been completely sequenced to date, and sequence analysis has revealed that the majority of proteins involved in replication, recombination, transcription and translation in archaea have homologues in eukaryotes (16,17). Regarding the pre-RC, homologues have been identified of the Orc/Cdc6 and MCM proteins. The euryarchaeon Archaeoglobus fulgidus is a hyperthermophilic, sulphate reducing, obligate anaerobe and encodes two homologues of Orc/Cdc6 and a single homologue of the Mcm 2–7 family. The two Orc/Cdc6 homologues cannot be clearly identified as being functional homologues of ORC rather than Cdc6 or vice versa simply by sequence alignments. The two proteins are AAA+ family members of similar size and share 23% identity at the amino acid level. Interestingly, sequencing of the Pyrobaculum aerophilum and several Pyrococcus sp. genomes have revealed that they only encode a single Orc/Cdc6 homologue (18–21).
The single Mcm protein of another euryarchaeon, Methanobacterium thermoautotrophicum has been shown previously to be an active ATP dependent 3′–5′ helicase (22–24). However, very little information is known about the substrate requirements and mechanism of action of the archaeal Mcm proteins. In order to gain more information on this subject, the Mcm homologue from A.fulgidus was purified and its activities investigated in vitro. The two Orc/Cdc6 homologues were also cloned to try to elucidate their function and their relationship to the Mcm protein, although only one of the two was amenable to purification. The results indicate that the Mcm protein is an active hexameric 3′–5′ helicase and shows a binding preference for substrates designed to resemble early replication intermediates (replication bubbles). The purified Orc/Cdc6 homologue also showed a distinct DNA binding preference, in this case for DNA bearing a 5′ tail. Furthermore, when the Mcm and Orc/Cdc6 homologues were added simultaneously to DNA, the Orc/Cdc6 protein was observed to displace Mcm from DNA.
MATERIALS AND METHODS
Expression and purification of A.fulgidus Mcm
The A.fulgidus Mcm gene (AF0517) was amplified by PCR from genomic DNA using primers bearing NdeI or EcoRI sites: 5′GGGAATCATCATATGGGTATAAGCAGTCCGGCAC3′ and 5′AGCCCGAATTCTTACTAAAGTTTGCTTACCAATTTG3′. The resulting product was cut with NcoI and the two restriction fragments gel purified. The larger fragment was then cut with EcoRI and the smaller with NdeI, followed by ligation of both fragments into pET22b (Novagen). The Mcm ΔN112 gene was cloned using an identical strategy except that a different primer for the N-terminus of the gene was used for the initial PCR reaction: 5′GGCATGCATATGGCGATAGAGGGGATTG3′. All restriction enzymes used were from New England Biolabs. Both plasmids were transformed into E.coli BL21 (DE3) bearing a plasmid encoding rare t-RNAs (pSJS1240, gift from S. J. Sandler). These cells were grown at 37°C in LB to an OD600 ≈ 0.5, then induced with 1 mM IPTG for 3 h at 37°C. Cells were harvested by centrifugation and disrupted by sonication, three 30 s pulses using a Sonifier 250 (Branson), in a buffer containing 50 mM Tris (pH 7.5), 100 mM NaCl, 20% sucrose, 1 mM EDTA, 140 ng/ml pepstatin, 400 ng/ml leupeptin, 1 mM phenylmethanesulfonyl fluoride, 1 mM dithiothreitol (DTT). The lysate was clarified by centrifugation at 20 000 g for 20 min, followed by heat treatment at 60°C for 10 min, then re-centrifugation. Ammonium sulphate (0.4 g/ml) was added to the resulting supernatant, followed by re-centrifugation. The pellet was then resuspended in buffer R [20 mM Tris (pH 7.5), 1 mM EDTA, 1 mM DTT] so that the conductivity was ∼10 mS/cm, and protein loaded onto a Cibacron Blue Sepharose column equilibrated with buffer A [20 mM Tris (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1 mM DTT], washed with several column volumes of buffer and then eluted with buffer A + 1 M NaCl. The eluant was diluted in buffer R so that the conductivity was 10 mS/cm and loaded onto a 5 ml hiTrap heparin column (Pharmacia). Protein was loaded in buffer A and eluted with a linear gradient to buffer A + 1 M NaCl. Eluant was run on a HiLoad 16/60 Superdex 200 gel filtration column (Pharmacia) equilibrated in 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM DTT (buffer S). The resulting peak was diluted with buffer R until the conductivity was ∼10 mS/cm and then loaded onto a 10 ml Mono Q column (Pharmacia) in buffer A and eluted by a linear gradient to buffer A + 1 M NaCl. The resultant protein was supplemented with glycerol to 10% and stored in aliquots at –70°C. Yields were typically 2–3 mg protein/l of culture.
Expression and purification of A.fulgidus Orc/Cdc6 homologues
AF0244 and AF0695 were each amplified by PCR and cloned into the NdeI/BamHI sites of pET22b. Expression tests were carried out in Escherichia coli BL21(DE3) bearing plasmids coding for rare t-RNAs [Codon+ (Stratagene) or pSJS1240 (gift from S. Sandler)]. AF0244 was then cloned into the NdeI/BamHI sites of pET28 to yield a protein with a six histidine N-terminal tag. This was expressed and treated as for the Mcm protein described above as far as re-suspension of the ammonium sulphate pellet. Following resuspension in buffer R, protein was passed over a metal ion chelating resin (Pharmacia) loaded with Ni2+ ions, in buffer N [containing 20 mM Tris (pH 7.5), 2 mM imidazole, 100 mM NaCl]. The column was washed with several column volumes of buffer and then protein eluted with buffer N + 250 mM imidazole. Eluate was passed over a 5 ml HiTrap heparin column as described above and then over a HiLoad 16/60 Superdex 75 (Pharmacia) in buffer S. Protein was supplemented with glycerol to 10% and stored in aliquots at –70°C.
DNA substrates
Oligonucleotides (Oswel or Cancer Research UK oligonucleotide synthesis service) were labelled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Amersham). Unincorporated nucleotide was removed using a Bio-Rad P6 spin column. For double stranded substrates the labelled oligonucleotide was annealed to a 2-fold molar excess of a cold complementary strand, or to a 1.5-fold molar excess of ssM13 DNA, by cooling at 1°C/min from 95 to 25°C in a PCR machine. Substrates containing a bubble made from duplex surrounding poly dT had the following sequences: 5′TCTACCTGGACGACCGGG(T)nGGGCCAGCAGGTCCATCA3′, 5′TGATGGACCTGCTGGCCC(T)nCCCGGTCGTCCAGGTAGA3′, where n = 0, 8, 10, 15 or 30. n = 0 produced the 36 bp duplex substrate. Substrates containing a bubble made from repeats of (GACT) were based on (25) and had the following sequences: 5′TCTACCTGGACGACCGGG(GACT)nGGGCCAGCAGGTCCATCA3′, 5′TGATGGACCTGCTGGCCC(GACT)nCCCGGTCGTCCAGGTAGA3′, where n = 2.5, 5 or 10. The first of these two oligonucleotides was also used for the ATPase assays (see below).
Tailed duplex and flayed duplex substrates were constructed by annealing the appropriate combinations of the following oligonucleotides: 5′GCTCGGTACCCGGGGATCCTCTAGA(T)n3′, 5′(T)nTCTAGAGGATCCCCGGGTACCGAGC3′, where n = 0 or 25. When n = 0 for both substrates, a duplex of 25 bp was produced.
Helicase substrates annealed to ssM13 DNA bore the sequences: 5′(T)nCGACTCTAGAGGATCCCCGGGTACC (T)m3′, where n = 0 or 25, and m = 0 or 25. When both n and m = 0 then a 25 bp duplex was produced. To test directionality of helicase action the 5′ labelled oligonucleotide 5′CAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGACTCGAATTCGTAATCATGG3′ was annealed to ssM13 and extended with Klenow (New England Biolabs) in the presence of 0.1 mM dTTP and [α-32P]dCTP to yield a substrate labelled at both ends. This was then cut with EcoRI to yield linearised ssM13 with a labelled oligonucleotide annealed at both ends.
For the test of processivity, oligonucleotides were labelled as above, mixed with ssM13, then extended by five cycles of PCR using Pfu polymerase (Stratagene). The extended product was then cut with EcoRI to yield linearised ssM13 with a labelled extension product of a known size. Oligonucleotides for the processivity substrates were: 200 nt 5′GCTGCGCAACTGTTGGG3′, 400 nt 5′GTCGGATTCTCCGTGGG3′, 600 nt 5′AATCAGAAAAGCCCCAA3′, 800 nt 5′GAGAAAGGCCGGAGACA3′, 1000 nt 5′CAGGCAAGGCAAAGAAT3′.
DNA binding assays
A.f Mcm or A.f AF0244, in the amounts indicated, were mixed with 5 fmol labelled DNA substrate in a buffer containing 20 mM Bis–Tris (pH 7.5), 20 mM MgCl2, 1 mM EDTA, in a total volume of 20 µl. Following incubation for 5 min at room temperature, complexes were separated by electrophoresis in 6 or 8% polyacrylamide gels (appropriate to the size of DNA substrate) in 1× TBE (90 mM Tris–borate, 1 mM EDTA). To test Mg2+ effects on binding, gels were also prepared and run in 90 mM Tris–borate + 10 mM MgCl2. Gels were dried down and quantified on a Molecular Dynamics Storm PhosphorImager, using ImageQuant software (Molecular Dynamics).
DNA helicase assays
Reactions were prepared as for the binding tests described above and incubated at the appropriate temperature (50–60°C) for 5 min. ATP was then added at the appropriate concentration to initiate the reaction. Reactions were stopped by addition of 2 µl stop buffer (10% glycerol, 1% SDS, 10 mM EDTA, bromophenol blue). Products were separated by electrophoresis in 5, 6 or 8% polyacrylamide gels run in 1× TBE + 0.1% SDS, polyacrylamide concentration being appropriate to the size of substrate and product to be separated. Following electrophoresis gels were dried down and quantified as above. Processivity tests were carried out as above except that the reaction was incubated for 10 min at 65°C after addition of ATP.
ATPase reactions
ATPase reactions were adapted from Lanzetta et al. (26). Malachite green solution (0.045%) was mixed immediately prior to use with 5% (NH4)6Mo7O24 in 4M H2SO4 in a 3:1 ratio. The resulting solution was filtered through a 0.2 µm syringe filter. Reactions were carried out at the indicated temperature in 50 µl vol for the indicated time. Reactions each contained 20 pmol hexamer in the buffer described for DNA binding, and were incubated at reaction temperature for 5 min prior to addition of ATP. At the appropriate time after addition of ATP, reactions were mixed with 800 µl malachite green solution for 1 min at room temperature. 100 µl of 34% w/v sodium citrate was then added and colour development allowed to proceed for 20 min. Optical densities at 660 nm were then taken and normalised to a control reaction of ATP in buffer without protein. For reactions with DNA either 40 pmol of oligonucleotide (see above) or an excess of sonicated salmon sperm DNA was added to the reaction mixture.
Exonuclease footprinting
Binding reactions were set up as above in 20 µl vol, using 20 fmol DNA, and incubated at 37°C for 5 min. Fifteen units of Exonuclease III (New England Biolabs) were then added and digestion continued at 37°C for either 30 s, 1, 2 or 4 min. Reactions were stopped by addition of 2 µl 1% SDS. H2O (80 µl) was added and the reactions extracted with 100 µl phenol, followed by extraction with 100 µl chloroform. DNA was precipitated with 10 µl 3 M sodium acetate and 300 µl ethanol. DNA was recovered by centrifugation and washed with 70% ethanol, dried and resuspended in formamide loading dye. DNA was electrophoresed on 15% polyacrylamide gels containing 7 M urea, run in 1× TBE at a constant temperature of 45°C. Following electrophoresis gels were dried down and exposed to a phosphorimager plate.
RESULTS AND DISCUSSION
Sequence of the A.fulgidus Mcm gene
A single Mcm homologue (AF0517) was identified in the genome database of A.fulgidus (27). However, sequence alignments showed that the Archaeoglobus protein was missing ∼100 amino acids at the N-terminus compared with other archaeal Mcm proteins. Although this region is not as well conserved as other regions of the protein, it is present in all other archaeal Mcm proteins examined to date. To investigate the absence of this part of the protein, the genomic DNA was re-sequenced, which revealed a single base omission in the database entry. The inclusion of this base provided a translational start codon (ATG) that was 112 amino acids upstream of the previous start site and the additional 112 residues showed significant homology with this region in other archaeal Mcm proteins. The corrected sequence encodes a 699 amino acid protein, which will be referred to as the full-length protein (A.f Mcm) and the original clone as a 112 amino acid deletion protein (Mcm ΔN112). Over its entire length A.f Mcm has 45% amino acid identity to the Methanobacterium thermoautotrophicum Mcm, its closest archaeal homologue.
Both the full-length and Mcm ΔN112 were over-expressed and purified from E.coli. Gel filtration on a Superdex 200 column showed that both proteins eluted at a size consistent with a hexamer (data not shown). This contrasts with the M.thermoautotrophicum Mcm, which was purified as a stable double hexamer (22–24).
ATPase activity
Our initial experiment was to test the ATPase activity of the full-length and truncated A.f Mcm proteins using a colorimetric assay detecting the release of free phosphate (26). Surprisingly, the full-length enzyme showed almost undetectable activity under the conditions tested whereas the Mcm ΔN112 mutant gave robust activity. Figure 1 shows the Mcm ΔN112 ATPase activity with varying concentrations of ATP in the presence or absence of DNA. The DNA used here was an excess of a 56mer single stranded oligonucleotide; although similar results were obtained using sonicated salmon sperm DNA. In the absence of DNA, maximum activity was obtained between 2 and 3 mM ATP whereas in the presence of DNA the maximum was between 5 and 8 mM ATP. The range over which activity was obtained is much broader than for many other helicases. Concentrations of ATP >10 mM began to inhibit the ATPase activity, with very little activity being detected at 20 mM ATP. This type of ATPase activity profile is very similar to the situation seen with DnaB, a bacterial hexameric replicative helicase (28).
Figure 1.

ATPase activity was measured by a colorimetric phosphate release assay. The results for the Mcm ΔN112 protein are shown here with or without ssDNA present as a function of ATP concentration. Each point is the average of three experiments.
The temperature dependence of the ATPase activity of both Mcm proteins was also investigated (Fig. 2) at an ATP concentration of 5 mM. Mcm ΔN112 is active over a range of temperatures between 50 and 85°C with maximal activity at 75°C and a rapid fall off in activity above this temperature. The full-length protein shows almost no activity across the entire temperature range.
Figure 2.

ATPase activity of the wt Mcm and Mcm ΔN112 mutant are compared over a range of temperatures in the presence of ssDNA, using 5 mM ATP. The wt Mcm protein shows very little activity although elevated temperature stimulates the weak activity. The Mcm ΔN112 shows a peak of activity between 75 and 80°C.
DNA helicase activity
Despite the very poor ATPase activity of the full-length protein, the helicase activity was analysed on various DNA substrates in the presence of ATP. Substrates contained a labelled oligonucleotide, either annealed to ssM13 DNA or to other oligonucleotides (as diagrammed in Fig. 3). Surpris ingly, the full-length protein showed robust helicase activity and, in common with other archaeal Mcm proteins, A.f. Mcm shows a 3′–5′ directionality (data not shown). All of the DNAs tested were substrates for Mcm helicase activity, except for the blunt duplex. The oligonucleotides annealed to the ssM13 were based on 25 bp of complementary sequence, with variants having a 20 base dT tail either at the 5′ end, the 3′ end or at both ends. Consequently, the untailed oligonucleotide bound to M13 gives a substrate in which there is a duplex region with ssDNA ‘tails’ of opposite polarity at each end, albeit part of a topologically closed circle. Addition of a poly dT tail to the oligonucleotides produces forked DNA substrates. Time courses of displacement of these oligonucleotides from the ssM13 by A.f Mcm were carried out and the results are shown in Figure 4A. Only the oligonucleotide bearing solely a 5′ tail showed any significant difference in its displacement, being unwound slightly less efficiently than the other substrates. This could be the result of non-productive binding to the 5′ poly dT tail. These results contrast to those obtained with the E.coli DnaB helicase, a 5′–3′ bacterial replicative helicase, which shows distinct preference for the presence of both a 3′ and a 5′ tail for activity (29).
Figure 3.

Diagrammatic representation of the oligonucleotide-based substrates used in this study (sequences of the oligonucleotides can be found in Materials and Methods). ssDNA used as a comparison was the 5′ tailed oligonucleotide without the complementary strand present.
Figure 4.


(A) Timecourse of wt Mcm helicase activity using labelled oligonucleotides annealed to ssM13 phage DNA. Substrates all contained the same 25 base sequence complementary to ssM13. As indicated the substrates were then varied by adding a 20 nt poly dT tail to either the 3′, 5′ or both ends of the DNA. (B) Helicase activity of wt Mcm as a function of varying (ATP). The substrate used was a flayed duplex, labelled on one strand. The control lanes are boiled DNA, which produces mostly single stranded product and a reaction without ATP added, which remains annealed at the substrate position. Arrows indicate the substrate and product positions. (C) A comparison of the wt Mcm and Mcm ΔN112 helicase activities on a flayed duplex substrate over a 10 min reaction. Again, the positions of substrate and product are indicated by arrows. The band that is higher than substrate in the zero time lane is believed to be a minor alternative annealing product of the oligonucleotides used.
Since there was little preference for the DNA substrate we utilised a synthetic flayed duplex substrate for further analysis. Figure 4B shows the variation of full-length Mcm activity on this substrate with different concentrations of ATP after a 10 min reaction. Controls consisted of a reaction without ATP, where no activity is observed, and a reaction where the substrate is boiled and placed immediately onto ice to maximise denaturation of the DNA. Helicase activity is very low when the ATP is <5 mM but higher activity is seen with the concentrations tested at or >5 mM.
The helicase activities of full-length Mcm and the Mcm ΔN112 mutant were compared using the flayed duplex substrate (Fig. 4C). The full-length protein has a higher helicase activity than the deletion protein under these conditions, despite having an apparently lower ATPase activity. These data suggest either that helicase activity has become uncoupled from the ATPase activity in the truncated protein, or that ssDNA only acts as co-factor for the ATPase activity of the truncated protein. The ΔN112 mutant was also tested on all the other substrates used with the wild-type protein, and showed the same substrate requirements as the wild-type despite having lower activity in every case.
Processivity of the A.f Mcm helicase
The processivity of the full-length and truncated Mcm proteins was tested on substrates produced by annealing labelled oligonucleotides to ssM13, then using these as primers in an extension reaction using Pfu polymerase. The resulting extended DNA was then cut at the unique EcoRI site to give a labelled piece of DNA of a known length, from 200 to 1000 bases, annealed to the linearised ssM13. This DNA was then incubated with either full-length Mcm or the Mcm ΔN112 mutant in the presence of ATP. Figure 5 shows the results of these reactions. Using these long substrates it is clear that the Mcm ΔN112 mutant is more processive than the full-length protein. Full-length Mcm can displace 200 bp, although the amount of the 400 bp fragment displaced by A.f Mcm is low but reproducible. In contrast, the ΔN112 protein can displace even a 1000 bp fragment, making it a more processive enzyme than either M.thermoautotrophicum Mcm or the S.pombe Mcm 4,6,7 complex (14,22,24). Although the full-length Mcm shows a lower processivity compared with the N-terminal deletion protein on these substrates, it can still displace up to 400 nt, which is comparable with the values recorded for other archaeal Mcm proteins (22,24).
Figure 5.

Processivity of the wt Mcm and Mcm ΔN112 proteins. Substrates contained a labelled extension product of the stated size annealed to ssM13 DNA. In all lanes there is some background of smaller extension products present but the major extension product of the expected size can be clearly seen in the boiled control lanes and is marked by an arrow. It is clear that after the 10 min reaction the Mcm ΔN112 protein is able to displace more of these long substrates than the wt Mcm, and that it can displace at least 1000 bases of DNA.
These data leave us with an apparent conundrum. The greater processivity of the truncated protein is surprising since the enzyme appears to have a lower activity on smaller substrates (Fig. 4C), yet the ATPase activity of the truncated protein appears to be greater. This latter phenomenon does not appear to be confined to A.fulgidus Mcm since truncated Aeropyrum pernix Mcm proteins also show enhanced ATPase activity compared with the full-length protein (data not shown). The recent crystal structure of an N-terminal fragment of the Mcm protein from M.thermoautotrophicum (30) may provide a clue to this problem. The structure revealed that residues equivalent to the first 110 or so residues of A.fulgidus Mcm form a discrete domain. It is possible that this domain provides a regulatory function to keep the enzyme inactive until correctly loaded onto an appropriate substrate and/or activated by the interaction with other proteins in the Pre-RC, and that by truncating the protein we have uncoupled the enzyme from this control mechanism.
DNA binding activity
DNA binding was assayed using a standard mobility shift assay: purified protein was added to oligonucleotide substrates and the bound complexes separated from unbound substrate on polyacrylamide gels (Fig. 6). Again, a variety of DNA substrates was used to assay the binding preference of the full-length A.f Mcm protein (see Fig. 3). These encompassed substrates of single stranded or double stranded DNA, or substrates containing both. All of the different substrates in the assays were bound by A.f Mcm, with the extent of binding being dependent upon concentration of Mcm. Furthermore, this binding was not affected by the presence of ATP or divalent metal ions; standard conditions contained 10 mM Mg2+ but binding was equally efficient when this was replaced by 10 mM EDTA. In substrates that contained only 25 bases of either single stranded or duplex DNA, a single species was observed on polyacrylamide gels (data not shown). However, when a 36 bp duplex was used, a second slower migrating species was also observed (Fig. 6A). Furthermore, all the other substrates tested were larger than 36 bases and showed the presence of this second slower migrating complex. The simplest interpretation of these results is the binding of first one, then two hexamers to a single substrate when the DNA is sufficiently long. Given that a 25 bp fragment can only bind a single Mcm complex, yet a 36 bp fragment can accommodate two hexamers, this suggests that a single Mcm complex spans a region of duplex DNA of between 13 and 18 bp.
Figure 6.


(A) DNA binding gel of A.f Mcm with two different DNA substrates. Note the two low mobility complexes produced upon binding: these are annotated as one or two hexamers binding DNA simultaneously. (B) Graph showing variation in DNA binding on various substrates at different concentrations of Mcm hexamer. The percent bound represents substrate shifted to either of the two complexes shown in (A). (C) The percentage of substrate shifted to the upper complex shown in (A) is shown against the concentration of hexamer present. Note that the bubble sizes of 20 or over give very similar response to Mcm binding, and are roughly an order of magnitude higher affinity than a bubble with only 10 nt of ssDNA. This smaller bubble is itself almost an order of magnitude higher affinity than blunt duplex or flayed duplex DNA.
Although binding to a variety of DNA structures was analysed, there was very little variation in binding efficiency between them, with the exception of those containing a single stranded bubble flanked by duplex DNA. Here the amount of the higher complex, possibly bound by two hexamers, was markedly increased compared with other substrates. Figure 6B shows plots of the percentage of bound substrate against the concentration of A.f Mcm hexamers for a flayed duplex, a blunt duplex (B0) and two bubble substrates with either 10 or 20 nt single stranded regions (B10 and B20, respectively). In this plot the amount bound includes both complexes. A.f Mcm has a roughly 10-fold higher affinity for the B20 substrate over the blunt duplex B0 (KDs of 10 and 100 nM, respectively), with the flayed duplex and the smaller bubble being of intermediate affinity (25 and 40 nM, respectively).
However, if the percentage shifted to the upper complex is plotted (Fig. 6C) then the differences between substrates become clearer. Even at the highest concentration used here, close to 1 µM hexamer, only 50% of the flayed and blunt duplexes were shifted to the upper complex. Conversely, B10 achieves a 50% shift to this complex at just over 100 nM hexamer and B20 at ∼20 nM. These data illustrate two important points. First, the DNA bubble substrate is a preferred ligand for the Mcm hexamer and, secondly, that there is co-operative binding between hexamers on the bubble that is absent on the other substrates. This latter point is evident from the gels that show relatively little DNA in the lower band and that binding of a second MCM molecule is facilitated once the first has bound so that the DNA is super shifted to the upper complex before the lower band has been saturated. Furthermore, the size of the single stranded region in the bubble affects this co-operativity. Substrates with a bubble size between 15 and 40 bases all behaved similarly to B20. However, the bubble size appears to become limiting at 10 bases reflected by the decreased affinity of B10 compared with B20 or larger bubbles.
Footprinting of the Mcm–DNA complex
A.f Mcm was bound to DNA and then probed with exonuclease III, a double strand specific, single stranded 3′–5′ exonuclease. The footprinting was carried out on a variety of different bubble substrates and upon a flayed duplex substrate. In order to compare our data with those obtained for other helicases, the bubble substrate shown in Figure 7 was based upon the sequence used to footprint the SV40 large T antigen (25) and contains a bubble with repeats of the sequence (GACT)n. In the absence of A.f Mcm, the DNA was degraded beyond the duplex and into the single stranded bubble region of the substrate. This is presumably because the DNA in the bubble is not completely single stranded at 37°C and can be degraded in the absence of protein although with several significant pause sites (compare this with the pattern obtained using a bubble made from poly dT; Fig. 9). With Mcm present there was an alteration of the digestion pattern with protection of 2–3 bases of the double stranded DNA before the bubble region but almost no detectable progression of the nuclease past this point. The observation that only 2–3 bases of the duplex region are protected suggests that the location of the protein is mainly across the single stranded region since the DNA binding data shown above suggests that each Mcm hexamer binding site spans 13–18 bp of DNA.
Figure 7.

Autoradiogram of a denaturing polyacrylamide gel showing exonuclease III digestion of a bubble substrate with and without Mcm present. The bubble in this substrate was made by repeats of d(GACT) on each strand. Digestion without Mcm present proceeds within the bubble region indicating that it is not fully single stranded in the reaction conditions. The frequent pause sites do indicate that it is not stable duplex DNA either. In the presence of Mcm digestion is halted 2–3 bases within the duplex region, indicating protection of this region by the bound protein.
Figure 9.

Exonuclease III digestion of a bubble and a flayed duplex DNA substrate, on either naked DNA or DNA bound by wt Mcm (400 nM) or AF0244 (200 nM). The bubble used here consisted of dT10 region on each strand, flanked by 18 bp duplex on either side. In the absence of protein, digestion proceeds up to the end of the duplex DNA and no further. In the presence of either AF0244 or Mcm there is protection of the final 2–3 bp of duplex DNA before the bubble region. A similar pattern is seen with the flayed duplex where digestion can proceed up to the end of the duplex DNA but is halted 3 bases earlier in the presence of Mcm or AF0244.
The amount of A.f Mcm used in these experiments was sufficient to completely shift the bubble to the upper complex proposed earlier to represent two hexamers bound. The complete protection of the bubble region was also observed when the other strand of the DNA substrate was labelled, suggesting that both junctions of single (ss)/double strand (ds)DNA are protected simultaneously, again consistent with two hexamers bound, one at either end of the bubble region. This binding is co-operative between the two hexamers, which could either reflect direct interaction of the two hexamers or stabilisation of a preferred DNA conformation. These data are the only indication to date that double hexamers, readily seen with the M.thermoautotrophicum Mcm (22–24), may be formed by the A.fulgidus protein too, albeit in a DNA dependent manner.
It is also apparent that at the concentration of A.f Mcm used there is significant protection of the DNA ends. However, the exonuclease is able to progress past this blockage so that cleavage increases with time. This probably reflects a weaker association of a further Mcm hexamer with the end of the DNA in addition to the more tightly bound hexamers at the ss/dsDNA boundary.
The two Orc/Cdc6 homologues of A.fulgidus
Archaeoglobus fulgidus codes for two proteins that are homologues of Orc/Cdc6 (AF0244 and AF0695). These proteins share 23% identity at the amino acid level and are both members of the AAA+ family of ATPases. Consequently, it is not easy to assign to either of them, a priori, a definite Orc-like or Cdc6-like function based on sequence similarities. It is perhaps relevant that P.aerophilum and various Pyrococcus sp. only have a single gene encoding an Orc/Cdc6 family protein (18–21). In light of these ambiguities, both genes were cloned into E.coli expression vectors and their expression tested. AF0695 gave very low levels of expression in all strains tested and no soluble protein could be detected. AF0244 expressed well but was largely insoluble and the protein aggregated when the salt was reduced below 500 mM. An N-terminal 6× His Tag was added to AF0244 to aid purification and, fortuitously, the presence of the tag also increased the solubility of the protein. The purified His-tagged Orc/Cdc6 homologue eluted as a single peak during gel filtration of a size consistent with a monomer. The addition of neither ATP nor Mg2+ to the buffers had any effect on the elution position of the protein (data not shown).
DNA binding of AF0244
Although the origin of A.fulgidus has yet to be identified, it is expected that one or both of the Orc/Cdc6 homologues will bind to origin DNA with some specificity. However, in the absence of any sequence information for specific binding sites in an origin, DNA association was assayed upon the previously described oligonucleotide substrates. Figure 8A shows the binding of AF0244 to a bubble containing substrate (B20). AF0244 bound the bubble to produce a set of discrete bands, which were independent of the presence of ATP in the reaction. The addition of Mg2+ appeared to produce more of the upper complex compared with the lanes without Mg2+, although the effect is not very great.
Figure 8.
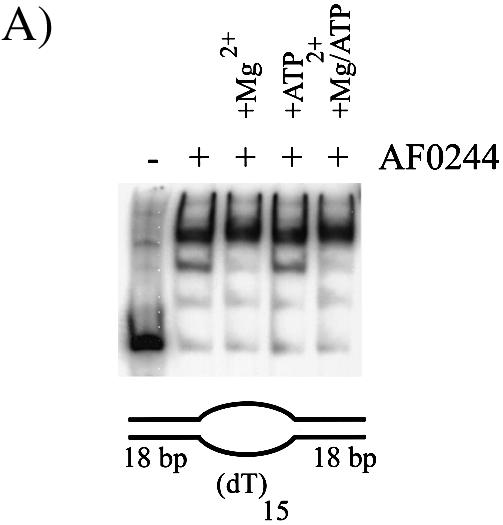

(A) Binding of AF0244 (AF0244) to a bubble DNA substrate. Binding was carried out with 200 nM AF0244 (monomer) and was also supplemented with 10 mM MgCl2, 5 mM ATP or both as indicated. (B) Schematic representation of the preferred binding substrates for AF0244. The ssDNA or 3′ tailed duplex are bound with roughly an order of magnitude lower affinity than the 5′ tailed duplex, and almost no binding was detectable for blunt duplex DNA. The bubble substrate and the flayed duplex are clearly preferred binding substrates for AF0244.
The relative affinity of binding of AF0244 to various oligonucleotide-based substrates is tabulated in Figure 8B. The binding was most efficient for the substrate containing a bubble. Binding to the flayed duplex was only marginally reduced compared with that seen with the bubble. A duplex with a 5′ single stranded tail was also bound by AF0244, albeit with about half of the affinity of the flayed duplex. The other substrates showed significantly weaker binding, with a blunt duplex having at least 1000-fold lower affinity than the bubble. Therefore, it appears that the binding of AF0244 to DNA shows structural specificity, with a preference for a 5′ tail or a fork.
Footprinting of AF0244
Exonuclease III footprinting was carried out on either bubble-containing or flayed duplex substrates bound by AF0244 protein (Fig. 9). A.f Mcm is shown alongside for comparison. The single stranded region of the bubble substrate shown in Figure 7 was dT10 on both strands. In contrast to the bubble shown in Figure 3, when ExoIII was added in the absence of A.f Mcm, the nuclease only degraded the DNA to the border of the expected dsDNA showing that this bubble substrate with a poly dT region was fully single stranded in character. Similarly, on the flayed duplex substrate, degradation by ExoIII stopped at the end of the duplex region as expected. However, when A.f Mcm was present the protection of the last 2–3 bases was still clear on both substrates. A very similar protection pattern was observed upon addition of AF0244 suggesting that the two proteins occupy similar positions on the DNA, namely at the border of the ds/ssDNA. It appears that the AF0244 may protect a couple of bases less than A.f Mcm from digestion by exonuclease III, although there is also somewhat more read-through of the nuclease to the single stranded region than seen with A.f Mcm, suggesting either incomplete binding or AF0244 dissociation during the reaction.
In the light of this discovery, we decided to investigate whether both proteins could be accommodated on the same DNA bubbles or whether their binding was mutually exclusive, or even cooperative. As demonstrated previously the addition of Mcm to the bubble produces a very retarded complex (Fig. 6A). However, when AF0244 was mixed with Mcm and added to DNA (Fig. 10), only AF0244 was observed to bind to the DNA. The same result was observed when the two proteins were added sequentially to DNA, in either order. Therefore, rather than observing any loading of Mcm onto DNA, it appears that AF0244 can displace Mcm from DNA and prevent the Mcm from binding. Again this effect was independent of both ATP and Mg2+. The simplest explanation is that both proteins compete for the same binding sites on the DNA, which would be consistent with the nuclease protection studies shown above. However, an alternative possibility is that the AF0244 protein plays a more active role in the displacement of A.f Mcm from DNA.
Figure 10.
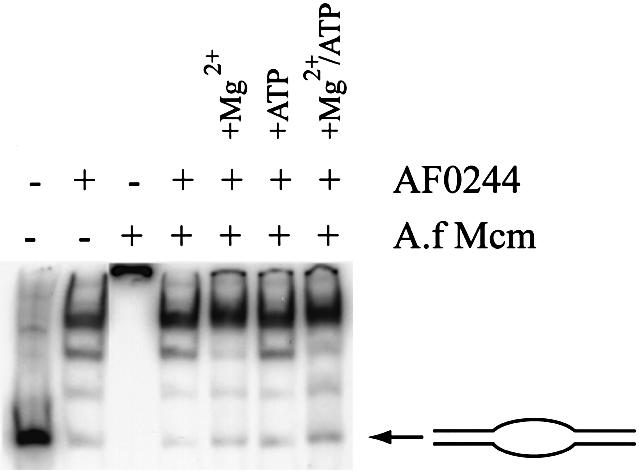
Binding competition between A.f Mcm (400 nM) and AF0244 (200 nM). Note that the presence of AF0244 precludes the binding of Mcm to DNA, and that this is independent of MgCl2 or ATP.
The data we present here open up some interesting possibilities about the activities and interactions between proteins of the Pre-RC in archaea. The unexpected activities of full-length Mcm protein compared with the Δ112 N-terminally truncated enzyme suggest that helicase activity is regulated until the helicase is loaded at an appropriate site. Other helicases show similar properties (e.g. DnaB is activated by binding of primase) (28). This mechanism ensures that helicase activity is directed to appropriate sites at appropriate times since unregulated activity could have disastrous consequences. The DNA binding specificity of Mcm also makes sense with what we understand to be the function of the protein, namely fork extension from a replication bubble. The observation that one of the Orc/Cdc6 homologues of A.fulgidus can displace Mcm from bubble DNA substrates opens up further questions about how Mcm can be loaded at real replication origins. Evidence from yeast suggests that the ORC complex occupies the origin throughout the cell cycle and recruits Cdc6, which then allows association of the MCM complex. It is expected that the archaeal Orc1/Cdc6 homologues will have some sequence specific binding activity to localise them to origin DNA. It is attractive to consider that the structure specific binding of AF0244 reflects a mechanism whereby the Orc1/Cdc6 complex can remain associated with the origin DNA when it is unwound either to load the helicase or while being replicated. However, it is clear that further work remains to be done on the interaction between Mcm and other proteins at the replication fork and/or origins.
Acknowledgments
ACKNOWLEDGEMENTS
We thank N. Raven for providing A.fulgidus cells and S. Sandler for plasmid pSJS1240.
REFERENCES
- 1.Bell S.P. and Dutta,A. (2002) DNA replication in eukaryotic cells. Annu. Rev. Biochem., 71, 333–374. [DOI] [PubMed] [Google Scholar]
- 2.Ogawa Y., Takahashi,T. and Masukata,H. (1999) Association of fission yeast Orp1 and Mcm6 proteins with chromosomal replication origins. Mol. Cell. Biol., 19, 7228–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aparicio O.M., Weinstein,D.M. and Bell,S.P. (1997) Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell, 91, 59–69. [DOI] [PubMed] [Google Scholar]
- 4.Santocanale C. and Diffley,J.F. (1996) ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae. EMBO J., 15, 6671–6679. [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka T., Knapp,D. and Nasmyth,K. (1997) Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell, 90, 649–660. [DOI] [PubMed] [Google Scholar]
- 6.Coleman T.R. Carpenter,P.B. and Dunphy,W.G. (1996) The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell, 87, 53–63. [DOI] [PubMed] [Google Scholar]
- 7.Nishitani H., Lygerou,Z., Nishimoto,T. and Nurse,P. (2000) The Cdt1 protein is required to license DNA for replication in fission yeast. Nature, 404, 625–628. [DOI] [PubMed] [Google Scholar]
- 8.Maiorano D., Moreau,J. and Mechali,M. (2000) XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature, 404, 622–625. [DOI] [PubMed] [Google Scholar]
- 9.Neuwald A.F., Aravind,L. and Spouge,J.L. (1999) AAA+: a class of chaperone-like ATPases associated with the assembly, operation and disassembly of protein complexes. Genome Res., 9, 27–43. [PubMed] [Google Scholar]
- 10.Labib K. and Diffley,J.F. (2001) Is the Mcm2-7 complex the eukaryotic DNA replication fork helicase? Curr. Opin. Genet. Dev., 11, 64–70. [DOI] [PubMed] [Google Scholar]
- 11.Taneja P., Gu,J., Peng,R., Carrick,R., Uchiumi,F., Ott,R.D., Gustafson,E., Podust,V.N. and Fanning,E. (2002) A dominant-negative mutant of human DNA helicase B blocks the onset of chromosomal DNA replication. J. Biol. Chem., 277, 40853–40861. [DOI] [PubMed] [Google Scholar]
- 12.You Z., Komamura,Y. and Ishimi,Y. (1999) Biochemical analysis of the intrinsic Mcm4-Mcm6-mcm7 DNA helicase activity. Mol. Cell. Biol., 19, 8003–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishimi Y. (1997) A DNA helicase activity is associated with an MCM4, –6 and –7 protein complex. J. Biol. Chem., 272, 24508–24513. [DOI] [PubMed] [Google Scholar]
- 14.Lee J.K. and Hurwitz,J. (2001) Processive DNA helicase activity of the minichromosome maintenance proteins 4, 6 and 7 complex requires forked DNA structures. Proc. Natl Acad. Sci. USA, 98, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labib K., Tercero,J.A. and Diffley,J.F. (2000) Uninterrupted MCM2-7 function required for DNA replication fork progression. Science, 288, 1643–1647. [DOI] [PubMed] [Google Scholar]
- 16.Edgell D.R. and Doolittle,W.F. (1997) Archaea and the origin(s) of DNA replication proteins. Cell, 89, 995–998. [DOI] [PubMed] [Google Scholar]
- 17.Bernander R. (1998) Archaea and the cell cycle. Mol. Microbiol., 29, 955–961. [DOI] [PubMed] [Google Scholar]
- 18.Fitz-Gibbon S.T., Ladner,H., Kim,U.J., Stetter,K.O., Simon,M.I. and Miller,J.H. (2002) Genome sequence of the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Proc. Natl Acad. Sci. USA, 99, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen G.N., Barbe,V., Flament,D., Galperin,M., Heilig,R., Lecompte,O., Poch,O., Prieur,D., Querellou,J., Ripp,R., Thierry,J.C., Van der Oost,J., Weissenbach,J., Zivanovic,Y. and Forterre,P. (2003) An integrated analysis of the genome of the hyperthermophilic archaeon Pyrococcus abyssi. Mol. Microbiol., 47, 1495–1512. [DOI] [PubMed] [Google Scholar]
- 20.Robb F.T., Maeder,D.L., Brown,J.R., DiRuggiero,J., Stump,M.D., Yeh,R.K., Weiss,R.B. and Dunn,D.M. (2001). Genomic sequence of hyperthermophile, Pyrococcus furiosus: implications for physiology and enzymology. Methods Enzymol., 330, 134–157. [DOI] [PubMed] [Google Scholar]
- 21.Kawarabayasi Y., Sawada,M., Horikawa,H., Haikawa,Y., Hino,Y., Yamamoto,S., Sekine,M., Baba,S., Kosugi,H., Hosoyama,A., Nagai,Y., Sakai,M., Ogura,K., Otsuka,R., Nakazawa,H., Takamiya,M., Ohfuku,Y., Funahashi,T., Tanaka,T., Kudoh,Y., Yamazaki,J., Kushida,N., Oguchi,A., Aoki,K. and Kikuchi,H. (1998). Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res., 5, 55–76. [DOI] [PubMed] [Google Scholar]
- 22.Chong J.P.J., Hayashi,M.K., Simon,M.N., Xu,R.-M. and Stillman,B. (2000). A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicase. Proc. Natl Acad. Sci. USA, 97, 1530–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelman Z., Lee,J.-K. and Hurwitz,J. (1999). The single minichromosome maintenance protein of Methanobacterium thermoautotrophicum ΔH contains DNA helicase activity. Proc. Natl Acad. Sci. USA, 96, 14783–14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shechter D.F., Ying,C.Y. and Gautier,J. (2000). The intrinsic DNA helicase activity of Methanobacterium thermoautotrophicum delta H minichromosome maintenance protein. J. Biol. Chem., 275, 15049–15059. [DOI] [PubMed] [Google Scholar]
- 25.Smelkova N.V. and Borowiec,J.A. (1998) Synthetic DNA replication bubbles bound and unwound with twofold symmetry by a simian virus 40 T-antigen double hexamer. J. Virol., 72, 8676–8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanzetta P.A., Alvarez,L.J., Reinach,P.S. and Candia,O.A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem., 100, 95–97. [DOI] [PubMed] [Google Scholar]
- 27.Klenk H.P., Clayton,R.A., Tomb,J.F., White,O., Nelson,K.E., Ketchum,K.A., Dodson,R.J., Gwinn,M., Hickey,E.K., Peterson,J.D., Richardson,D.L., Kerlavage,A.R., Graham,D.E., Kyrpides,N.C., Fleischmann,R.D., Quackenbush.J., Lee,N.H., Sutton,G.G., Gill,S., Kirkness,E.F., Dougherty,B.A., McKenney,K., Adams,M.D., Loftus,B., Venter,J.C. et al. (1998) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature, 390, 364–370. [DOI] [PubMed] [Google Scholar]
- 28.Bird L.E., Pan,H., Soultanas,P. and Wigley,D.B. (2000) Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry, 39, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan D.L. and Steitz,T.A. (1999) DnaB from Thermus aquaticus unwinds forked duplex DNA with an asymmetric tail length dependence. J. Biol. Chem., 274, 6889–6897. [DOI] [PubMed] [Google Scholar]
- 30.Fletcher R.J., Bishop,B.E., Leon,R.P., Sclafani,R.A., Ogata,C.M. and Chen,X.S. (2003). The structure and function of MCM from archaeal M. Thermoautotrophicum. Nature Struct. Biol., 10, 160–167. [DOI] [PubMed] [Google Scholar]